The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering
- University of California, Berkeley, United States
Abstract
Cohesin tethers together regions of DNA, thereby mediating higher order chromatin organization that is critical for sister chromatid cohesion, DNA repair and transcriptional regulation. Cohesin contains a heterodimeric ATP-binding Cassette (ABC) ATPase comprised of Smc1 and Smc3 ATPase active sites. These ATPases are required for cohesin to bind DNA. Cohesin’s DNA binding activity is also promoted by the Eco1 acetyltransferase and inhibited by Wpl1. Recently we showed that after cohesin stably binds DNA, a second step is required for DNA tethering. This second step is also controlled by Eco1 acetylation. Here, we use genetic and biochemical analyses to show that this second DNA tethering step is regulated by cohesin ATPase. Furthermore, our results also suggest that Eco1 promotes cohesion by modulating the ATPase cycle of DNA-bound cohesin in a state that is permissive for DNA tethering and refractory to Wpl1 inhibition.
https://doi.org/10.7554/eLife.11315.001eLife digest
The bulk of the genetic material in cells from yeast to humans is organized into chromosomes. These chromosomes must be duplicated and the copies need to be segregated every time cells divide. Cohesin is a protein complex that helps to organize the structure of chromosomes by tethering together two regions of DNA, either within a chromosome or between chromosomes. Problems with cohesin have been linked to cancer and birth defects, but it is not clear how cohesin binds DNA and how it makes a tether between two DNA regions. It is also unclear how cohesin’s activity is coordinated with the series of events that allow cells to divide (known as the cell cycle).
Cohesin has two active sites that can break down molecules of ATP. Previous research had suggested that these active sites (called ATPases) controlled cohesin’s activity by regulating whether or not it could bind to DNA. However, Çamdere et al. now reveal that cohesin’s ATPases do not simply provide an ‘on/off switch’ for DNA binding. The experiments, which involved a combination of genetic, cell biology and biochemical techniques in budding yeast, instead revealed that one of cohesin’s ATPases regulates structural rearrangements in cohesin that is already bound to DNA. These structural rearrangements fine-tune the complex’s ability to tether two regions of DNA.
Further experiments then revealed that two cohesin regulators (namely Eco1 and Wpl1) altered this ATPase active site to control cohesin’s DNA tethering and DNA binding activities. These findings provide a molecular explanation for how these regulators control cohesin’s activity to make sure that the chromosomes have the correct structure during cell division.
The next challenge is to identify the structural changes in cohesin that are triggered by cohesin’s two ATPases and to understand how these structural changes promote DNA binding followed by DNA tethering.
https://doi.org/10.7554/eLife.11315.002Introduction
Multi-subunit protein complexes called Structural Maintenance of Chromosomes (SMC) mediate most aspects of higher-order chromosome organization (Hirano, 2006). Cohesin is an SMC complex that was first identified as being essential for sister chromatid cohesion, the process of holding together the sister chromatids from their synthesis in S phase until their segregation in anaphase (Guacci et al., 1997; Michaelis et al., 1997). Cohesin is also involved in mitotic condensation, meiotic chromosome condensation and structure, post-replicative DNA repair, and transcriptional regulation (Onn et al., 2008). Cohesin performs these different functions by tethering together two regions of DNA, either within a single DNA molecule (condensation and regulation of gene expression) or between two DNA molecules (sister chromatid cohesion and DNA repair). How cohesin-mediated DNA tethering is regulated is one of the critical unanswered questions in the field.
The process of sister chromatid cohesion involves the binding of cohesin to chromosomes prior to DNA synthesis, and the subsequent tethering of sister chromatids by the DNA-bound cohesin to generate cohesion during S phase (Onn et al., 2008). Cohesin appears to topologically entrap DNA through its ring-like architecture that results from the assembly of its four core subunits, Smc1, Smc3, Mcd1/Scc1, and Scc3/Irr1 (Nasmyth and Haering, 2009) (Figure 1A). Cohesin’s loading onto DNA is controlled by its ATP Binding Cassette (ABC) ATPase domain (Arumugam et al., 2003; Heidinger-Pauli et al., 2010b; Murayama and Uhlmann, 2014). Cohesin’s ATPase domain is composed of two ATPase active sites each containing four conserved motifs: Walker A, Walker B, signature motif, and D-loop. The Smc1 ATPase active site is a hybrid site containing the Walker A and Walker B motifs encoded by Smc1, and the D-loop and signature motifs encoded by Smc3. Likewise, the Smc3 ATPase active site contains the Smc3-encoded Walker A and Walker B motifs, and Smc1-encoded D-loop and signature motifs (Arumugam et al., 2006; Haering et al., 2004; Hopfner et al., 2000) (Figure 1A inset). Characterization of mutations that block ATP binding (Walker A) or hydrolysis (Walker B) suggest that ATP binding and hydrolysis by both ATPases are necessary for the DNA binding of cohesin (Arumugam et al., 2003; Heidinger-Pauli et al., 2010b). Cohesin’s ATPase activity is also stimulated by the Scc2/Scc4 complex (Murayama and Uhlmann, 2014), which is required for the DNA binding of cohesin in vivo (Ciosk et al., 2000). These observations led to the hypothesis that Scc2/Scc4 complex stimulates ATP hydrolysis to open the cohesin ring and allow the entry of DNA. Subsequent ATP binding closes the ring, entrapping the DNA (Arumugam et al., 2003). Presumably, in order for cohesin to remain stably bound to DNA, its ATPase activity would have to be suppressed to prevent ring reopening and DNA escape. However, the ATPase activity of cohesin in its stable DNA-bound form had never been determined.
Figure 1 with 4 supplements see all
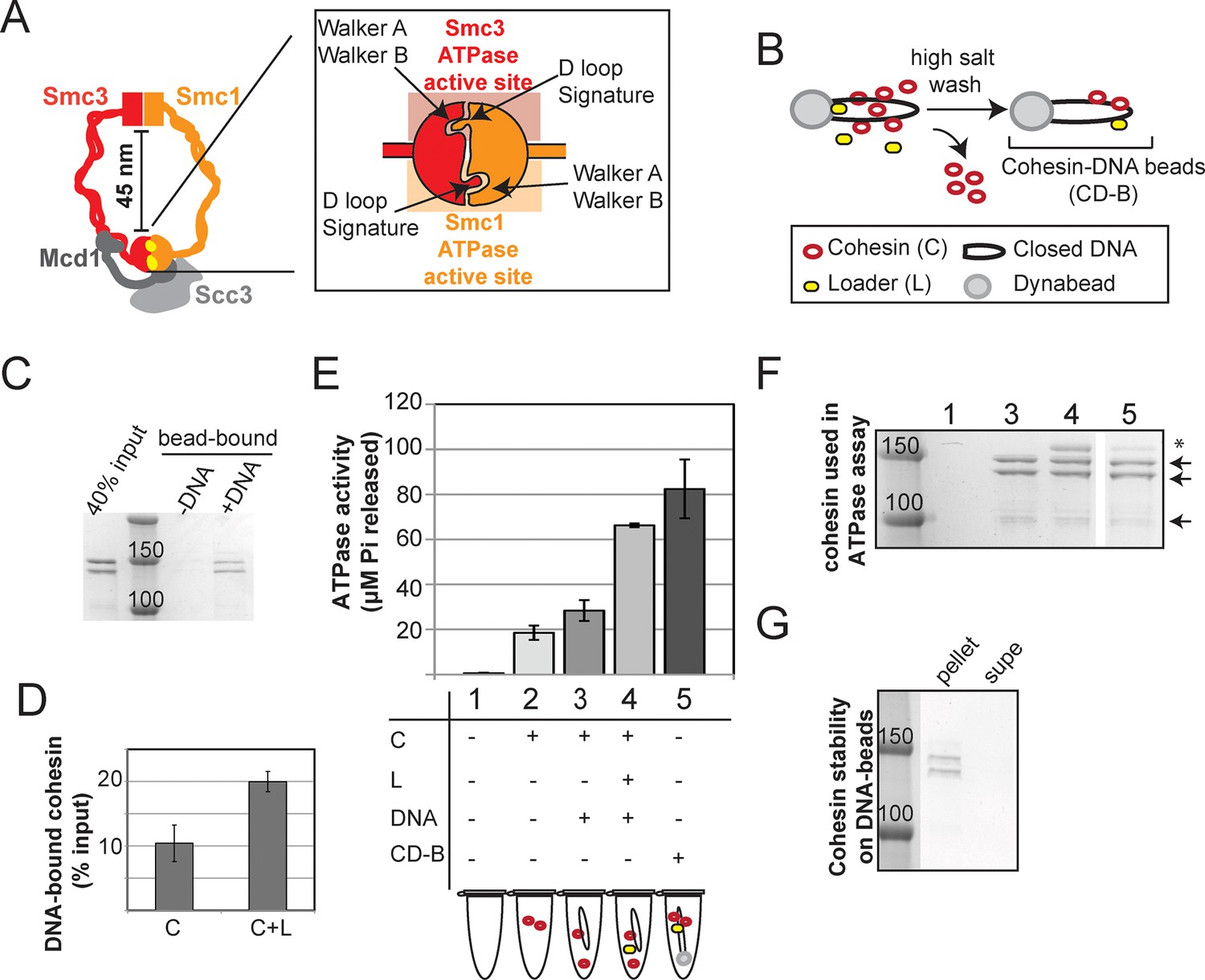
Stable DNA binding does not suppress ATPase activity of cohesin.
(A) Cartoon representation of the cohesin complex. Inset shows a model of the ABC ATPase domain of cohesin complex, based on the X-ray crystal structure of Rad50 in the presence of ATP (Hopfner et al., 2000), and of Smc1 head domain in the presence of ATP and Mcd1 C-terminus (Haering et al., 2004). This model has not been experimentally verified for the cohesin complex heterodimeric ATPase head domain. (B) Schematic showing in vitro assembly of stable cohesin-DNA bead complexes. DNA bearing CARC1 sequence was attached to dynabeads via biotin-streptavidin interaction at both ends. Cohesin was incubated with bead-bound DNA and loader in buffer containing 25 mM KCl and 25 mM NaCl, then washed in 500 mM KCl to wash off salt-sensitive cohesin. The remaining DNA-bound cohesin (and small amount of loader) is referred to as cohesin-DNA-beads (CD-B). (C) Cohesin assembly on DNA-beads. S. pombe cohesin and loader complexes were purified from Y4443 and Y4483, respectively. Purified cohesin and loader were incubated with dynabeads-DNA or dynabeads alone for 1 hour at 30°C, then cohesin was washed off as described in B. Cohesin bound DNA-beads (DNA) but failed to bind beads lacking DNA (-). (D) Cohesin binding to DNA is stimulated by the loader complex. DNA binding was done as described in B & C, except loader was omitted in one sample. Percent cohesin bound was calculated by quantifying bands on Coomassie-stained SDS-PAGE. Data from two independent experiments, error bars represent standard deviation. (E) Effect of stable DNA binding on cohesin ATPase activity. ATPase activity of cohesin alone (2) was compared to cohesin with DNA (3), cohesin in presence of loader complex and DNA (4), and cohesin in stable cohesin-DNA complexes (CD-B, 5). Proteins were incubated in ATPase buffer 2 spiked with hot ATP for 1 hour at 30°C. Released Pi was calculated and plotted as described in Methods. Error bars represent standard deviation from two independent experiments. (F) Equal concentrations of cohesin were used in the ATPase reactions. Arrows point to S. pombe homologs of cohesin subunits, Smc1, Smc3 (Psm1 and Psm3 in S. pombe, ~150 kD), Mcd1 and Scc3 (Rad21 and Psc3 in S. pombe, ~100 kD). Asterisks mark subunits of the S. pombe homolog of the loader complex, Scc2/Scc4 (Mis4/Ssl3 in S. pombe). Due to the lower ability of the loader complex to bind to DNA under these conditions, there is less loader complex present in sample 5 than in sample 4. (G) Stably DNA bound cohesin remained bound to DNA-beads throughout the course of the ATPase experiment. Cohesin was incubated with DNA-beads in the presence of loader and ATP as described before and washed in 0.5 M KCl, then resuspended in ATPase buffer. Samples were incubated for 1 hour at 30°C. Supernatant and beads were separated and visualized on SDS-PAGE. Protein gels are representative of at least 2 independent experiments. Bands in some panels were spliced from the same gel for representation purposes. Please see Figure supplements 1–4 for further characterization of cohesin’s ATPase activity while stably bound to DNA.
