Sam68/KHDRBS1-dependent NF-κB activation confers radioprotection to the colon epithelium in γ-irradiated mice
- Bloomberg School of Public Health, Johns Hopkins University, United States
- School of Medicine, Johns Hopkins University, United States
- Johns Hopkins University, United States
Abstract
Previously we reported that Src-associated-substrate-during-mitosis-of-68kDa (Sam68/KHDRBS1) is pivotal for DNA damage-stimulated NF-κB transactivation of anti-apoptotic genes (Fu et al., 2016). Here we show that Sam68 is critical for genotoxic stress-induced NF-κB activation in the γ-irradiated colon and animal and that Sam68-dependent NF-κB activation provides radioprotection to colon epithelium in vivo. Sam68 deletion diminishes γ-irradiation-triggered PAR synthesis and NF-κB activation in colon epithelial cells (CECs), thus hampering the expression of anti-apoptotic molecules in situ and facilitating CECs to undergo apoptosis in mice post whole-body γ-irradiation (WBIR). Sam68 knockout mice suffer more severe damage in the colon and succumb more rapidly from acute radiotoxicity than the control mice following WBIR. Our results underscore the critical role of Sam68 in orchestrating genotoxic stress-initiated NF-κB activation signaling in the colon tissue and whole animal and reveal the pathophysiological relevance of Sam68-dependent NF-κB activation in colonic cell survival and recovery from extrinsic DNA damage.
https://doi.org/10.7554/eLife.21957.001Introduction
Nuclear factor kappa B (NF-κB) plays a crucial function in a variety of human disorders, in particular inflammatory diseases and cancers (Hayden and Ghosh, 2008; Scheidereit, 2006; Sun et al., 2013; Vallabhapurapu and Karin, 2009; Wan and Lenardo, 2010; Wu and Miyamoto, 2007). Accumulating evidence highlights an important role of NF-κB signaling pathway in cellular responses to various genotoxic stresses and DNA damage-stimulated NF-κB signaling cascade in the nucleus that leads to NF-κB activation has been recently revealed (McCool and Miyamoto, 2012; Miyamoto, 2011). In particular, ataxia telangiectasia mutated (ATM), inhibitor of NF-κB kinase gamma subunit (IKKγ), protein inhibitor of activated STATy (PIASy), and poly (ADP-ribose) polymerase 1 (PARP1) were reported to be indispensible for genoxic stress-induced NF-κB activation (Huang et al., 2003; Li et al., 2001; Mabb et al., 2006; Piret et al., 1999; Stilmann et al., 2009). Moreover, we recently revealed that Sam68/KHDRBS1 (Src-associated substrate during mitosis of 68 kDa/KH domain containing, RNA binding, signal transduction associated 1, encoded by KHDRBS1 gene), a versatile single-strand nucleic acid binding protein (Lukong and Richard, 2003; Richard, 2010), is an important molecule in orchestrating genotoxic stress-initiated NF-κB signaling in the nucleus (Fu et al., 2016). Specifically, Sam68 is essential for DNA damage-triggered PARP1 activation and the subsequent polymers of ADP-ribose (PAR) synthesis (Fu et al., 2016). Sam68 deletion dampens the PAR-dependent NF-κB signaling and transcription of an array of anti-apoptotic genes, thus sensitizing Sam68-deficient mouse embryonic fibroblasts (MEFs) and colon epithelial cells (CECs) in culture to genotoxicity caused by DNA-damaging agents (Fu et al., 2016). The levels of Sam68, PAR, NF-κB activation, and anti-apoptotic molecules B-cell lymphoma-extra large (Bcl-XL) and X-linked inhibitor of apoptosis protein (XIAP) are elevated and positively correlated in colon tumors compared to adjacent normal tissue derived from either the tumor-laden Apcmin716/+ mice or human colon cancer patients. Moreover, downregulation of Sam68 substantially sensitizes human colon cancer cells to spontaneous and genotoxic stress-induced cell death and retards colon tumor burden in Apcmin716/+ mice (Fu et al., 2016). These findings suggest that upregulated Sam68 is crucial in orchestrating DNA damage-initiated NF-κB activation signaling in cultured cells and conferring the PAR-dependent NF-κB activation to respond to the intrinsic DNA damage frequently occurred in cancerous cells. However, the in vivo impact of physiological Sam68 levels on extrinsic genotoxic stress-induced NF-κB signaling and activation in normal cells at the organ and even the whole animal levels has not been fully understood.
