Bioinformatics: Selecting against accidental RNA interactions
- University of North Carolina, United States
Translation is the process by which the genetic information in a molecule of messenger RNA (mRNA) produces a protein, and the rate at which protein is produced from a given mRNA molecule is called the translation efficiency. This number is different for different mRNA molecules (Maier et al., 2009; Guo et al., 2008), which is why researchers are trying to determine which features of these molecules affect their translation efficiency (Tuller et al., 2010; Ferreira et al., 2013; Kozak, 2005; Gingold and Pilpel, 2011).
Now, in eLife, Paul Gardner of the University of Canterbury and colleagues – including Sinan Umu (as first author), Anthony Poole and Renwick Dobson – report that the translation efficiency in bacteria and archaea is influenced by a phenomenon called "avoidance" (Umu et al., 2016). Avoidance is the degree to which an mRNA molecule avoids random interactions with noncoding RNA molecules in the cell. Noncoding RNAs, as their name suggests, do not code for proteins, but they still make up a majority of the RNA in any given cell. Indeed, the researchers show that the levels of noncoding RNAs in bacterial cells are two orders of magnitude greater than the levels of mRNAs.
To estimate the probability of random base-pairing interactions taking place between mRNAs and noncoding RNAs, consider a five-base region in a single mRNA. This region can have any one of a possible 4∧5=1024 sequences. If the total number of bases from all the noncoding RNAs in the cell is S, then the number of noncoding RNAs in the cell that have a perfectly complementary five-base region is approximately S/1024. Umu et al. studied 325 noncoding RNAs so, assuming an average length of 200 bases for these, we have S ≈ 325*200 ≈ 65000. Therefore, on average, the number of these noncoding RNAs that have a five-base region that is perfectly complementary to the five-base region in the mRNA will be 65000/1024 ≈ 63. Given the number of mRNAs and noncoding RNAs that are found in cells, random interactions between the two are inevitable. However, if we find that a given mRNA has base-pairing interactions with fewer noncoding RNAs than expected, then this is avoidance (Figure 1A).
Figure 1
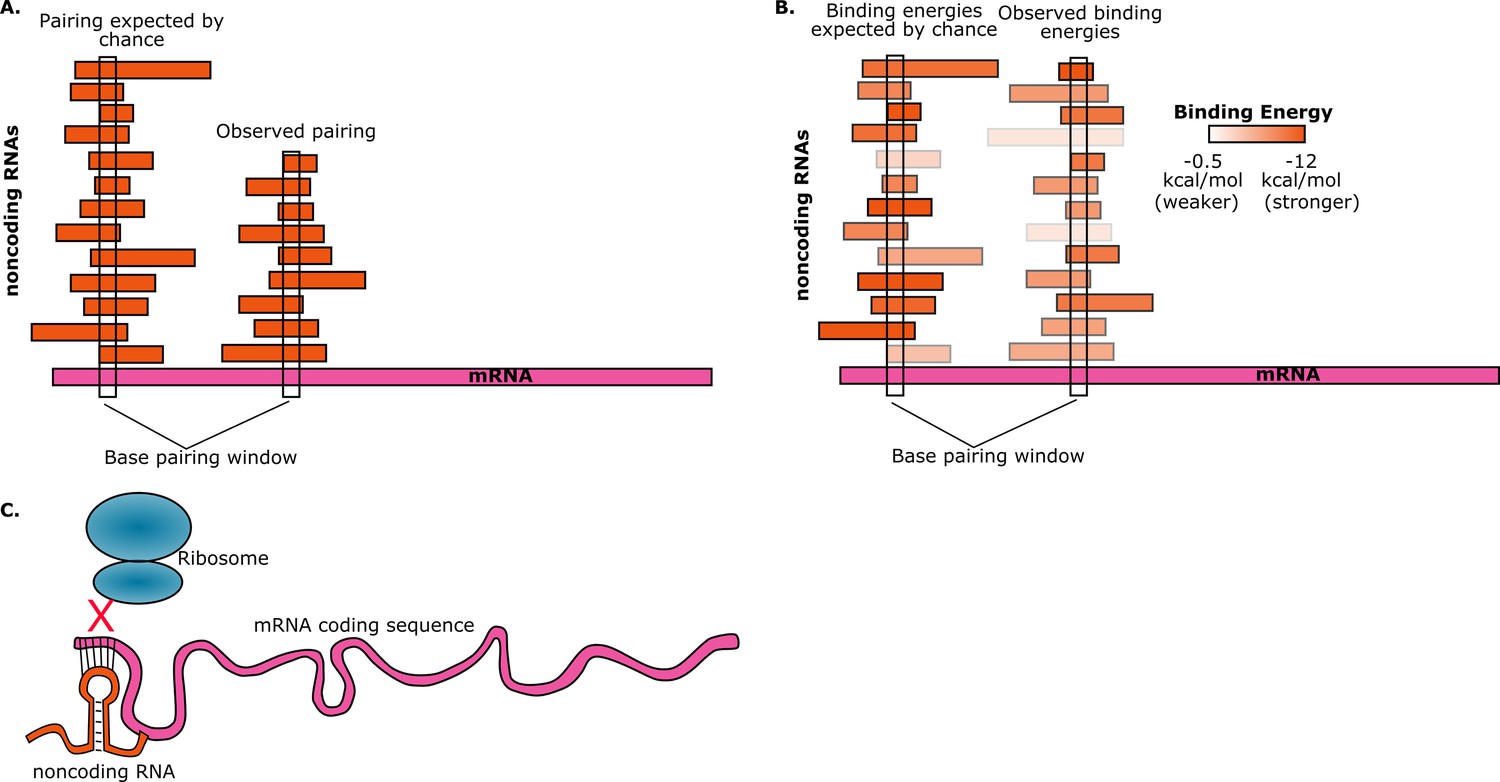
Random interactions between mRNAs and noncoding RNAs.
(A) A given region of mRNA (pink) should undergo random base-pairing interactions with a certain number of noncoding RNAs (orange). However, a phenomenon called "avoidance" results in the number of observed pairings (right) being fewer than the number expected by chance (left). This simplified picture does not allow for the influence of binding energies and other effects. (B) The ability of RNA molecules to fold into complex structures means that mRNA-noncoding RNA interactions can have a range of binding energies (see color bar). "Avoidance" results in the observed binding energies (right) being, on average, weaker than the binding energies expected by chance (left). Umu et al. report a significant difference in binding energy distributions in 73% of bacterial and archeal species. (C) If a random base-pairing interaction results in a noncoding RNA pairing with part or all of a start codon in an mRNA, the ribosome will not be able to translate the mRNA.