Cohesin binding to DNA is also regulated by the antagonistic activites of Wpl1 and Eco1. Wpl1 is thought to destabilize cohesin binding to DNA (Chan et al., 2012; Kueng et al., 2006). Wpl1 is antagonized by the Eco1 acetyl-transferase through its acetylation of lysines 112 and 113 of Smc3. The key evidence supporting this view is that the deletion of WPL1 (wpl1∆) suppresses the inviability of cells lacking ECO1 (eco1∆) or expressing acetyl null alleles of Smc3 (smc3-K112R, K113R) (Rolef Ben-Shahar et al., 2008; Sutani et al., 2009; Unal et al., 2008; Zhang et al., 2008).
Recent evidence suggests that Eco1 acetylation promotes cohesion by additional mechanisms besides stabilizing cohesin binding to DNA. First, while eco1∆ wpl1∆ cells are viable and have stable cohesin-DNA interaction, they have cohesion defects as severe as mutants of ECO1 or cohesin subunits (Guacci and Koshland, 2012; Rowland et al., 2009; Sutani et al., 2009). Second, other mutations identified in cohesin and its regulators demonstrate that stable binding of cohesin to DNA is not sufficient for cohesion (Eng et al., 2014; Guacci et al., 2015). Together, these data strongly argue that cohesion is a two-step process: First, cohesin associates with DNA in a stable form. Then, cohesin undergoes a second transition to tether sister chromatids together. This transition could entail conformational changes involving oligomerization (Eng et al., 2015), or the activation of a second, independent DNA binding activity through rearrangements of the coiled coils (Soh et al., 2015).
How is cohesin-mediated DNA tethering regulated? One hypothesis is that Eco1-mediated acetylation of Smc3 regulates this second, post-DNA binding step by modulating the cohesin ATPase (Guacci et al., 2015). This hypothesis appears to contradict the finding that Walker A and Walker B mutations in either cohesin ATPase blocks DNA binding (Arumugam et al., 2003; Heidinger-Pauli et al., 2010b). However, this observation does not preclude a specialized role for the Smc3 ATPase active site in regulating DNA tethering after DNA binding. Indeed, the acetylated K112 and K113 residues in Smc3 are proximal to the Smc3 ATPase active site (Gligoris et al., 2014; Haering et al., 2004). Moreover, a recently identified suppressor mutation located near the Smc3 ATPase active site bypasses the requirement for Smc3 acetylation in cohesion establishment (Guacci et al., 2015). Led by these observations, we reconsider the role of the ATPase domain of cohesin as a potential regulator of the second, post-DNA binding step of cohesion establishment.
Here, we present in vitro and in vivo evidence that the ATPase domain of cohesin plays a role after the initial stable DNA binding of cohesin. We provide evidence suggesting that the Smc1 ATPase active site is involved only in regulating DNA binding, whereas the Smc3 ATPase active site functions in DNA tethering as well as DNA binding. We characterize an Smc3 ATPase active site mutant in Saccharomyces cerevisiae that bypasses the requirement for Eco1 acetylation in cohesion generation, and uncouples the level of ATPase activity from cohesin’s DNA binding and tethering activities. We propose that cohesin’s ATPase has two distinct functions in regulating DNA binding and subsequent DNA tethering. We suggest that Eco1 promotes cohesion by slowing or trapping the ATPase cycle of DNA-bound cohesin to promote a conformation that is permissive for DNA tethering and refractory to Wpl1 inhibition.
Results
Cohesin that is stably bound to DNA retains its ATPase activity
Earlier models suggest that cohesin’s ATPase head domain is only involved in the initial DNA binding step, and that ATP hydrolysis releases the DNA from cohesin. These models predict that stably DNA-bound cohesin should not show ATPase activity. However, recent literature suggests that Eco1 might promote cohesion by regulating the cohesin ATPase after the stable DNA binding of cohesin. If ATPase activity is required to regulate this second step of cohesion establishment, we should be able to observe ATPase activity for purified cohesin-DNA complexes. To test this possibility, we purified Schizosaccharomyces pombe cohesin and loader complexes as described previously (Murayama and Uhlmann, 2014). Purified cohesin showed basal ATPase activity that was stimulated by the presence of the loader complex and DNA, and abolished by a Walker A mutation in Smc3 (Figure 1—figure supplement 1), similar to published results (Murayama and Uhlmann, 2014). Cohesin binding to chromosomes in vivo is enriched at centromeres and at cohesion-associated regions (CARs) along the chromosome arms (Laloraya et al., 2000; Onn et al., 2008), and is highly salt resistant (Ciosk et al., 2000). To purify stable cohesin-DNA complexes that are physiologically relevant, we assembled cohesin on DNA molecules that contained CARC1 sequence and were coupled by both ends to dynabeads. Cohesin and its loader were incubated with DNA-beads under low salt conditions (25 mM KCl, 25 mM NaCl). The cohesin-DNA bead mix was washed with high salt (500 mM KCl) to remove any free cohesin or cohesin not stably bound to DNA (Figure 1B). The cohesin that remained bound to the DNA-beads was then eluted and quantified by Coomassie staining or Western blots. In the presence of the loader, 20% of the input cohesin was bound to DNA-beads after the high salt wash (Figure 1C,D). In the absence of the loader, 2-fold less cohesin bound to DNA (Figure 1D). Cohesin did not bind to beads that lack DNA (Figure 1C). In addition, this stable population of cohesin on DNA-beads could be eluted from the beads by either a restriction enzyme digest or a DNase treatment (Figure 1—figure supplement 2). These results suggest that cohesin bound specifically to the DNA that was coupled to beads, and did so in a salt-resistant and loader-inducible manner. These DNA binding features recapitulated the properties of stable cohesin-DNA complexes assembled in vivo (Ciosk et al., 2000).
To address whether stably DNA-bound cohesin was an active ATPase, we next assessed whether cohesin in our purified, stable cohesin-DNA complexes (CD-B, Figure 1B) retained its ATPase activity. DNA-beads were incubated with cohesin and loader, then washed with high salt buffer to remove free and unstably bound cohesin, as described above. Cohesin-DNA beads (CD-B) were then resuspended in buffer containing ATP to measure ATPase activity of CD-B, compared to basal and loader/DNA-stimulated activities of cohesin. No ATPase activity was detected when cohesin was omitted from the reaction (Figure 1E and Figure 1—figure supplement 1). As shown before (Murayama and Uhlmann, 2014), cohesin’s basal ATPase activity (sample 2) was stimulated about 3-fold by the presence of loader and DNA (sample 4), but only subtly increased by DNA alone (sample 3, Figure 1E). Our purified cohesin-DNA complexes on beads (CD-B, sample 5) showed about 4-fold increased ATPase activity compared to the basal activity of equal amounts of cohesin (sample 2, Figure 1E,F). Furthermore, the ATPase activity of CD-B was at least as high, if not higher, as the activity of equal concentrations of loader/DNA-stimulated cohesin in solution, where additional rounds of cohesin loading were possible (sample 4 compared to sample 5, Figure 1E,F).
The ATPase activity in the fraction containing purified cohesin-DNA complexes might have been derived from DNA-free cohesin that had dissociated from DNA-beads. To test this possibility, a parallel sample of CD-B was treated similarly to the ATPase assay to assess the amount of cohesin that remained bound to DNA through the course of the ATPase experiment. DNA-beads were separated from the supernatant after the 30-minute incubation with ATP and the amount of cohesin in each fraction was visualized by SDS-PAGE. Very little, if any, cohesin was found in the supernatant at the end of the incubation, suggesting that most of the ATPase activity was derived from cohesin bound to the DNA-beads (Figure 1G). Even in the presence of excess competitor DNA in solution, cohesin remained stably bound to the DNA-beads (Figure 1—figure supplement 3). Thus in vitro, cohesin can have robust ATPase activity while remaining stably bound to DNA.
In our preparations of purified cohesin-DNA complexes, a small amount of the loader complex was also present, due to the ability of the loader complex to bind DNA. Given that the loader complex stimulates cohesin’s ATPase activity in the presence of DNA, it was possible that the ATPase activity in our purified cohesin-DNA complexes depended upon the presence of the loader complex. To test this possibility, we repeated these experiments but omitted the loader from the binding reaction. Under these conditions, a smaller percentage of cohesin could be assembled on DNA in a salt-resistant manner (Figure 1D and Figure 1—figure supplement 4). Cohesin in these purified cohesin-DNA complexes that have no co-purifying loader complex also retained ATPase activity equivalent to that seen in the mixture of cohesin and DNA in solution (Figure 1—figure supplement 4). Thus, the persistence of ATPase activity in stable cohesin-DNA complexes was not dependent upon the loader. The presence of cohesin’s ATPase activity in the stable cohesin-DNA complex is consistent with a regulatory role for cohesin ATPase in cohesion establishment in vivo after cohesin stably binds to DNA.
Mutations proximal to the Smc3 ATPase active site bypass the requirement for Eco1 in cohesion establishment
Cohesin exhibited robust ATPase activity after stably binding DNA, which encouraged us to re-examine the in vivo role of cohesin ATPases and Eco1 in cohesin function. Cohesin-DNA complexes in eco1∆ wpl1∆ cells are stably bound to DNA but fail to establish cohesion, indicating a post-DNA binding step is required for cohesion. We postulated that this failure to generate cohesion was due to a misregulation of the cohesin ATPase active sites. If so, a subset of mutations altering the Smc1 or Smc3 ATPase active sites might mimic the state of the ATPase upon Eco1 acetylation, and thereby might suppress the cohesion defect of eco1∆ wpl1∆ cells.
To identify such suppressors, we exploited the fact that eco1∆ wpl1∆ cells remain viable in the absence of cohesion because of an unusual feature of budding yeast cell cycle that gives rise to a cohesin-independent mechanism of sister chromatid segregation (Guacci and Koshland, 2012). The reliance of eco1∆ wpl1∆ cells on this cohesin-independent segregation makes them sensitive to the microtubule depolymerizing drug benomyl. A screen for suppressors of the benomyl sensitivity of eco1∆ wpl1∆ cells should identify a subset of mutations that restore cohesion and the cohesin-dependent benomyl-resistant mechanism of segregation. Indeed, we recently reported one such benomyl-resistant suppressor allele, smc3-D1189H (Guacci et al., 2015), that restored cohesion. smc3-D1189H is located in the ATPase head domain, near the Smc3 ATPase active site. Due to this restoration of cohesion to eco1∆ wpl1∆ cells, we termed this mutation a cohesion activator mutation.
Here, we present two new SMC1 alleles from our screen, smc1-D1164E and smc1-Y1128C. Re-introduction of these mutations into the parent eco1∆ wpl1∆background generated benomyl-resistance identical to the initial suppressor eco1∆ wpl1∆ isolates and close to the resistance of wild type cells (Figure 2A), demonstrating their causal role for this phenotype. The SMC1 suppressor mutations are even more proximal to the Smc3 ATPase active site than smc3-D1189H (Figure 2B). smc1-Y1128C is adjacent to the signature motif, which is thought to modulate ATP binding, but this residue is not conserved. smc1-D1164E alters the invariant aspartate that is part of the conserved D-loop motif found in ABC and SMC ATPases (Figure 2C). Studies from other ABC ATPases suggest that this aspartate interacts with the catalytic H loop and Walker A (Hohl et al., 2012; la Rosa and Nelson, 2011), and mediates communications between the two ATPase active sites within the ATPase head domain (Furman et al., 2013). Taken together, it is likely that these suppressor mutations modulate the cohesin ATPase to promote cohesion.