Radiotherapy and chemotherapy are extensively used in current-day cancer treatments. It has been recognized that γ-irradiation induced DNA damage triggers rapidly-proliferating tumor cells to undergo apoptosis; whereas non- and slowly-dividing cells rarely die of γ-irradiation. Of note, CECs in colon crypts are among the most rapidly dividing cells in the body, which makes them susceptible to γ-irradiation-induced cell death. Indeed, colon tissue injury remains one of the major adverse effects of radiotherapy, when γ-irradiation is employed to treat colon cancer and other intra-abdominal cancers (Egan et al., 2004). Although greater antitumor effects could be produced by higher doses of γ-irradiation, the tolerance of patients to the acute side effects caused by γ-irradiation to the colon limits the administered dose (Egan et al., 2004). Given the essential role of DNA damage-induced NF-κB transactivation of anti-apoptotic genes in cell fate determination post genotoxic stresses, it will be extremely important to understand genotoxic stress-induced NF-κB activation signaling pathway not only in cancerous cells but also in normal cells under pathophysiological conditions. Besides the recently revealed critical function of Sam68-dependent NF-κB activation to overcome intrinsic DNA damage for the development and survival of colon cancer (Fu et al., 2016), whether Sam68-dependent NF-κB signaling is crucial in normal colon epithelium in response to extrinsic γ-irradiation remains elusive. We therefore examined the hypothesis that Sam68-dependent NF-κB activation offers an anti-apoptotic response in the γ-irradiated colon epithelium in vivo hence providing radioprotection to the organ and the animal following whole-body γ-irradiation (WBIR).
Results
Sam68 confers genotoxic stress-induced NF-κB signaling in the γ-irradiated colon
To assess the in vivo impact of Sam68 on genotoxic stress-induced NF-κB signaling and transactivation, Khdrbs1+/− (Sam68 heterozygote) and Khdrbs1−/− (Sam68 knockout) mice were subjected to a sublethal dose of WBIR and we examined the γ-irradiation-initiated NF-κB activation signaling cascade in the derived colons at defined times post WBIR (Figure 1A). As expected, vigorous PAR production, as illustrated by immunofluorescence staining on colon tissue sections, occurred in Khdrbs1+/− colon 20 min post WBIR; whereas such an acute response was almost abolished in the colon from Khdrbs1−/− mice (Figure 1B). In support, immunoblot analyses showed that robust PAR chair formation in whole cell lysates of CECs from Khdrbs1+/− mice at 20 min post WBIR, which was markedly tempered in Khdrbs1−/− mice post WBIR (Figure 1C). These results suggest that Sam68 is crucial for facilitating genotoxic stress-induced PAR production in the γ-irradiated colon tissue in vivo. Consistently, WBIR-induced p65 phosphorylation, one of the biochemical hallmarks of NF-κB activation, was profound in the CECs from γ-irradiated Khdrbs1+/− mice, but was substantially tempered in Khdrbs1−/− animals (Figure 1D). Moreover, WBIR triggered nuclear translocation of p65, as assayed by immunohistostaining and subcellular fractionation, in Khdrbs1+/− CECs; whereas p65 nuclear accumulation was greatly attenuated in the γ-irradiated Khdrbs1−/− cells (Figure 1E–F). Of note, the levels of PAR, total p65, and nuclear accumulated p65 were comparable in CECs derived from mock-irradiated Khdrbs1+/− and Khdrbs1−/− animals (Figure 1B–F), suggesting that Sam68 deletion does not affect the physiological PAR synthesis and NF-κB signaling in the colon without any stimulation. In contrast, Sam68 deletion almost abolished genotoxic stress-triggered PAR formation and the signaling events that lead to NF-κB activation in the colon following WBIR (Figure 1B–F). Consistent with our recent report that Sam68 plays a key role in DNA damage-initiated PAR synthesis and the PAR-dependent NF-κB signaling in the isolated and in vitro cultured CECs (Fu et al., 2016), these results further support the crucial function of Sam68 in genotoxic stress-triggered PAR production and signaling to NF-κB activation in the γ-irradiated colon from whole animals in vivo.
