Contagious Cancers: A ‘devil’ of a problem
The discovery of a second facial tumor disease in the Tasmanian devil has provided insights into the emergence of contagious cancers.
- United States Department of Agriculture/Agricultural Research Service, United States
- University of New Mexico, United States
Times are tough for the Tasmanian devil – nature's largest carnivorous marsupial. Over the past two decades, a contagious form of cancer called devil facial tumor disease has been wreaking havoc on the Tasmanian devil and may even be driving it to extinction (McCallum and Jones, 2006).
Cancers are not usually contagious, but devil facial tumor disease (DFTD) is, with tumor cells being passed between individual devils, much like viruses or bacteria. The disease is transmitted when devils bite each other during territorial disputes or courtship, making them most susceptible as they reach sexual maturity. Once the tumors are evident, the life expectancy of a devil is less than six months (Hawkins et al., 2006). One consequence of DFTD is that devils are reaching sexual maturity at a younger age, illustrating the strong selective pressure the disease is putting on this species (Jones et al., 2008).
So far, transmissible cancers have been found in just a few species besides the Tasmanian devil – notably dogs and soft-shell clams (Metzger et al., 2015; Murgia et al., 2006). DFTD is remarkable in that two – apparently independent – cancers have arisen in wild devil populations over a relatively short period of time. Tasmanian devils with tumors and lesions in the face caused by the highly contagious DFT1 were first sighted in 1996 (Hawkins et al., 2006); DFT2, which is thought to be less pathogenic, was discovered in 2014 (Pye et al., 2016).
One transmissible cancer in a species may be unfortunate, but two suggest that there is something more going on. Now, in eLife, Hannah Siddle and colleagues – including Alison Caldwell as first author – report that DFT2 may be evolving tactics to evade the immune system that are similar to those used by DFT1 (Caldwell et al., 2018).
The ability to distinguish self from non-self molecules is at the core of immunology, and the underlying mechanisms are generally well established. In the immune system of vertebrates, a cluster of genes called the major histocompatibility complex (MHC) enables the body to recognize and destroy abnormal tissues (such as infected or tumorous tissues) and foreign tissues (like organ transplants; see Schwartz, 1985 for a review).
One group of MHC genes, the class I (MHC-I), encodes molecules that play an important role in this process. Most cells display MHC-I molecules on their surfaces, and these molecules present peptides which reveal if the cell is self or not: if a peptide indicating a mutated-self of pathogenic cell is detected, a specific type of immune cell, called cytotoxic T cell, is alerted and sent to destroy the cell.
However, cancer cells or cells infected with a virus, often escape death at the hands of these T cells by losing their MHC-I molecules, which is one of the mechanisms employed by DFT1 (Siddle et al., 2013). Consequently, when MHC-I expression is reduced or lost, T cells are unable to see the tumor or infected cell. By studying DFT2 cancer cells taken from biopsies or grown in the laboratory, Caldwell et al. – who are based at the universities of Southampton, Tasmania, Cambridge and Southern Denmark – discovered that DFT2 appears to use a similar tactic (Figure 1). While it still has MHC-Is, DFT2 seems to be slowly evolving to express fewer of them on its surface – potentially becoming as contagious as DFT1.
Figure 1
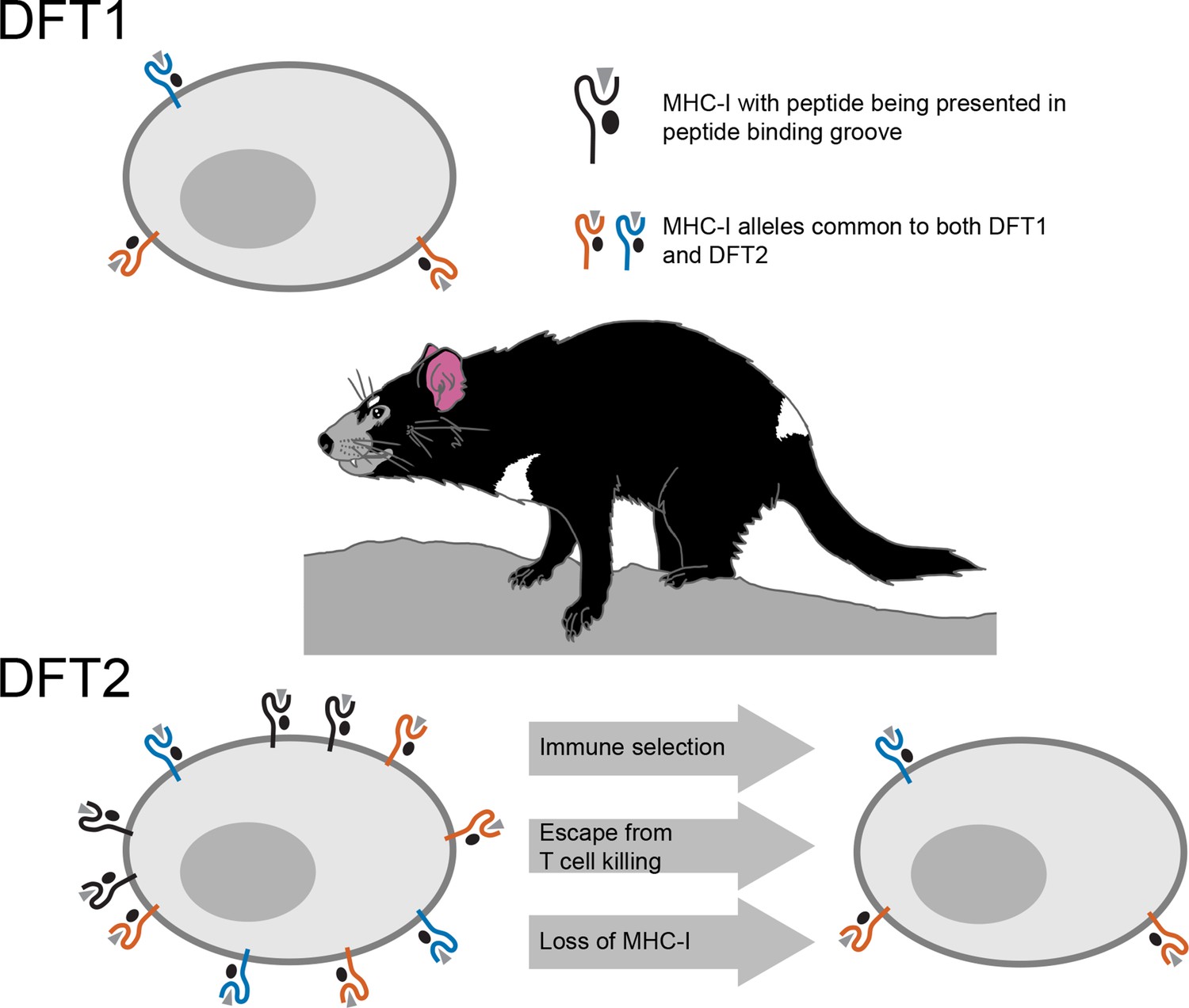
Schematic illustrating the mechanisms underlying devil facial tumor disease.