Figure 2
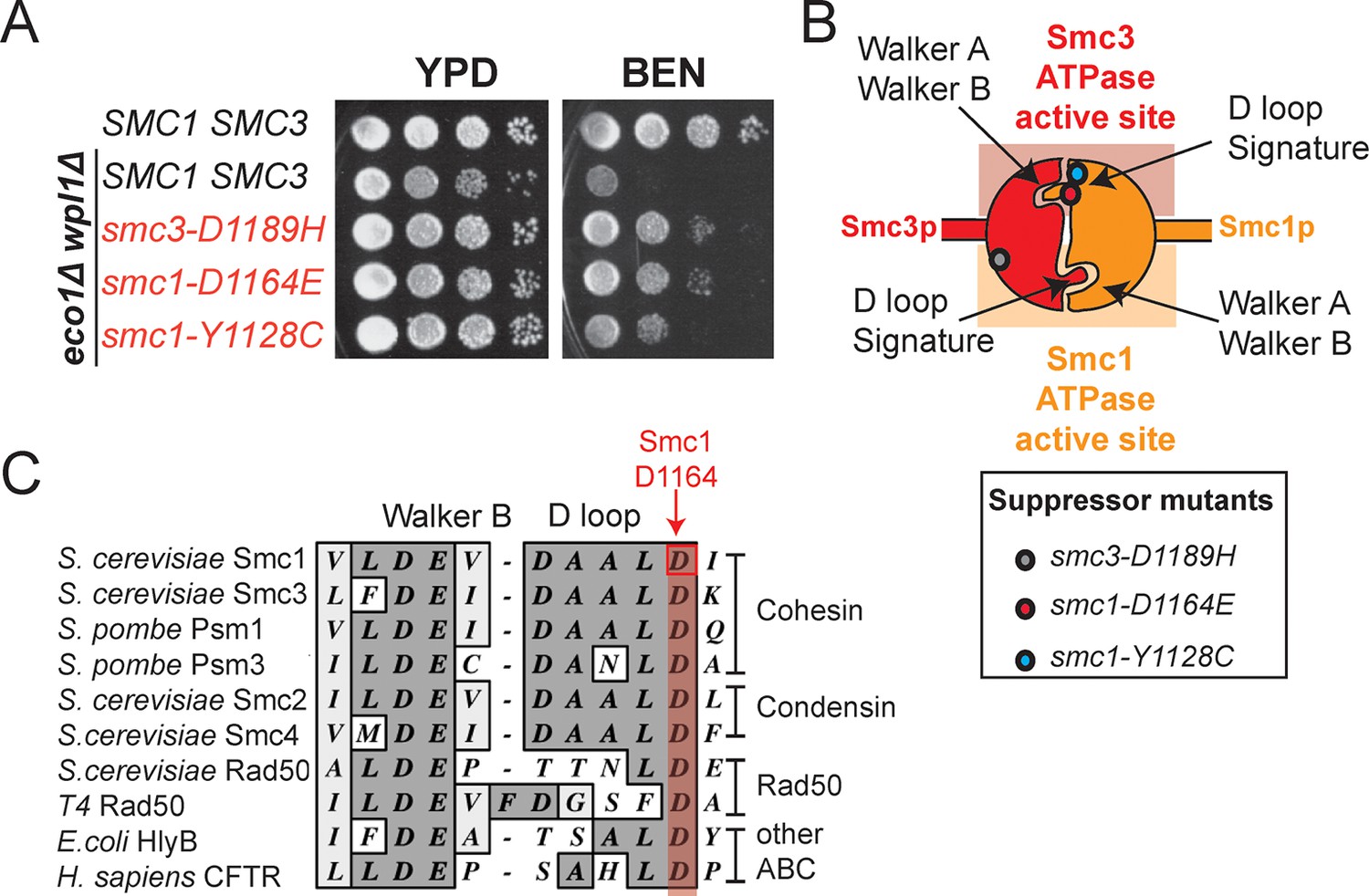
smc1-D1164E and smc1-Y1128C mutations suppress the benomyl sensitivity of eco1△ wpl1△ cells and are part of the Smc3 ATPase active site.
(A) Assessing effects of smc1-D1164E and smc1-Y1128C on eco1∆ wpl1∆ benomyl sensitivity. Haploid wild-type cells (VG3460-2A), and eco1∆ wpl1∆ cells alone (VG3502-1A) or containing smc1-D1164E (VG3574-5A), smc1-Y1128C (VG3576-1C) or smc3-D1189H (VG3547-3B) were grown to saturation in YPD at 23°C, plated as 10-fold serial dilutions on YPD alone, or containing benomyl at 12.5 μg/mL (BEN) then incubated at 23°C for 3 days. (B) Cartoon depicting the Smc1 and Smc3 ATPase active sites along with the position of three suppressor mutations shown in A. All three suppressor mutations are in the vicinity of the Smc3 ATPase active site. Note that the Smc1 D-loop and signature motifs form part of the Smc3 ATPase active site. (C) The conservation of residues around the D-loop in distant ABC ATPases.
To begin to test this hypothesis, we first asked whether smc1-D1164E or smc1-Y1128C were also cohesion activator mutations, i.e. suppressed the severe cohesion defect of eco1∆ wpl1∆ cells. Haploid wild type cells and eco1∆ wpl1∆ parent cells containing SMC1, smc1-D1164E or smc1-Y1128C alleles were arrested in G1, then released into media containing nocodazole to arrest them in M phase (Figure 3A). To assay for cohesion, these cells contained a LacO array integrated at a CEN-proximal locus (TRP1), and a GFP-LacI fusion protein that bound to the array. In this assay, sister chromatids that have cohesion generate a single GFP spot, whereas chromatids lacking cohesion generate 2 GFP spots (Figure 3A). Wild type cells had robust cohesion as shown by few M phase cells with two GFP spots (Figure 3B and Figure 3—figure supplement 1A). eco1∆ wpl1∆ cells exhibited a severe cohesion defect (more than 60% of cells had two GFP spots), which was reduced by half by the smc1-D1164E suppressor (Figure 3B and Figure 3—figure supplement 1A). The smc1-Y1128C suppressor also reduced the eco1∆ wpl1∆ cohesion defect, but less effectively (Figure 3—figure supplement 2). Thus, both of these new suppressor mutations, like smc3-D1189H, suppressed significantly but not completely the cohesion defect of eco1∆ wpl1∆ cells. The increased CEN-proximal cohesion generated by smc1-D1164E or smc1-Y1128C was likely responsible for the increased benomyl resistance in the eco1∆ wpl1∆ background. Subsequent studies focused on smc1-D1164E, because its alters a critical and absolutely conserved D-loop aspartate, and it more robustly suppresses the cohesion defect of eco1∆ wpl1∆ cells.
Figure 3 with 6 supplements see all

smc1-D1164E allele restores cohesion in the absence of both Eco1 and Wpl1.
(A) Regimen used to assess sister chromatid cohesion in cells. Mid-log phase cultures of asynchronously growing cells at 23°C were arrested in G1 with alpha factor for 3 hours, then released into media containing nocodazole for 3 hours to arrest cells in M phase. Representative images of M phase arrested cells are shown with cells being visualized by Nomarski (NOM) and cohesion (GFP), which marks a CEN-proximal TRP1 locus. Left side shows a cell where cohesion exists (one GFP spot.) Right side shows a cell where sisters have separated (2 GFP spots). (B,C) smc1-D1164E partially restored cohesion in eco1△ wpl1△ cells at the CEN-proximal TRP1 locus. Haploid wild type (WT, VG3460-2A), eco1∆ wpl1∆ (VG3502-2A) and smc1-D1164E eco1∆ wpl1∆ (VG3574-5A) cells were released from G1 and arrested in M phase using nocodazole as described in A. The percentage of cells with two GFP spots was plotted. (B) Cohesion loss at a CEN-proximal locus (TRP1) in M phase arrested cells. DNA content of these cells is shown in Figure 3—figure supplement 1, panel A. (C) Time course to assess kinetics of cohesion loss at a CEN-proximal locus (TRP1). Cell aliquots were fixed in G1 and at 20-minute intervals after release. Grey box shows S phase (based on DNA content shown in Figure 3—figure supplement 1, panel B). Please see Figure supplements 1–6 for further characterization of cohesin activator mutants.
To better understand the impact of the smc1-D1164E suppressor on cohesion, we monitored cohesion at the CEN-proximal TRP4 locus at 20 minute intervals as cells progressed from G1 to M (Figure 3—figure supplement 1B). As expected, wild type cells showed robust cohesion where very few cells had separated sisters, and eco1∆ wpl1∆ cells showed sister chromatid separation starting in S phase (Figure 3C), consistent with a cohesion establishment defect that was shown previously for this strain (Guacci and Koshland, 2012; Guacci et al., 2015). The small amount of cohesion loss in smc1-D1164E eco1∆ wpl1∆ cells also began during S phase, indicating that the residual cohesion defect in these cells resulted from a failure in cohesion establishment. However, these cells displayed fewer separated sisters throughout the time course compared to eco1∆ wpl1∆ cells. These results indicated that (1) smc1-D1164E promotes cohesion establishment in the eco1∆ wpl1∆, albeit incompletely, and (2) once cohesion is formed in smc1-D1164E eco1∆ wpl1∆ cells, it is maintained. smc1-D1164E also partially restored cohesion establishment in eco1∆ wpl1∆ cells at the CEN-distal locus, LYS4 (Figure 3—figure supplement 3). We conclude that smc1-D1164E promotes cohesion establishment in the eco1∆ wpl1∆, albeit incompletely.
Since cohesin is already stably bound to DNA in eco1∆ wpl1∆ cells, the restoration of cohesion in smc1-D1164E eco1∆ wpl1∆ cells should occur without altering the DNA binding of cohesin. To test this prediction, we performed chromosome immunoprecipitation (ChIP) to compare cohesin binding to chromosomes in eco1∆ wpl1∆ cells containing SMC1 or smc1-D1164E allele. Cohesin subunits colocalize on chromosomes, making the analysis of any cohesin subunit a surrogate for cohesin binding (Glynn et al., 2004; Heidinger-Pauli et al., 2010a; Lengronne et al., 2004). Here we used anti-Mcd1 antibodies as a means to assess cohesin binding to chromosomes. Cohesin binding at CARs has been shown to be reduced 2-3 fold in eco1∆ wpl1∆ cells as compared to wild type cells (Guacci et al., 2015; Sutani et al., 2009). In eco1∆ wpl1∆ cells containing either SMC1 or smc1-D1164E allele, there was little or no difference in Mcd1 binding at the centromere-proximal CARC1 or centromere-distal CARL1 (Figure 3—figure supplement 4). This similarity in chromosomal binding indicates that cohesion restoration in eco1∆ wpl1∆ cells by the smc1-D1164E allele was not due to an altered level of cohesin binding to chromosomes, but instead was due to a change in cohesin function at a post-DNA binding step.
smc1-D1164E has constitutive but reduced cohesion independent of Eco1 and Wpl1
The restoration of cohesion in smc1-D1164E eco1∆ wpl1∆ cells indicated that the smc1-D1164E could bypass one or more aspects of cohesion regulation normally imposed by Eco1 and Wpl1. To understand better how smc1-D1164E, and by inference the Smc3 ATPase active site, interfaced with cohesin regulators, we examined its impact on the phenotypes of wpl1∆ and eco1∆single mutants. In a wpl1∆ background, SMC1and smc1-D1164E alleles had similar viability and the same moderate cohesion defect characteristic of wpl1∆ mutants (Figure 3—figure supplement 5). The smc1-D1164E allele suppressed the essential ECO1 function, as smc1-D1164E eco1∆ cells showed good viability (Figure 3—figure supplement 6). smc1-D1164E eco1∆ cells showed a 35% cohesion defect (Figure 3—figure supplement 6), which was greatly reduced compared to 65-70% cohesion defect that was observed previously for eco1 mutant cells or cells lacking both Eco1 and Wpl1 (Guacci et al., 2015; Sutani et al., 2009). Taken together, these results suggest that smc1-D1164E restored cohesion by phenocopying both Eco1’s antagonism of Wpl1 and its ability to promote stably DNA-bound cohesin to tether sister chromatids.