Figure 1
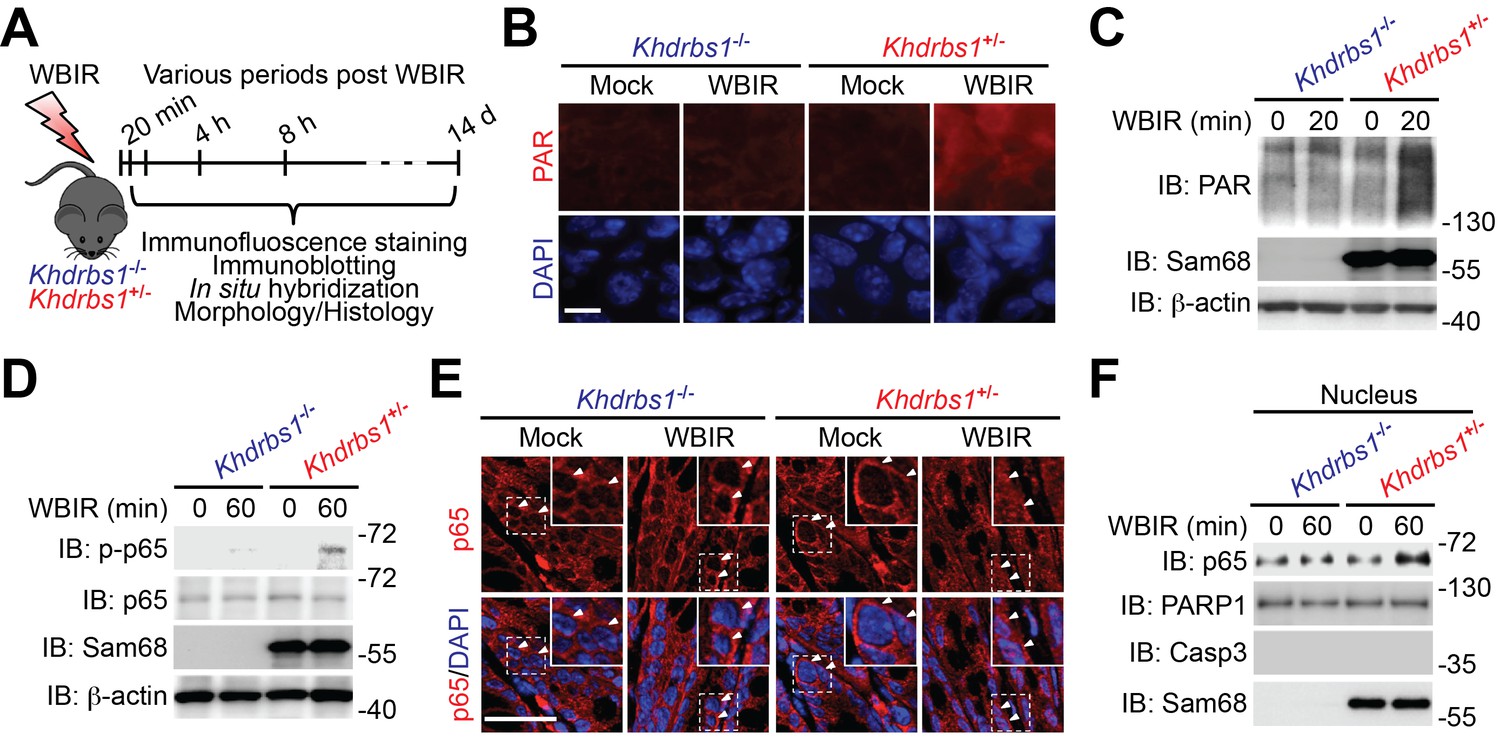
Sam68 deletion diminishes genotoxic stress-induced NF-κB signaling in the γ-irradiated colon.
(A) A schematic of the experimental timeline for the impact of Sam68 deletion on DNA damage-induced NF-κB signaling pathway in γ-irradiated mice. Khdrbs1+/− and Khdrbs1−/− mice subjected to a sublethal dose (6.5 Gy) of whole body γ-irradiation (WBIR) or mock irradiation were euthanized at the indicated periods post WBIR, followed by the analyses as indicated. (B) Immunofluorescence micrographs of PAR in colon tissue collected from Khdrbs1+/− and Khdrbs1−/− mice at 20 min following WBIR or mock irradiation, with nuclei counterstained by DAPI. Scale bar, 25 μm. (C and D) Colon epithelial cells (CECs) were isolated from Khdrbs1+/− and Khdrbs1−/− mice at the indicated periods post WBIR, and whole cell lysates were derived and immunoblotted (IB) for indicated proteins, with β-actin as a loading control. p-p65, phosphorylated p65. (E) Immunofluorescence micrographs of p65 in colon tissue collected from Khdrbs1+/− and Khdrbs1−/− mice at 60 min post WBIR. Scale bar, 50 μm. (F) CECs were collected from Khdrbs1+/− and Khdrbs1−/− mice as treated in (E) and nuclear fractions were derived and IB for indicated proteins. Caspase-3 (Casp3) and PARP1 served as loading controls and cytosolic and nuclear markers, respectively.