The immune system identifies cells that should not be in an organism based on foreign or abnormal peptides (grey triangles) presented in the binding groove of MHC-I molecules on the surface of the cell. DFT1 cells (top) avoid the immune system of the Tasmanian devil by losing expression of their MHC-I molecules (orange and blue). DFT2 cells (bottom left) appear to be developing a similar trick, but still express a number of MHC-I molecules (shown in black) that can be recognized by the immune system. However, Caldwell et al. found that DFT2 cells are starting to lose MHC-I molecules (bottom right), which suggests that this form of tumor could soon become as pathogenic as DFT1. DFT1 and DFT2 also appear to share gene alleles of MHC-I genes (orange and blue), suggesting there may be something about these variants associated with the emergence of a contagious tumor.
Usually, the MHC differs between the individuals of a species due to genetic variation. MHC molecules encoded by different gene variants bind and present different sets of peptides. This enhances the killing abilities of T cells by providing a diversity of peptides for T cells to recognize and respond against. This also contributes to the need for genetic matches between an organ donor and a recipient. The MHC-I molecules of DFT2, however, are genetically similar among Tasmanian devil populations, and are therefore less likely to be seen as foreign by T-cells. Caldwell et al. found that DFT1 and DFT2 share MHC-I genes, suggesting that something about these particular variants may be conducive to avoiding the devil immune system.
The emergence of DFT2 provides new insights into the early evolution of transmissible tumors. Nevertheless, many questions persist. It remains to be seen whether these tumors are more common in nature than we assume. If they were indeed so rare, why did they arise twice in the same species? Could specific features of their immune system or their (loss of) environment make the Tasmanian devil more susceptible to the emergence of transmissible tumors?
However, an increasing number of animal species are experiencing both a reduction in number (and, therefore, a reduction in genetic diversity) and a loss of natural habitats. If the emergence of transmissible tumors is dictated by a set of rules rather than plain bad luck, we may discover more cases of contagious cancers that take advantage of common MHC-I alleles. Tasmanian devils and other endangered species may be particularly vulnerable to cancers exploiting loop-holes in their immune systems.
References
Article and author information
Author details
Publication history
- Version of Record published: August 14, 2018 (version 1)
Copyright
© 2018, Hansen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,719
- views
-
- 181
- downloads
-
- 1
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Contagious Cancers: A ‘devil’ of a problem
eLife 7:e39976.
https://doi.org/10.7554/eLife.39976
Further reading
-
Transmissible tumors threaten the existence of the Tasmanian devil.
-
- Cancer Biology
Tyrosine kinase inhibitors (TKI) directed against MET have been recently approved to treat advanced non-small cell lung cancer (NSCLC) harbouring activating MET mutations. This success is the consequence of a long characterization of MET mutations in cancers, which we propose to outline in this review. MET, a receptor tyrosine kinase (RTK), displays in a broad panel of cancers many deregulations liable to promote tumour progression. The first MET mutation was discovered in 1997, in hereditary papillary renal cancer (HPRC), providing the first direct link between MET mutations and cancer development. As in other RTKs, these mutations are located in the kinase domain, leading in most cases to ligand-independent MET activation. In 2014, novel MET mutations were identified in several advanced cancers, including lung cancers. These mutations alter splice sites of exon 14, causing in-frame exon 14 skipping and deletion of a regulatory domain. Because these mutations are not located in the kinase domain, they are original and their mode of action has yet to be fully elucidated. Less than five years after the discovery of such mutations, the efficacy of a MET TKI was evidenced in NSCLC patients displaying MET exon 14 skipping. Yet its use led to a resistance mechanism involving acquisition of novel and already characterized MET mutations. Furthermore, novel somatic MET mutations are constantly being discovered. The challenge is no longer to identify them but to characterize them in order to predict their transforming activity and their sensitivity or resistance to MET TKIs, in order to adapt treatment.




{kind=link}