The partial restoration of cohesion in smc1-D1164E eco1∆ wpl1∆ cells could reflect the inability of smc1-D1164E to fully compensate for the loss of Eco1 or Wpl1. Alternatively, it might reflect an inherent cost of uncoupling cohesin from its regulators. To differentiate between these possibilities, we characterized the smc1-D1164E allele when both ECO1 and WPL1 were present. In plasmid shuffle assays (Materials and methods), the viability of cells containing the smc1-D1164E or SMC1 alleles was indistinguishable (Figure 4—figure supplement 1). However, otherwise wild type cells carrying the smc1-D1164E allele had a moderate cohesion defect at both CEN-proximal and CEN-distal loci, which arose around the time of replication (Figure 4—figure supplement 2). This defect was similar to that observed for the smc1-D1164E eco1∆ wpl1∆ cells. Thus, cohesin in smc1-D1164E cells had an inherent moderate cohesion establishment defect independent of cohesin regulators. Taken together, our results suggest that the smc1-D1164E allele was constitutively cohesive as it uncoupled cohesin function from its regulators, which reduced the efficiency of cohesion generation.
Analogous D-loop mutations in Smc1 and Smc3 ATPase active sites differentially affect cohesin’s DNA-binding and DNA-tethering activities
Our two new cohesion activator mutations mapped to residues intimately involved in the Smc3 ATPase active site, and the previously characterized third cohesion activator was in close proximity (Guacci et al., 2015). This observation suggested that the Smc3 but not the Smc1 ATPase active site might play a specialized role in activating stably DNA-bound cohesin to tether sister chromatids.
To test more directly whether the two active sites played distinct roles in cohesin regulation, we compared the phenotypes of smc1-D1164E to smc3-D1161E, the analogous glutamate substitution of the D-loop aspartate of the Smc1 ATPase active site (Figure 4A). We used Auxin Inducible Degron (AID) system to obtain conditional alleles of SMC1 and SMC3, SMC1-AID and SMC3-AID, respectively, which can be degraded upon the addition of auxin (Figure 4—figure supplement 3). In strains containing either SMC1-AID or SMC3-AID alleles as sole source of Smc1 or Smc3, we integrated our test alleles, smc1-D1164E and smc3-D1161E, respectively. The smc1-D1164E allele robustly suppressed the inviability associated with depletion of Smc1-AID, whereas smc3-D1161E could not support viability when Smc3-AID was depleted (Figures 4B,C). Similar results in viability were seen when these alleles were tested by plasmid shuffle assays (Figure 4—figure supplement 1 & 4). This difference in viability between the smc1-D1164E and smc3-D1161E alleles suggests that Smc1 and Smc3 ATPase active sites have distinct functions.
Figure 4 with 6 supplements see all
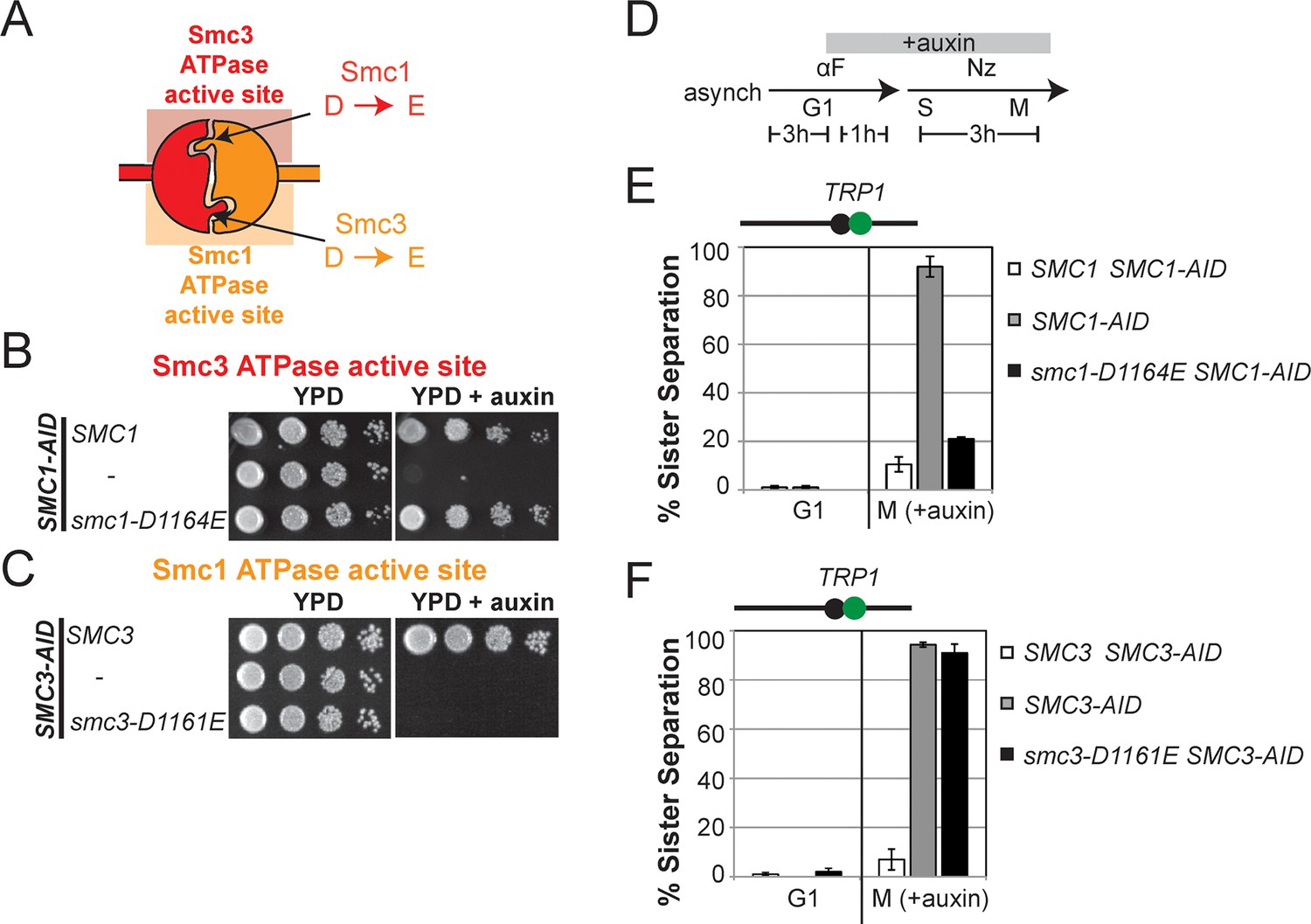
D-loop mutations in Smc1 and Smc3 ATPase active sites reveal that the two sites are not functionally equivalent.
(A) Cartoon representation of cohesin’s ATPases. The Smc1-encoded D-loop is part of the Smc3 ATPase active site and the Smc3-encoded D-loop is part of the Smc1 ATPase active site. (B) Assessing whether the Smc3 ATPase active site D-loop mutant smc1-D1164E promotes cell viability. Haploid SMC1 SMC1-AID (VG3764-3A), SMC1-AID (VG3711-5D), and smc1-D1164E SMC1-AID (VG3765-3D) cells were grown and plated on as described in Figure 2A onto YPD alone or YPD + auxin and incubated 3 days at 23°C. SMC1-AID was depleted in media containing auxin, which allows assessment of whether smc1-D1164E promotes viability. (C) Assessing whether the Smc1 ATPase active site D-loop mutant smc3-D1161E promotes cell viability. Haploid SMC3 SMC3-AID(VG3771-10C), SMC3-AID (VG3651-3D), and smc3-D1161E SMC3-AID (VG3773-16D) were grown and plated dilution as described in C. (D) Regimen used to assess sister chromatid cohesion in cells containing AID tagged proteins. Asynchronous cells were arrested in G1, depleted for AID tagged proteins by the addition of auxin, then released from G1 and arrested in M phase in the presence of auxin. (E,F) Cohesion loss at the CEN-proximal locus (TRP1) in M phase cells depleted for AID tagged proteins. SMC1-AID or SMC3-AID was depleted in strains from G1 through M phase as described in D. The percentage of cells with two GFP spots (sister separation) is plotted. (E) smc1-D1164E promotes cohesion in SMC1-AID depleted cells. Haploid SMC1 SMC1-AID (VG3794-2E), SMC1-AID (VG3711-5D) and smc1-D1164E SMC1-AID (VG3795-2C) assayed for cohesion. DNA content analysis of these cells can be seen in Figure 4—figure supplement 4. (F) smc3-D1161E fails to promote cohesion in to promote cohesion SMC1-AID depleted cells. Haploid SMC3 SMC3-AID (VG3797-1A), SMC3-AID (VG3651-3D) and smc3-D1161E SMC3-AID (VG3799-3C) strains assayed for cohesion. DNA content analysis of these cells can be seen in Figure 4—figure supplement 4. Please see Figure supplements 1–6 for further characterization of smc1-D1164E and smc3-D1161E alleles.
We next assessed cohesion at CEN-proximal and CEN-distal loci in smc1-D1164E SMC1-AID and smc3-D1161E SMC3-AID strains under conditions in which AID tagged subunits remain depleted from G1 through M phase (Figure 4D). As expected, SMC1-AID and SMC3-AID cells showed severe cohesion defects that could be rescued by SMC1 and SMC3, respectively (Figure 4E and Figure 4—figure supplement 5A). smc1-D1164E SMC1-AID cells had only a modest cohesion defect, in line with our previous results for the smc1-D1164E in an otherwise wild type background. In contrast, in the presence of auxin, the smc3-D1161E SMC3-AID cells were severely compromised for cohesion, at levels comparable to cells that only have the SMC3-AID allele (~90% separated sisters, Figure 4F and Figure 4—figure supplement 5B). Similar results were obtained when cohesion was assessed at the CEN-distal locus (Figure 4—figure supplement 6). Thus, smc1-D1164E and smc3-D1161E differ in their ability to promote viability and cohesion.
To understand the molecular basis for this difference in cohesion, we performed ChIP on this panel of strains to assess cohesin binding to chromosomes under conditions in which the AID-tagged proteins were depleted. The level of cohesin binding to chromosomes in smc1-D1164E SMC1-AID cells was reduced about twofold compared to SMC1 SMC1-AID (Figure 5A and Figure 5—figure supplement 1). In contrast, cohesin binding to chromosomes in smc3-D1161E SMC3-AID cells was essentially at the background levels observed in SMC3-AID-only cells (Figure 5B and Figure 5—figure supplement 1). Taken together, these data suggest that the Smc1 ATPase active site regulates cohesin ATPase to modulate cohesin’s DNA binding, whereas the Smc3 active site regulates cohesin ATPase to modulate cohesin-mediated DNA tethering after its stable binding to DNA.
