Disease: Deciphering the triad of infection, immunity and pathology
The factors which drive and control disease progression can be inferred from mathematical models that integrate measures of immune responses, data from tissue sampling and markers of infection dynamics.
- BioQuant (Center for Quantitative Biology) at Heidelberg University, Germany
A fever, a cough, a splitting headache… Being sick often comes with tell-tale signs which worsen as the disease progresses and tissues become damaged. These symptoms result from complex interactions between the infecting pathogen, the inflammation process, and the response from the immune system. Tracking these mechanisms and how they interact, as well as identifying which factors determine when the disease recedes or progresses, is essential for establishing better treatment strategies.
In this effort, a more refined understanding of infection and immune responses has emerged from combining experimental and clinical measurements with mathematical models (Perelson, 2002). However, it is still difficult to link tissue pathology and disease severity with viral load or immune cell counts, which respectively measure the amount of virus and of certain immune actors in the body. Now, in eLife, Amber Smith and colleagues at St. Jude Children’s Research Hospital, the University of Tennessee Health Science Center and the Washington University School of Medicine – including Margaret Myers and Amanda Smith as joing first authors – report how viral infection, counteracting immune responses and lung pathology interact as mice fight off influenza A (Myers et al., 2021).
First, the team tracked how viral load and the number of CD8+ T cells, an important immune actor that helps to clear infected cells, progressed over time. In combination with mathematical models, these measurements allowed Myers et al. to estimate several parameters that reflect the pace at which the virus replicates, the strength of the immune response, and the interactions between these processes. While this had already been achieved in previous studies (e.g. Baccam et al., 2006), Myers et al. also analyzed the anatomy of the lung tissue over time, assessing the damage caused by infection and inflammation as well as how much the organ eventually regenerates.
Then, the team compared these data to values from their mathematical model that described viral load and CD8+ T cells counts, thereby linking viral load dynamics and specific immune responses to disease pathology and severity (Figure 1). In particular, the analysis shed light on how the relative number of immune cells correlates with the level of lung tissue cleared from the virus and, thus, the mice’s ability to recover from infection. These quantitative relationships could help to assess how well the virus is controlled within tissues simply by relying on easily accessible markers that are, for example, present in the blood. This would reduce the need for invasive tissue samples.
Figure 1
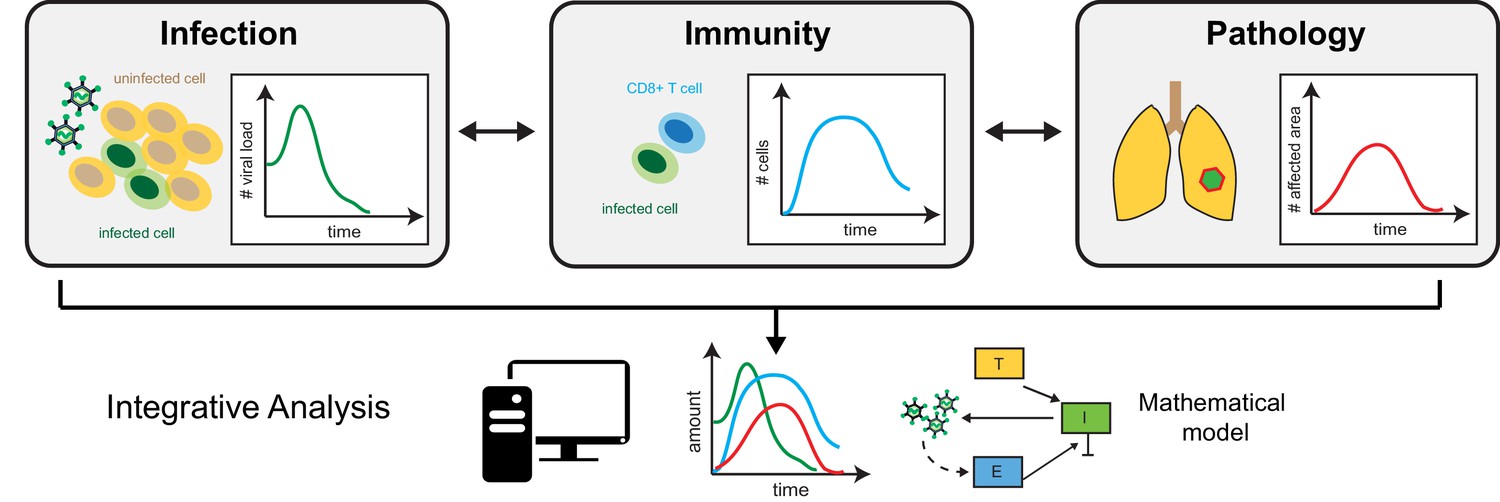
The triad of infection, immunity and disease pathology.
Separate data, such as viral load (left) – the quantity of virus present in a specific volume of fluid – immune cell counts (middle) and histological assessment of tissue sections (right) provide information on the dynamics of infection, immune responses and disease pathology. Mathematical modelling that integrates these measurements makes it possible to assess how the individual processes are connected, and to identify relevant prognostic markers that allow prediction of disease progression.