Realistically, RNA binding interactions are governed by thermodynamics and do not always follow strict pairing rules. Furthermore, RNA molecules can pair with themselves in intra-molecular interactions and must “unfold” a given region in order to pair with another molecule. Thus it is important to quantify mRNA-noncoding RNA interactions with net "binding energy" calculations. The binding energy quantifies the thermodynamics of RNA base-pairing, with low binding energies indicating very stable interactions. To explore the phenomenon of avoidance, Umu et al. used a computational RNA interaction model to estimate the binding energies for interactions between mRNAs and noncoding RNAs. The RNAs include a core set of 114 mRNAs that are well conserved across bacteria (including 40 that are also conserved across archaea) and 325 noncoding RNAs from six families of RNA that are also well conserved across bacterial and archaeal species.
They found that, on average, the interactions between the core noncoding RNAs and mRNAs were weaker than the interactions between the noncoding RNAs and a control set. In other words, they found that the average mRNA "avoids" interactions due to less stable pairing with noncoding RNAs (Figure 1B). This trend holds true for over 70% of the bacteria and archaea that they tested. There are, of course, noncoding RNAs whose primary function is to bind to mRNAs, but these were excluded from the study. Instead, the goal was to observe selection against accidental interactions between mRNAs and the large and diverse set of noncoding RNAs that are resident in the cell.
Umu et al. hypothesize that avoidance is due to the negative effect that the interactions between mRNAs and noncoding RNA could have on translation efficiency: for example, if a noncoding RNA pairs with a start codon in an mRNA, it will prevent translation from taking place because the ribosome will not be able to bind to that mRNA (Figure 1C). To test this hypothesis, the researchers designed and synthesized a set of mRNAs with sequences that have high levels of avoidance, and a set of mRNAs with low levels of avoidance. When they measured the translation efficiency for both sets of mRNAs, they found that it was much higher for the highly-avoidant set.
Umu et al. also synthesized different sets of mRNAs to explore two other factors that are thought to influence translation efficiency: codon bias and the intra-mRNA folding energy (Kudla et al., 2009; Tuller et al., 2010). Both factors did cause some variation in the production of protein, but avoidance was responsible for the most variation. They also found the same correlation with avoidance when they studied previously published measurements of bacterial translation efficiency. This suggests that the ability of an mRNA to avoid interactions with noncoding RNAs is a hitherto unknown, yet important factor affecting translation efficiency.