Figure 5 with 2 supplements see all
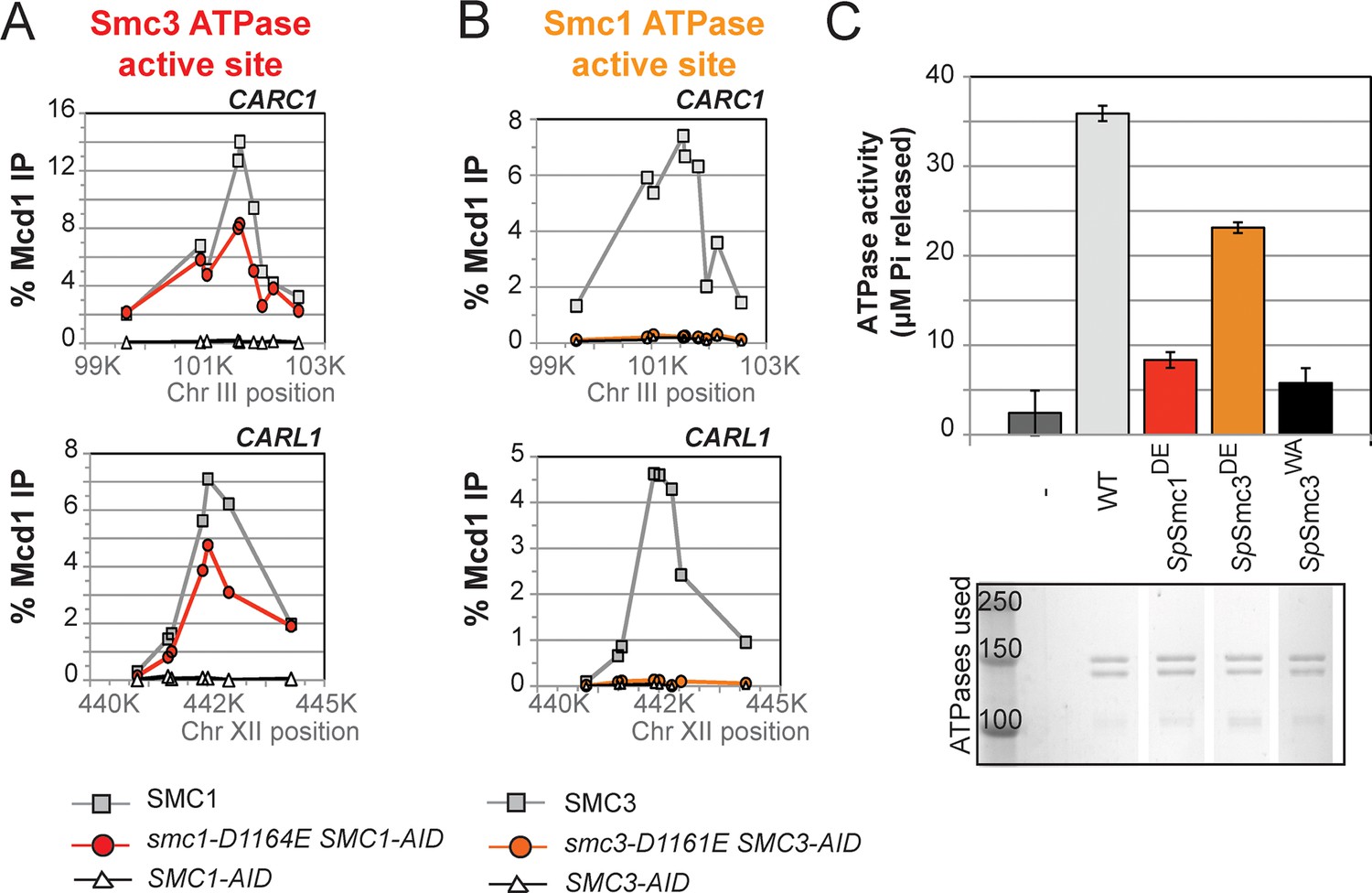
D-to-E mutations in Smc1 and Smc3 ATPase active sites uncouple the level of ATPase activity from chromosome binding.
(A,B) ChIP of Mcd1 in M phase cells depleted for AID tagged proteins. G1 cells were depleted for AID tagged proteins then released under depletion condition and arrested in M phase as described in Figure 4D. M phase cells were fixed and processed for ChIP using Mcd1 antibodies, and the% Mcd1 binding plotted as described in Figure 3—figure supplement 4. (A) ChIP of Mcd1 in smc1-D1164E cells at centromere-proximal CARC1 (top panel) and centromere-distal CARL1 (bottom panel). Haploid M phase cells from Figure 4B, expressing Smc13FLAG Smc1AID (SMC1; light grey line, light grey squares), Smc1-D1164E3FLAG Smc1AID (smc1-D1164E SMC1-AID; red line, red circles), and Smc1AID alone (SMC1-AID; black line, open triangles) were used for ChIP under conditions in which AID-tagged proteins were degraded. (B) ChIP of Mcd1 in M phase in smc3-D1161E cells at centromere-proximal CARC1 (top panel) and centromere-distal CARL1 (bottom panel). Haploid M phase cells from Figure 4C expressing Smc36HA Smc3AID (SMC3; light grey line, light grey squares), Smc3-D1161E6HA Smc3AID (smc3-D1161E SMC3-AID; orange line, orange circles), and Smc3AID alone (SMC3-AID; black line, open triangles) were used for ChIP under conditions in which AID-tagged proteins were degraded. (C) ATPase activity of purified S. pombe cohesin bearing D-loop mutations Psm1-D1167E or Psm3-D1132E, analogous to smc1-D1164E or smc3-D1161E, respectively. Same amount of cohesin was used in ATPase experiments (lower panel). Cohesin complexes were purified from cells overexpressing wild type S. pombe cohesin (WT) or S. pombe cohesin with mutations analogous with Smc3 ATPase active site D-loop-E mutation (SpSmc1DE) or Smc1 ATPase active site D-loop mutation (SpSmc3DE). ATPase assays were carried out in ATPase buffer 1 for 2 hours at 30°C. Cohesin with a K-to-I mutation in the Walker A motif of Smc3 ATPase active site (SpSmc3WA, Psm3 K38I in S. pombe) abolished most, if not all, ATPase activity. Coomassie-stained protein bands were spliced from the same gel for representation purposes. Please see figure supplement 1 for further characterization of chromosomal association of cohesin in smc1-D1164E or smc3-D1161E. Figure supplement 2 shows ATPase activity of Rad50 protein when the D-loop residue is mutated to an E or an A.
The D-loop mutants of Smc1 and Smc3 ATPase active sites uncouple the level of ATPase activity from DNA binding and tethering
Precedent that the D-loop mutations might impact ATPase activity come from studies of the homodimeric SMC protein Rad50. The mutation of the D-loop aspartate in Rad50 to an alanine (Rad50DA) dramatically reduced its ATPase activity (la Rosa and Nelson, 2011) (Figure 5—figure supplement 2). Furthermore, the substitution of the D-loop aspartate with a glutamate (Rad50DE) led to a 3-fold reduction in ATPase activity (Figure 5—figure supplement 2).
To address how the cohesin ATPase activity would be affected by these substitutions, we purified the S. pombe cohesin complex with the analogous mutations (Murayama and Uhlmann, 2014) and assayed the ATPase activity (a sum of the ATP hydrolysis by Smc1 and Smc3 active sites) for equivalent amounts of wild type and mutant cohesin complexes. The mutation in the S. pombe homolog of cohesin analogous to smc1-D1164E (SpSmc1DE), which was competent for cohesion and viability in S. cerevisiae, reduced the ATPase activity of cohesin to levels close to the Smc3 walker A mutant (SpSmc3WA, Figure 5C). In contrast, the D-loop-E mutation in the Smc1 ATPase active site (SpSmc3DE), analogous to the smc3-D1161E allele that abolished chromosomal association of cohesin, led to a subtler reduction in cohesin’s total ATPase activity. These results suggested that the substitution of the D-loop aspartate with a glutamate in Smc3 and Smc1 ATPase active sites affect cohesin in unique ways that uncouple the level of ATPase activity from DNA tethering and DNA binding.
The novel and distinct phenotypes of smc1-D1164E and smc3-D1161E might result from a subtle change that alters rather than abrogates D-loop function, given the chemical similarity of aspartate and glutamate. To test whether these unusual phenotypes persisted with more radical substitutions of the D-loop aspartate, we changed it to alanine in the Smc1 or Smc3 ATPase active sites. We introduced these smc1-D1164A or smc3-D1161A alleles into strains bearing SMC1-AID or SMC3-AID alleles, respectively (Figure 6A). We then characterized these alleles under conditions in which the AID-tagged proteins were degraded (Figure 6—figure supplement 1). Neither smc1-D1164A nor smc3-D1161A allele was able to sustain viability on auxin plates (Figures 6B,C). Similar results in viability were seen using plasmid shuffle assays (Figure 6—figure supplement 2). Moreover, the smc1-D1164A or smc3-D1161A cells were both severely compromised for cohesion, although the cohesion defect in smc3-D1161A was more severe (Figures 6D,E, Figure 6—figure supplements 3 and 4). ChIP using anti-Mcd1 antibodies showed that smc1-D1164A had 7-10 fold less cohesin binding than wild type, whereas smc3-D1161A reduced cohesin binding to chromosomes to background levels seen with the SMC3-AID alone (Figures 7A,B). Similar results were obtained when tagged smc1-D1164A and smc3-D1161A proteins were assessed by ChIP (Figure 7—figure supplement 1). Therefore, the glutamate substitution in the D-loop of the Smc3 ATPase active site is unique in its ability to support cohesion suggesting it alters rather than abolishes the in vivo function of the Smc3 ATPase active site.
Figure 6 with 4 supplements see all

D-loop-A (DA) mutations perturb cohesin function more severely than D-loop D to E (DE) mutations
(A) Cartoon representation of the D-to-A substitution mutants in the Smc3 ATPase active site (smc1-D1164A) and Smc1 ATPase active site (smc3-D1161A). (B,C) Assessing whether the DA D-loop mutants can support viability. (B) Smc3 ATPase active site D-loop-A mutant, smc1-D1164A, failed to promote viability. SMC1-AID smc1-D1164A (VG3766-3C) cells were grown and dilution plated as described in Figure 4B. SMC1-AID SMC1(VG3764-3A), SMC1-AID (VG3311-5D) cells were re-plated here for comparison. The smc1-D1164A SMC1-AID was moved from a different region of this same plate for clarity of presentation. (C) Smc1 ATPase active site D-loop-A mutant, smc3-D1161A, failed to promote viability. SMC3-AID smc3-D1161A (VG3772-13A) cells were grown and dilution plated. SMC3-AID SMC3(VG3726-6A) and SMC3-AID (VG3711-5D) cells were re-plated here for comparison. The smc3-D1161A SMC3-AID was moved from a different region of this same plate for clarity of presentation. (D,E) Cohesion loss at the CEN-proximal locus (TRP1) in M phase cells depleted for AID tagged proteins from G1 through M phase as described in Figure 4D. The percentage of cells with two GFP spots (sister separation) was plotted. (D) Cohesion loss in smc1-D1164A cells at the CEN-proximal TRP1 locus. Haploid strains SMC1 SMC1-AID (VG3794-2E), SMC1-AID (VG3711-5D) and smc1-D1164A SMC1-AID (VG3796-1F) assayed for cohesion. (E) Cohesion loss in smc3-D1161A cells at the CEN-proximal TRP1 locus. Haploid strains SMC3 SMC3-AID (VG3797-1A), SMC3-AID (VG3651-3D) smc3-D1161A SMC3-AID (VG3798-2B) assayed for cohesion. Note: smc1-D1164A SMC1-AID and smc3-D1161A SMC3-AID cells were analyzed in the same experiments as Figure 4D,E, respectively. smc-DA data was omitted from Figure 4 but presented here with controls from those experiments for clarity of presentation. DNA content analysis of cells in Figure 6D,E is shown in Figure 6—figure supplement 3. Please see Figure supplements 1–6 for further characterization of smc1-D1164A and smc3-D1161A alleles.