Sam68 is critical for anti-apoptotic gene transcription in the γ-irradiated colon
It has been well established that NF-κB mediates the gene transcription of a panel of anti-apoptotic molecules in cells following genotoxic stress (Fu et al., 2016; Kim et al., 2005; Stilmann et al., 2009). We therefore assessed the impact of Sam68 on γ-irradiation-induced expression of NF-κB target gene Bcl2l1, which encodes B-cell lymphoma-extra large (Bcl-XL). As illustrated by digoxigenin-labeled messenger RNA (mRNA) in situ hybridization, Bcl2l1 mRNA levels were elevated in colon tissue sections derived from whole-body γ-irradiated Khdrbs1+/− mice (Figure 2A). In contrast, WBIR-induced transcription of Bcl2l1 was substantially tempered in the γ-irradiated Khdrbs1−/− colon tissue (Figure 2A). In line with the nearly abolished γ-irradiation-initiated NF-κB signaling cascade in the colon of Khdrbs1−/− mice post WBIR (Figure 1B–F), these results demonstrate that Sam68 deletion suppresses the inducible transcription of NF-κB target genes in the colon in situ following WBIR. Mirroring the robust transcription of Bcl2l1 triggered by γ-irradiation (Figure 2A), Bcl-XL protein levels were also induced in the colon tissue derived from Khdrbs1+/− mice at 4 hr post WBIR (Figure 2B). In striking contrast, WBIR-induced Bcl-XL upregulation was diminished in the colon derived from the whole-body γ-irradiated Khdrbs1−/− animals (Figure 2B). Moreover, these results were further supported by immunoblot of Bcl-XL and another anti-apoptotic protein B-cell lymphoma 2 (Bcl2), encoded by the NF-κB target gene Bcl2, in the CEC lysates isolated from the whole-body γ-irradiated mice (Figure 2C). Hence Sam68 is essential for genotoxic stress-induced and NF-κB-mediated expression of anti-apoptotic genes in the γ-irradiated colon epithelium.
Figure 2
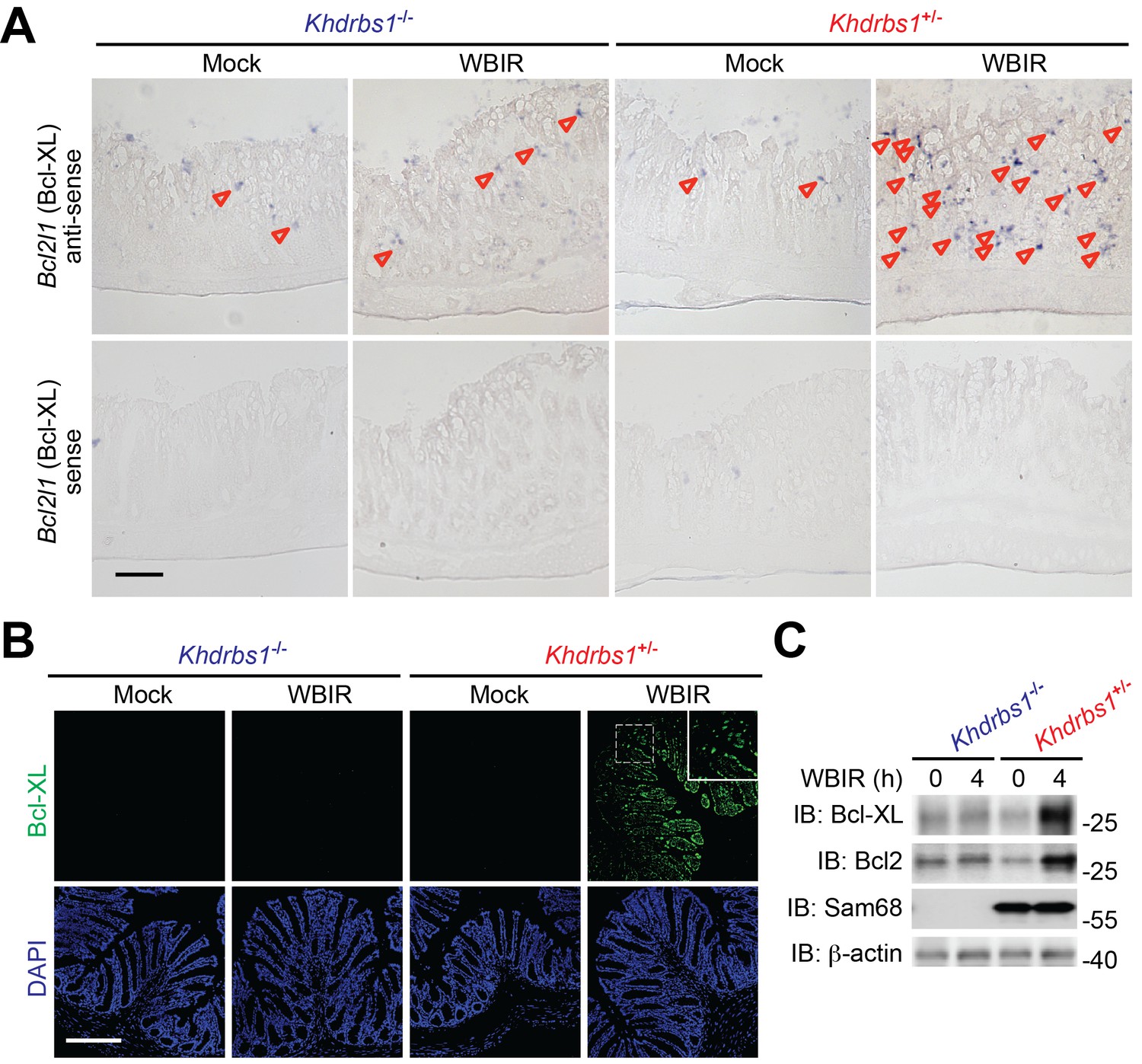
Sam68 is pivotal for NF-κB-mediated anti-apoptotic gene expression in the γ-irradiated colon.
(A) Colon tissue sections derived from Khdrbs1+/− and Khdrbs1−/− mice at 4 hr post whole body γ-irradiation (WBIR) or mock irradiation were stained by in situ hybridization with in vitro synthesized anti-sense probe targeting Bcl2l1 mRNA (purple dots as indicated by triangles), with Bcl2l1 mRNA sense probe as a negative control. Scale bar, 100 μm. (B) Immunofluorescence micrographs of Bcl-XL (encoded by Bcl2l1) in colon tissue collected from mice treated as in (A), with nuclei counterstained by DAPI. Scale bar, 200 μm. (C) Colon epithelial cells were isolated from mice treated as in (A) and whole cell lysates were derived and immunoblotted (IB) for indicated proteins, with β-actin as a loading control.