Individual molecular processes and specific aspects of viral replication can be studied extensively within in vitro cell culture systems. However, the full triad of infection, immunity and especially tissue pathology can only be reliably assessed within conditions that are physiologically relevant (Fackler et al., 2014). Indeed, simple cell culture systems insufficiently address the impact tissue structure can have on infection dynamics, immune activation and clearing mechanisms (Fackler et al., 2014; Imle et al., 2019).
Myers et al. used frequent samples and histological analyses to infer how infected tissues change over time. Yet, imaging technologies may continue to improve so that it becomes possible to observe the interactions between host and pathogen within tissues in real-time (Coombes and Robey, 2010). These approaches could help to investigate whether quantitative relationships as highlighted by Myers et al. also play a role in other infections and in other tissues. The expanding field of organoids – whereby simple, miniature organs are grown in the laboratory – also represents a promising step towards understanding how cells interact within structured, tissue-related environments (Gosselin et al., 2018; Bar-Ephraim et al., 2020). Combined with new technologies such as single-cell sequencing methods (Triana et al., 2021; Youk et al., 2020), these approaches will help to determine the molecular processes that govern disease progression, and how these might differ between patients.
Despite these new experimental and diagnostic technologies, data-driven mathematical modeling and analytical methods will continue to fulfil a key role for deciphering the interplay between infection, tissue pathology and disease severity. Using these models makes it possible to integrate different types of measurements from various places and times, and to disentangle the contributions of individual processes to the infection dynamics. It is only by understanding exactly how individual processes interact over time that scientists will be able to find and validate prognostic markers which predict disease progression.
References
-
Kinetics of influenza A virus infection in humansJournal of Virology 80:7590–7599.https://doi.org/10.1128/JVI.01623-05
-
Organoids in immunological researchNature Reviews Immunology 20:279–293.https://doi.org/10.1038/s41577-019-0248-y
-
Dynamic imaging of host-pathogen interactions in vivoNature Reviews Immunology 10:353–364.https://doi.org/10.1038/nri2746
-
Adding new dimensions: towards an integrative understanding of HIV-1 spreadNature Reviews Microbiology 12:563–574.https://doi.org/10.1038/nrmicro3309
-
Designing natural and synthetic immune tissuesNature Materials 17:484–498.https://doi.org/10.1038/s41563-018-0077-6
-
Modelling viral and immune system dynamicsNature Reviews Immunology 2:28–36.https://doi.org/10.1038/nri700
-
Single‐cell analyses reveal SARS‐CoV‐2 interference with intrinsic immune response in the human gutMolecular Systems Biology 17:e10232.https://doi.org/10.15252/msb.202110232
Article and author information
Author details
Publication history
- Version of Record published: September 1, 2021 (version 1)
Copyright
© 2021, Graw
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 781
- views
-
- 92
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Disease: Deciphering the triad of infection, immunity and pathology
eLife 10:e72379.
https://doi.org/10.7554/eLife.72379
Further reading
-
- Computational and Systems Biology
- Genetics and Genomics
We propose a new framework for human genetic association studies: at each locus, a deep learning model (in this study, Sei) is used to calculate the functional genomic activity score for two haplotypes per individual. This score, defined as the Haplotype Function Score (HFS), replaces the original genotype in association studies. Applying the HFS framework to 14 complex traits in the UK Biobank, we identified 3619 independent HFS–trait associations with a significance of p < 5 × 10−8. Fine-mapping revealed 2699 causal associations, corresponding to a median increase of 63 causal findings per trait compared with single-nucleotide polymorphism (SNP)-based analysis. HFS-based enrichment analysis uncovered 727 pathway–trait associations and 153 tissue–trait associations with strong biological interpretability, including ‘circadian pathway-chronotype’ and ‘arachidonic acid-intelligence’. Lastly, we applied least absolute shrinkage and selection operator (LASSO) regression to integrate HFS prediction score with SNP-based polygenic risk scores, which showed an improvement of 16.1–39.8% in cross-ancestry polygenic prediction. We concluded that HFS is a promising strategy for understanding the genetic basis of human complex traits.
-
- Computational and Systems Biology
Revealing protein binding sites with other molecules, such as nucleic acids, peptides, or small ligands, sheds light on disease mechanism elucidation and novel drug design. With the explosive growth of proteins in sequence databases, how to accurately and efficiently identify these binding sites from sequences becomes essential. However, current methods mostly rely on expensive multiple sequence alignments or experimental protein structures, limiting their genome-scale applications. Besides, these methods haven’t fully explored the geometry of the protein structures. Here, we propose GPSite, a multi-task network for simultaneously predicting binding residues of DNA, RNA, peptide, protein, ATP, HEM, and metal ions on proteins. GPSite was trained on informative sequence embeddings and predicted structures from protein language models, while comprehensively extracting residual and relational geometric contexts in an end-to-end manner. Experiments demonstrate that GPSite substantially surpasses state-of-the-art sequence-based and structure-based approaches on various benchmark datasets, even when the structures are not well-predicted. The low computational cost of GPSite enables rapid genome-scale binding residue annotations for over 568,000 sequences, providing opportunities to unveil unexplored associations of binding sites with molecular functions, biological processes, and genetic variants. The GPSite webserver and annotation database can be freely accessed at https://bio-web1.nscc-gz.cn/app/GPSite.




{kind=link}