Figure 7 with 1 supplement see all
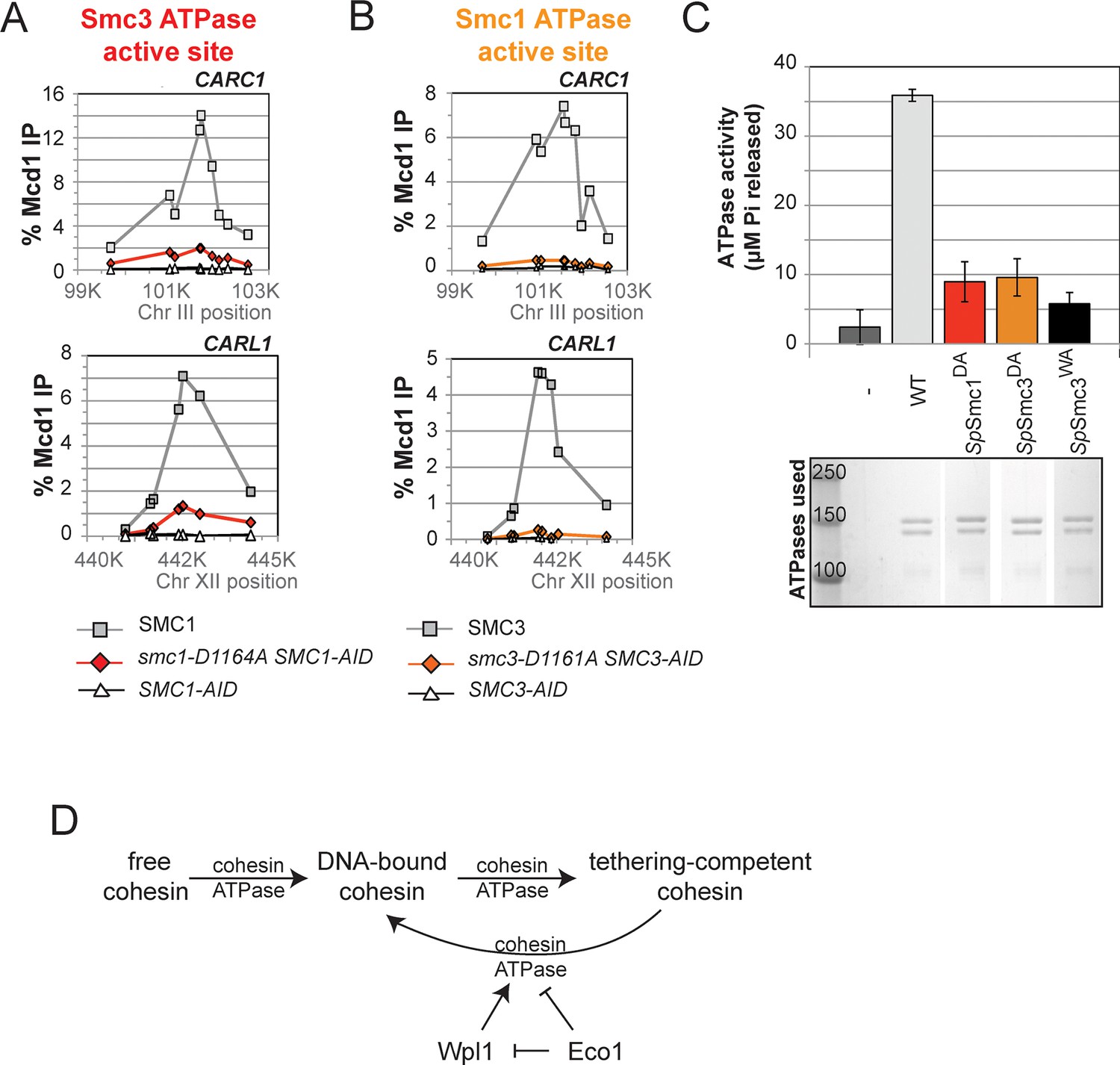
DA mutations in Smc1 and Smc3 ATPase active sites abolish chromosome binding and ATPase activity.
(A) ChIP of Mcd1 in M phase smc1-D1164A cells grown in auxin-containing media. Haploid M phase cells from Figure 6B were fixed and processed for ChIP using Mcd1 antibodies.% Mcd1 binding plotted as described in Figure 3—figure supplement 4. Mcd1 ChIP at centromere-proximal CARC1 (top panel) and centromere-distal CARL1 (bottom panel). (B) ChIP of Mcd1 in M phase smc3-D1161A cells grown in auxin-containing media. Haploid M phase cells from Figure 6C were fixed and processed for ChIP. Mcd1 ChIP at centromere-proximal CARC1 (top panel) and centromere-distal CARL1 (bottom panel). (C) ATPase activity of purified cohesin complexes from S. pombe bearing D-loop-A mutations in Smc3 and Smc1 ATPase sctive sites. Psm1 D1167A (SpSmc1DA) and Psm3 D1132A (SpSmc1DA), analogous to smc1-D1164A and smc3-D1161A, respectively, were purified from overexpression strains listed in Supplementary file 1. Same amount of cohesin was used in the ATPase experiments (lower panel). ATPase assays were carried out in ATPase buffer 1 for 2 hours at 30°C. ATPase activity of wild type (WT) and Walker A-mutant (SpSmc3WA) cohesin was represented here for comparison. Bands were spliced from the same gel for representation purposes. (D) Model for how the two ATPase active sites regulate cohesin function. Free cohesin is converted to a stable DNA-bound form by the action of the loader complex (not shown) and ATP binding/hydrolysis by ATPase active sites. A particular unknown nucleotide state at the Smc3 ATPase active site induces the tethering (cohesive) form. Upon finding the correct sister to be paired with, the Eco1-mediated acetylation of Smc3 leads to the stabilization of this cohesive state. Absent this stabilization, the Smc3 ATPase active site destabilizes the tethering form or induces cohesin dissociation from chromosomes. Wpl1 could either promote this Smc3 ATPase active site function or destabilize the cohesin bound to DNA in the non-tethering form. Figure supplement 1 shows cohesin binding to DNA in smc1-D1164E and smc3-D1161E strains.
Finally, we asked whether analogous aspartate-to-alanine substitution mutations altered the ATPase activity of purified S. pombe cohesin. Both mutations (SpSmc1DA and SpSmc3DA) reduced the ATPase activity of cohesin to levels comparable to the walker A mutation (SpSmc3WA, Figure 7C). The similar severe defects in ATPase activities for the S. pombe Smc1DE and Smc1DA cohesin complexes was striking given the dramatic differences in the analogous complexes to promote viability, cohesion and cohesin binding to DNA in S. cerevisiae (Figures 4–7). This functional difference reinforces the conclusion that the cohesin’s DNA binding and DNA tethering activities can be uncoupled from the level of cohesin ATPase activity.
Discussion
The cohesin ATPase functions in tethering DNA after cohesin’s stable binding to DNA
Until this study, there was no evidence for a role for the ATPase regulating post-DNA binding steps. Here, we show that cohesin retains its ATPase activity after stably binding to DNA in vitro, implying ATP hydrolysis plays an additional role(s) after cohesin binds DNA. We also show that cohesion activator mutations in key residues of the Smc3 ATPase active site, smc1-D1164E and smc1-Y1128C, suppress the severe cohesion defect of eco1∆ wpl1∆ cells, similar to the previously described smc3-D1189H allele (Guacci et al., 2015). These cohesion activators restore cohesion in eco1∆ wpl1∆ cells without increasing the amount of cohesin stably bound to DNA (this study, Guacci et al., 2015). Lastly, a mutation analogous to one of these activator mutations, smc1-D1164E, abolishes most, if not all, cohesin ATPase activity when introduced in purified S. pombe cohesin complex. Taken together, our results suggest the robust ATPase activity of DNA-bound cohesin is physiologically relevant, likely acting as an inhibitor of the conversion of stably bound cohesin to a form capable of tethering sister chromatids.
Smc3 ATPase active site has a unique function in DNA tethering
We also show that the cohesion activator phenotype of the smc1-D1164E allele is a unique feature of the D-loop of the Smc3 ATPase active site. The analogous substitution in the D-loop of the Smc1 ATPase active site, smc3-D1161E, not only fails to act as a cohesion activator but also is unable to support viability, fails to load cohesin to DNA, and cannot generate cohesion. These results suggest that the Smc1 and Smc3 ATPase active sites differentially affect cohesin ATPase function and that the Smc3 ATPase active site has a distinct function in the generation of cohesion after stable DNA binding of cohesin. This distinct function provides an explanation for the evolutionary diversion of the SMC subunits. Indeed, use of asymmetrical ATPases for regulation purposes is common to ABC transporters (Antony and Hingorani, 2004; Furman et al., 2013; Yang et al., 2003). We suspect the differential roles of cohesin ATPase active sites and a specialized role for the Smc3 active site in DNA tethering was missed previously, in part due to their composite nature and the particular mutations that have been analyzed to date. How the D-loop of the Smc3 ATPase active site executes its specialized function in regulating the cohesin ATPase remains unclear. The D-loop may impact the nucleotide state only in the Smc3 ATPase active site. Alternatively, it could alter the nucleotide state of both active sites. Support for the latter comes from studies on the ABC ATPase domains in ABC transporters, which suggest that ATP hydrolysis by the two sites is cooperative (Holland and Blight, 1999), and that D-loops are involved in mediating the communicaton between the two active sites within the ATPase (Furman et al., 2013; Hohl et al., 2012, 2014).
Cohesin functions can be separated from overall ATPase activity levels
The phenotypes of the D-loop mutants in the Smc1 and Smc3 active sites did not correlate with the ATPase activities of the corresponding purified S. pombe cohesin complexes. For example, cohesin complexes with aspartate-to-glutamate substitution in the D-loop of the Smc1 active site had significant ATPase activity in vitro, but were severely perturbed for DNA binding in S. cerevisiae. In addition, cohesin complexes with aspartate-to-glutamate substitution in the D-loop of the Smc3 ATPase had low levels of ATPase activity indistinguishable from the walker A mutant, but could promote DNA binding and cohesion in S. cerevisiae. While it is possible that this lack of correlation could be explained by our use of the ATPase activity of the S. pombe wild type and mutant cohesins as an approximation of the S. cerevisiae cohesin, this explanaition seems unlikely. The aspartate of the D-loop is absolutely conserved in all SMC complexes and highly conserved among very diverse ABC ATPase modules. Our analysis of analogous substitutions in the homodimeric Rad50 protein shows similar reductions in the ATPase activity. Finally, many published biochemical and structural studies have used cohesin complexes from different organisms as a proxy for S. cerevisiae cohesin to provide physiologically relevant insights. Intriguingly, the uncoupling of ATPase activity and function by D-loop mutations has been observed for several other ABC ATPases (Furman et al., 2013; Grossmann et al., 2014).
Therefore, we favor a model in which cohesin’s functions and conformations could be coordinated by the different states in the hydrolysis cycle of ATPase active sites. For example, the tethering-competent state of cohesin could require that the Smc3 active site be bound with ADP+Pi. The smc1-D1164E mutation may mimic the ADP-Pi state, whereas the smc1-D1164A mutant could be blocked elsewhere in the ATPase cycle (ATP bound or nucleotide free). Both the smc1-D1164E and smc1-D1164A would dramatically reduce overall ATPase activity but only the smc1-D1164E would promote cohesin function. A similar case of trapping the ADP+Pi state is hypothesized for the MRP1 ABC transporter, for a glutamate substitution of an aspartate residue (Qin and Cao, 2008). Elucidating the exact relationship between the particular nucleotide states of the Smc1 and Smc3 ATPase active sites and cohesin functions will likely require in vitro reconstitution of cohesin’s various functions and detailed structural studies.