One notable aspect of this study is that it relied almost entirely on publicly available data sets. This underscores the importance of open data for exploring basic biological questions that apply to many different organisms. Using this data, which no single lab could have collected alone, Umu et al. have shown that mRNA sequences are optimized to minimize interactions with noncoding RNAs and have demonstrated why such avoidance is so desirable. And the need to avoid spurious interactions is not unique to RNA: the emergence of complex life depends on optimizing molecular interactions that lead to reproduction in the midst of molecular chaos. Although networks of highly specific molecular interactions are a hallmark of evolution, in many cases it is just as important to avoid accidental interactions.
References
-
Determinants of translation efficiency and accuracyMolecular Systems Biology 7:481.https://doi.org/10.1038/msb.2011.14
-
How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytesActa Biochimica Et Biophysica Sinica 40:426–436.https://doi.org/10.1111/j.1745-7270.2008.00418.x
-
Correlation of mRNA and protein in complex biological samplesFEBS Letters 583:3966–3973.https://doi.org/10.1016/j.febslet.2009.10.036
Article and author information
Author details
Publication history
- Version of Record published: September 20, 2016 (version 1)
- Version of Record updated: October 3, 2016 (version 2)
Copyright
© 2016, Corley et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,500
- Page views
-
- 218
- Downloads
-
- 1
- Citations
Article citation count generated by polling the highest count across the following sources: Crossref, PubMed Central, Scopus.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Bioinformatics: Selecting against accidental RNA interactions
eLife 5:e20686.
https://doi.org/10.7554/eLife.20686
Further reading
-
- Computational and Systems Biology
Revealing protein binding sites with other molecules, such as nucleic acids, peptides, or small ligands, sheds light on disease mechanism elucidation and novel drug design. With the explosive growth of proteins in sequence databases, how to accurately and efficiently identify these binding sites from sequences becomes essential. However, current methods mostly rely on expensive multiple sequence alignments or experimental protein structures, limiting their genome-scale applications. Besides, these methods haven’t fully explored the geometry of the protein structures. Here, we propose GPSite, a multi-task network for simultaneously predicting binding residues of DNA, RNA, peptide, protein, ATP, HEM, and metal ions on proteins. GPSite was trained on informative sequence embeddings and predicted structures from protein language models, while comprehensively extracting residual and relational geometric contexts in an end-to-end manner. Experiments demonstrate that GPSite substantially surpasses state-of-the-art sequence-based and structure-based approaches on various benchmark datasets, even when the structures are not well-predicted. The low computational cost of GPSite enables rapid genome-scale binding residue annotations for over 568,000 sequences, providing opportunities to unveil unexplored associations of binding sites with molecular functions, biological processes, and genetic variants. The GPSite webserver and annotation database can be freely accessed at https://bio-web1.nscc-gz.cn/app/GPSite.
-
- Cell Biology
- Computational and Systems Biology
Computer models of the human ventricular cardiomyocyte action potential (AP) have reached a level of detail and maturity that has led to an increasing number of applications in the pharmaceutical sector. However, interfacing the models with experimental data can become a significant computational burden. To mitigate the computational burden, the present study introduces a neural network (NN) that emulates the AP for given maximum conductances of selected ion channels, pumps, and exchangers. Its applicability in pharmacological studies was tested on synthetic and experimental data. The NN emulator potentially enables massive speed-ups compared to regular simulations and the forward problem (find drugged AP for pharmacological parameters defined as scaling factors of control maximum conductances) on synthetic data could be solved with average root-mean-square errors (RMSE) of 0.47 mV in normal APs and of 14.5 mV in abnormal APs exhibiting early afterdepolarizations (72.5% of the emulated APs were alining with the abnormality, and the substantial majority of the remaining APs demonstrated pronounced proximity). This demonstrates not only very fast and mostly very accurate AP emulations but also the capability of accounting for discontinuities, a major advantage over existing emulation strategies. Furthermore, the inverse problem (find pharmacological parameters for control and drugged APs through optimization) on synthetic data could be solved with high accuracy shown by a maximum RMSE of 0.22 in the estimated pharmacological parameters. However, notable mismatches were observed between pharmacological parameters estimated from experimental data and distributions obtained from the Comprehensive in vitro Proarrhythmia Assay initiative. This reveals larger inaccuracies which can be attributed particularly to the fact that small tissue preparations were studied while the emulator was trained on single cardiomyocyte data. Overall, our study highlights the potential of NN emulators as powerful tool for an increased efficiency in future quantitative systems pharmacology studies.




{kind=link}