Eco1 and Wpl1 regulate cohesin’s tethering activity through cohesin ATPase
The phenotypes of smc1-D1164E cells suggest that Eco1 modulation of cohesin’s ATPase impacts two distinct modes of cohesin regulation. First, smc1-D1164E suppresses the inviability of eco1∆ cells, which normally die because they are unable to inhibit Wpl1 (Rolef Ben-Shahar et al., 2008; Rowland et al., 2009; Sutani et al., 2009). This result suggests that Eco1 acetylation alters the Smc3 ATPase active site to make cohesin resistant to Wpl1. Second, smc1-D1164E also suppresses the cohesion defect in eco1△ wpl1△ cells, suggesting that it phenocopies the second function of Eco1 acetylation, the activation of DNA-bound cohesin to tether sister chromatids.
A clue to how Eco1 modulates cohesin ATPase to promote DNA tethering comes from our in vitro and in vivo characterization of smc1-D1164E. Since smc1-D1164E suppresses the eco1∆ cohesion defect, we propose that smc1-D1164E mimics the Eco1 acetylation of Smc3 by blocking the cohesin ATPase at a particular stage in its hydrolysis cycle, promoting DNA tethering. In the absence of acetylation, continued cycles of ATP hydrolysis prevent cohesin from maintaining the conformation necessary for tethering (Figure 7D). The completion of the ATPase cycle may render cohesin in a conformation capable of being recognized by and acted upon Wpl1, which leads to cohesin dissociation from DNA.
This model predicts that Eco1-mediated acetylation of the Smc3 may have an impact on the ATPase activity of cohesin. However, previous studies on in vitro acetylated cohesin or acetyl-mimic mutants of cohesin suggest that Eco1 acetylation does not impact cohesin’s total ATPase activity (Ladurner et al., 2014). This could be due to (1) the inherent inefficiency of acetylation both in vitro and in vivo, resulting in only a small subset of cohesin being acetylated, (2) the inability of glutamine substitutions of K112 and K113 in Smc3 to mimic the acetylated state, (3) the failure to assay cohesin ATPase activity on DNA. Alternatively, Eco1-mediated acetylation may stabilize an intermediate state of cohesin in a way that makes subsequent cycles of ATP hydrolysis unable to alter the tethering conformation. Either way, the result is that acetylation traps cohesin in its tethering form to stabilize cohesion during or soon after establishment. Testing whether Eco1-mediated acetylation directly downregulates the cohesin ATPase cycle when bound to DNA and how the ATPase cycle affects cohesin conformation will require better reconstitution of cohesin function in vitro.
Our model of transient conformational changes that must be stabilized provides a plasticity to cohesin-mediated DNA tethering. Cohesin that is bound to a locus could form transient tethers with a number of different DNA loci and at different times. The spatial and temporal control of Eco1 would ensure that only the proper tethers are stabilized, allowing a very tight regulation for cohesion establishment in sister chromatid cohesion. This plasticity of cohesin tethering and its Eco1-dependent regulation may be critical for other aspects of cohesin function, such as condensation, DNA repair and transcriptional regulation.
Materials and methods
Protein expression and purification
Request a detailed protocolS.pombe cohesin complex was over-expressed and purified from S. cerevisiae as described (Murayama and Uhlmann, 2014) with minor modifications. Briefly, over-expressing cells were harvested and lysed with a cryomill. For cohesin purifications, 50 mL of cell powder was thawed in the presence of 100 mL CLH300 buffer (50 mM HEPES, pH 7.5, 1 mM DTT, 300 mM NaCl, 20% glycerol, and protease inhibitor cocktail from Roche) and the clarified by centrifugation. The clarified supernatant was bound to 2 mL IgG beads (Invitrogen) overnight in the presence of RNase A (10 μg/mL). The beads were washed 300 mL in H300 (300 mM NaCl, 25 mM HEPES pH 7.5, 0.5 mM TCEP, 10% glycerol). To elute protein off the beads, 5 mL of elution buffer (H300 with 2 mM MgCl2, 5 U/mL Prescission protease, 10 μg/mL RNase A) was added. Eluted protein was diluted to achieve 100 mM NaCl and then bound to heparin column. The column was washed in 4xColumn Volume (CV) H100M (25 mM HEPES pH 7.5, 0.5 mM TCEP, 10% glycerol, 100 mM NaCl and 2 mM MgCl2), 4xCV H300 with 2 mM MgCl2. Cohesin was eluted from the column at 600 mM NaCl. Cohesin containing fractions were pooled, concentrated and frozen in same buffer containing 200 mM NaCl. Mutants of cohesin were generated by site-directed mutagenesis and over-expression strains were generated as described (Murayama and Uhlmann, 2014). Mutant proteins were purified using the same protocol as described above. The loader complex from S.pombe was expressed and purified as described (Murayama and Uhlmann, 2014). Rad50 protein from T4 bacteriophage was purified as described (la Rosa and Nelson, 2011).
Assembly of stable cohesin-DNA complexes in vitro
Request a detailed protocolCARC1 DNA substrates were prepared as described (Onn and Koshland, 2011). Briefly, for each binding reaction, 500 ng biotin-labeled DNA was assembled on 20 μL streptavidin-conjugated dynabeads. Beads were washed 6x 30 μL in CL1 buffer (35 mM Tris pH 7.5, 1 mM TCEP, 25 mM NaCl, 25 mM KCl, 1 mM MgCl2, 15% glycerol and 0.003% Tween 20) and resuspended in 60 μL CL1. 80 nM cohesin complex and 0.5 mM ATP (with or without 80 nM loader) was added to beads and incubated at 30°C for 1 hour in a reaction volume of 100 μL. Assembled cohesin-DNA complexes were washed in 100 μL buffer once in CL1, three times in CL1 with 500 mM KCl, and once more with CL1. Resuspending beads in 30 μL SDS sample buffer eluted bead-bound cohesin-DNA complexes. Samples were analyzed by SDS-PAGE followed by Coomassie staining or western blotting.
Elution of cohesin from DNA-beads with DNase or restriction enzyme digests
Request a detailed protocolStable cohesin-DNA complexes assembled as described above were resuspended in CL1 buffer in the presence of 2U DNase or 5U Mnl I at 30°C for 30 minutes. Eluted proteins (supernatant) and beads were subjected to SDS-PAGE and proteins were visualized by Coomassie staining or Western blotting.
ATPase assays
Request a detailed protocolFor basal ATPase activity of cohesin, 150 nM cohesin was diluted in 100 μL ATPase reaction buffer 1 (25 mM HEPES pH 7.5, 100 mM NaCl, 10% glycerol, 1 mM TCEP, 5 mM MgCl2). 10 μL of this sample was run on SDS-PAGE to visualize cohesin used in ATPase experiments. The ATPase reaction was started by the addition of 20 μL of 5x ATP hot mix (2.5 mM ATP, 1 μL of 10 μCi/ μL ATP-γ-P32 in 96.5 μL 5xATP buffer) to 80 μL cohesin mix. Thus, in our ATPase reactions, the final concentration of cohesin was 120 nM, and ATP was 0.5 mM. At appropriate time points, 20 μL of this reaction mix was taken out and mixed with 380 μL of 0.25 mg/mL BSA. 300 μL of STOP buffer (100 μL KPO4 1 M, 100 μL 1N HCl, 100 μL 20% Norit) was immediately added to the samples. Samples were then spun at 4°C 10000 rpm for 3 minutes. 500 μL of the supernatant was taken out in a new tube and spun again at 4°C 10000 rpm for 3 minutes. 350 μL of the supernatant of this second spin was counted in 5 mL of scintillation cocktail using a scintillation counter. Reads at time zero were counted as background and subtracted from later time points. To get% ATP hydrolysis, 10 μL of 1:5 diluted 5xATP buffer was counted to represent 100% ATP hydrolysis. For loader- and DNA-stimulated ATPase activity of cohesin, 80 nM cohesin, 80 nM loader, and 2.5 μg plasmid DNA containing the sequence for CARC1 (pIO2) were incubated in 50 μL ATPase reaction buffer 2 (25 mM HEPES pH 7.5, 25 mM NaCl, 25 mM KCl, 10% glycerol, 1 mM TCEP, 5 mM MgCl2). To measure the activity of stably DNA-bound cohesin complexes (CD-B), 80 nM cohesin, 80 nM loader and 0.5 mM ATP was incubated with 100 μL dynabeads coupled to 2.5 μg DNA in 500 μL total volume of CL1 at 30°C for 1 hour. Unbound/unstably bound cohesin was washed off using the following washes: 1x500 μL CL1, 3 x 500 μL in CL1 with 500 mM KCl, then 1x500 μL CL1. CD-B were then resuspended in 50 μL ATPase buffer 2. This sample of CD-B contained about% 20 of the initial input cohesin, which was approximately the same amount of cohesin as the other samples in this experiment (Figure 1E), as could be judged by the protein gel. The reaction was started by the addition of 10 μL 5xATP hot mix to 40 μL protein/DNA mix for 2 hours at 30°C and samples were processed as described above. The ATPase activity of T4 Rad50 and mutants was measured in ATPase buffer 1 for 2 hours at 30°C as described above, except 1 μM Rad50 was used. Error bars represent standard error of data from at least two independent experiments.
Yeast strains and media
Request a detailed protocolYeast strains used in this study are A364A background, and their genotypes are listed in Supplementary file 1. SC minimal and YPD media were prepared as described (Guacci et al., 1997). Benomyl (a gift from Dupont) and camptothecin (Sigma) plates used to assess drug sensitivity were prepared as previously described (Guacci and Koshland, 2012). Preparation of auxin (Sigma) containing media for depletion of AID tagged proteins was as previously described (Eng et al., 2014).
Dilution plating assays
Request a detailed protocolCells were grown to saturation in YPD media at 23°C then plated in 10-fold serial dilutions. Cells were incubated on plates at relevant temperatures or containing drugs as described. For plasmid shuffle assays, cells were grown to saturation in YPD media to allow loss of covering plasmid, then plated in 10-fold serial dilutions on YPD or FOA media.
G1 arrest and release into M phase arrest
G1 arrest
Request a detailed protocolAsynchronous cultures of cells were grown to mid-log phase at 23°C in YPD media, then α factor (Sigma) added to 10−8 M. Cells were incubated for 3 h to induce arrest in G1 phase. This incubation time was increased to 3.5 h for all strains in any experiment where an eco1∆ wpl1∆ background strain was used. For depletion of AID tagged proteins, auxin was added (500 mM final) to G1 arrested cells, and then incubated an additional 1 h in α factor containing media.
Release from G1 into M phase arrest
Request a detailed protocolG1 arrested cells were washed 3x in YPD containing 0.1 mg/ml Pronase E (Sigma), once in YPD, then resuspended in YPD containing nocodozale (Sigma) at 15 μg/ml final. Cells were incubated at 23°C for 3 h to arrest in M phase. For depletion of AID tagged proteins, auxin was added (500 mM final) in all wash media and in resuspension media containing nocodazole to ensure depletion at all times.
Monitoring cohesion using LacO-GFP assay
Request a detailed protocolCohesion was monitored using the LacO-LacI system where cells contained a GFP-LacI fusion and tandem LacO repreats integrated at one chromosomal locus, which recruits the GFP-LacI (Straight et al., 1996). CEN-distal cohesion was monitored by integrating LacO repeats at LYS4, located 470 kb from CEN4. CEN-proximal cohesion is monitored by integrating LacO at TRP1, located 10 kb from CEN4. Cells were fixed, and processed to allow the number of GFP signals in each cell to be scored and the percentage of cells with 2-GFP spots determined as previously described (Guacci and Koshland, 2012). Data is from 2 independent experiments and 200-300 cells scored for each data point in each experiment.
Plasmid constructs
Request a detailed protocolSite directed mutagenesis using the Stratagene Quick-change kit was employed to generate all mutants described. Mutations were confirmed by sequencing the entire ORF to ensure it was the only change.
Strain construction
SMC1 and SMC3 Shuffle strain construction
Request a detailed protocolHaploids containing pTH2 (SMC1 URA3 CEN) or pEU42 (SMC3 URA3 CEN) plasmid had their endogenous SMC1 or SMC3 gene deleted and replaced by the HPH cassette (encodes resistance to Hygromycin B) using standard PCR mediated homology-based recombination.
Plasmid shuffle to assess SMC1 and SMC3 mutant “test” alleles by integration at the LEU2 locus
Request a detailed protocolSMC1 or SMC3 WT of mutant alleles were cloned onto an integrating LEU2 vector. For SMC1 test alleles, these were plasmid pVG444 (SMC1 LEU2), pVG456 (smc1-D1164E LEU2) and pVG4 (smc1-D1164A LEU2), linearized within LEU2 by PpuMI. For SMC3 test alleles, these were plasmid pVG419 (SMC3 LEU2), pVG419 D1161A (smc3-D1161A LEU2) and pVG458 (smc3-D1161E LEU2) linearized within LEU2 using BstEII. Linearized plasmids were transformed into an SMC1 or SMC3 shuffle strain to integrate the vector at the LEU2 locus and LEU+ clones isolated. These “test alleles” were assayed for their ability to support viability as the sole SMC1 or SMC3 as sole source as follows. LEU+ clones were grown to saturation in YPD media at 23°C to allow loss of either plasmid pTH2 or pEU42, then plated in 10-fold serial dilutions on media containing 5-fluoroorotic acid (FOA). FOA selectively kills URA3 cells, thereby selecting for loss of pTH2 or pEU42, which allows assessment of test allele ability to support viability as the sole SMC1 or SMC3 in cells, respectively. As a control, cells were also plated on either YPD or URA- media.
Insertion of SMC1 and SMC3 alleles at the endogenous locus
Request a detailed protocolTwo different strategies were employed. One utilized shuffle strains described above for one step-gene replacement. A linear DNA fragment containing the desired SMC1 or SMC3 ORF, promoter and 3’ UTR were transformed into shuffle strains, plated on YPD and grown overnight. Plates were replica plated to FOA and FOA resistant clones selected, tested for sensitivity to Hygromycin B, which occurs when smc1∆::HPH or smc3∆::HPH is replaced by the transformed linear SMC1 or SMC3 allele, respectively. Transplacement alleles were confirmed by PCR screening and PCR sequencing.
The second strategy used to insert SMC1 and SMC3 alleles at the endogenous locus in haploid eco1∆ wpl1∆ cells, wpl1∆ cells, or in WT cells bearing TIR1 was as follows. Plasmids encoding the desired alleles were linearized within the ORF. Linearized plasmids were transformed into haploid strains. URA+ colonies contain the SMC1 URA3 or SMC3 URA3 plasmid integrated at the SMC1 or SMC3 locus to create tandem SMC1 or SMC3 genes. URA+ transformants were replica plated onto YPD then dilution streaked on FOA to excise the URA3 marker, and thereby select for loss of one SMC1/3 allele. PCR mediated sequencing was used to identify clones containing only the desired alleles.
Strains containing AID tagged proteins
Request a detailed protocolDetails about the Auxin mediated destruction of AID tagged proteins in yeast was previously described (Eng et al., 2014). Briefly, the TIR1 E3-ubiquiting ligase placed under control of the GPD promoter and marked by C. Albicans TRP1 replaced the TRP1 gene on chromosome IV. SMC1, SMC3 and ECO1 were internally tagged with 3V5-AID2 sequences and transformed into yeast strains bearing TIR1 to replace SMC1, SMC3 and ECO1 at the endogenous locus. PCR screening and auxin mediated sensitivity were used to identify clones containing AID tagged genes.
Genetic screen of eco1∆ wpl1∆ cells for cohesion activator suppressors was done as previously described (Guacci et al., 2015).
Chromatin Immunoprecipitation (ChIP) was performed as previously described (Eng et al., 2014; Wahba et al., 2013).
Microscopy. Images were acquired with a Zeiss Axioplan2 microscope (100X objective, NA=1.40) equipped with a Quantix CCD camera (Photometrics).
Flow cytometry analysis was performed as previously described (Eng et al., 2014).
References
-
ATP hydrolysis is required for cohesin's association with chromosomesCurrent Biology 13:1941–1953.https://doi.org/10.1016/j.cub.2003.10.036
-
Interallelic complementation provides functional evidence for cohesin-cohesin interactions on DNAMolecular Biology of the Cell 26:4224–4235.https://doi.org/10.1091/mbc.E15-06-0331
-
The deviant ATP-binding site of the multidrug efflux pump Pdr5 plays an active role in the transport cycleJournal of Biological Chemistry 288:30420–30431.https://doi.org/10.1074/jbc.M113.494682
-
Cohesin-independent segregation of sister chromatids in budding yeastMolecular Biology of the Cell 23:729–739.https://doi.org/10.1091/mbc.E11-08-0696
-
A novel mechanism for the establishment of sister chromatid cohesion by the ECO1 acetyltransferaseMolecular Biology of the Cell 26:117–133.https://doi.org/10.1091/mbc.E14-08-1268
-
At the heart of the chromosome: SMC proteins in actionNature Reviews Molecular Cell Biology 7:311–322.https://doi.org/10.1038/nrm1909
-
Crystal structure of a heterodimeric ABC transporter in its inward-facing conformationNature Structural & Molecular Biology 19:395–402.https://doi.org/10.1038/nsmb.2267
-
Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporterProceedings of the National Academy of Sciences of the United States of America 111:11025–11030.https://doi.org/10.1073/pnas.1400485111
-
Chromosomal addresses of the cohesin component Mcd1pThe Journal of Cell Biology 151:1047–1056.https://doi.org/10.1083/jcb.151.5.1047
-
Cohesin: its roles and mechanismsAnnual Review of Genetics 43:525–558.https://doi.org/10.1146/annurev-genet-102108-134233
-
Sister chromatid cohesion: a simple concept with a complex realityAnnual Review of Cell and Developmental Biology 24:105–129.https://doi.org/10.1146/annurev.cellbio.24.110707.175350
-
In vitro assembly of physiological cohesin/DNA complexesProceedings of the National Academy of Sciences of the United States of America 108:12198–12205.https://doi.org/10.1073/pnas.1107504108
-
Synthesis of a novel pH-sensitive methacrylate amphiphilic polymer and its primary application in aqueous two-phase systemsApplied Biochemistry and Biotechnology 150:171–183.https://doi.org/10.1007/s12010-008-8139-x
-
An interaction between the walker a and d-loop motifs is critical to ATP hydrolysis and cooperativity in bacteriophage T4 Rad50Journal of Biological Chemistry 286:26258–26266.https://doi.org/10.1074/jbc.M111.256305
-
ATP binding to the first nucleotide binding domain of multidrug resistance-associated protein plays a regulatory role at low nucleotide concentration, whereas ATP hydrolysis at the second plays a dominant role in ATP-dependent leukotriene C4 transportJournal of Biological Chemistry 278:30764–30771.https://doi.org/10.1074/jbc.M304118200
Article and author information
Author details
Funding
National Institutes of Health (GM092813)
- Douglas Koshland
Damon Runyon Cancer Research Foundation (DRG-2137-12)
- Gamze Çamdere
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Hugo Tapia, Thomas Eng, Jeremy Amon, Michelle Bloom, Orna Cohen-Fix and Jill Heidinger-Pauli for critically reading the manuscript, and other members of the Koshland lab for fruitful discussions. We thank Dr. Frank Uhlmann for sharing cohesin and loader expression strains and plasmids, Dr. Scott Nelson for sharing Rad50 expression plasmids, Dr. Stuart Lynn for advice on ATPase assays, the laboratory of David Drubin and Georjana Barnes for use of equipment. This work was funded by the National Institute of Health (GM092813). GC was supported by a fellowship from the Damon Runyon Cancer Research Foundation (DRG-2137-12).
Version history
- Received: September 2, 2015
- Accepted: November 18, 2015
- Accepted Manuscript published: November 19, 2015 (version 1)
- Version of Record published: December 31, 2015 (version 2)
Copyright
© 2015, Çamdere et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,309
- Page views
-
- 697
- Downloads
-
- 56
- Citations
Article citation count generated by polling the highest count across the following sources: Scopus, Crossref, PubMed Central.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering
eLife 4:e11315.
https://doi.org/10.7554/eLife.11315
Further reading
-
- Biochemistry and Chemical Biology
- Medicine
Type 2 diabetes (T2D) is associated with higher fracture risk, despite normal or high bone mineral density. We reported that bone formation genes (SOST and RUNX2) and advanced glycation end-products (AGEs) were impaired in T2D. We investigated Wnt signaling regulation and its association with AGEs accumulation and bone strength in T2D from bone tissue of 15 T2D and 21 non-diabetic postmenopausal women undergoing hip arthroplasty. Bone histomorphometry revealed a trend of low mineralized volume in T2D (T2D 0.249% [0.156–0.366]) vs non-diabetic subjects 0.352% [0.269–0.454]; p=0.053, as well as reduced bone strength (T2D 21.60 MPa [13.46–30.10] vs non-diabetic subjects 76.24 MPa [26.81–132.9]; p=0.002). We also showed that gene expression of Wnt agonists LEF-1 (p=0.0136) and WNT10B (p=0.0302) were lower in T2D. Conversely, gene expression of WNT5A (p=0.0232), SOST (p<0.0001), and GSK3B (p=0.0456) were higher, while collagen (COL1A1) was lower in T2D (p=0.0482). AGEs content was associated with SOST and WNT5A (r=0.9231, p<0.0001; r=0.6751, p=0.0322), but inversely correlated with LEF-1 and COL1A1 (r=–0.7500, p=0.0255; r=–0.9762, p=0.0004). SOST was associated with glycemic control and disease duration (r=0.4846, p=0.0043; r=0.7107, p=0.00174), whereas WNT5A and GSK3B were only correlated with glycemic control (r=0.5589, p=0.0037; r=0.4901, p=0.0051). Finally, Young’s modulus was negatively correlated with SOST (r=−0.5675, p=0.0011), AXIN2 (r=−0.5523, p=0.0042), and SFRP5 (r=−0.4442, p=0.0437), while positively correlated with LEF-1 (r=0.4116, p=0.0295) and WNT10B (r=0.6697, p=0.0001). These findings suggest that Wnt signaling and AGEs could be the main determinants of bone fragility in T2D.
-
- Biochemistry and Chemical Biology
Heat stress can cause cell death by triggering the aggregation of essential proteins. In bacteria, aggregated proteins are rescued by the canonical Hsp70/AAA+ (ClpB) bi-chaperone disaggregase. Man-made, severe stress conditions applied during, e.g., food processing represent a novel threat for bacteria by exceeding the capacity of the Hsp70/ClpB system. Here, we report on the potent autonomous AAA+ disaggregase ClpL from Listeria monocytogenes that provides enhanced heat resistance to the food-borne pathogen enabling persistence in adverse environments. ClpL shows increased thermal stability and enhanced disaggregation power compared to Hsp70/ClpB, enabling it to withstand severe heat stress and to solubilize tight aggregates. ClpL binds to protein aggregates via aromatic residues present in its N-terminal domain (NTD) that adopts a partially folded and dynamic conformation. Target specificity is achieved by simultaneous interactions of multiple NTDs with the aggregate surface. ClpL shows remarkable structural plasticity by forming diverse higher assembly states through interacting ClpL rings. NTDs become largely sequestered upon ClpL ring interactions. Stabilizing ring assemblies by engineered disulfide bonds strongly reduces disaggregation activity, suggesting that they represent storage states.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}