Sam68-deleted colon epithelial cells are more sensitive to whole-body γ-irradiation
The balance between severe DNA damage-triggered programmed cell death and genotoxic stress-induced NF-κB-mediated anti-apoptotic transcription is pivotal for cell fate determination in cellular responses to DNA-damaging agents (Fu et al., 2016; Kim et al., 2005; Stilmann et al., 2009). We barely detected the cleavage of Caspase-3, one well-established biochemical hallmark for apoptosis, in the colon tissue derived from Khdrbs1+/− mice at 8 hr post WBIR (Figure 3A), as supported by the evidence that WBIR triggered profound NF-κB activation signaling (Figure 1) and expression of anti-apoptotic molecules Bcl-XL and Bcl2 (Figure 2) in Sam68-sufficient CECs. In contrast, Caspase-3 cleavage was substantially augmented in the γ-irradiated Khdrbs1−/− colon (Figure 3A), which correlates with the diminished NF-κB signaling in the nucleus (Figure 1) and inefficient anti-apoptotic gene expression (Figure 2) in the absence of Sam68. Moreover, immunoblot analyses of the CEC lysates further ascertained that the elevation in cleaved Caspase-3 and cleaved PARP1, another known biochemical hallmark for apoptosis, occurred in the whole-body γ-irradiated Khdrbs1−/− mice, but not Khdrbs1+/− controls (Figure 3B). Such an inverse correlation between NF-κB-mediated anti-apoptotic gene expression and DNA damage-triggered apoptosis underscores the crucial function of Sam68 in genotoxic stress-induced NF-κB signaling and transactivation in the γ-irradiated colon epithelium. Consistently, far more apoptotic cells, as assayed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), were observed in the colon tissue sections in situ from Khdrbs1−/− mice post WBIR, compared to those from Khdrbs1+/− controls (Figure 3C–D). The amount of apoptotic cells on the colon tissue sections from the mock-γ-irradiated animals was comparable, regardless of Sam68 presence (Figure 3C–D). These results thus demonstrate that Sam68 deletion expedites CECs to undertake apoptosis in vivo, in parallel to the substantially dampened NF-κB signaling and anti-apoptotic gene expression caused by genotoxic stress, in the mice subjected to WBIR.
Figure 3
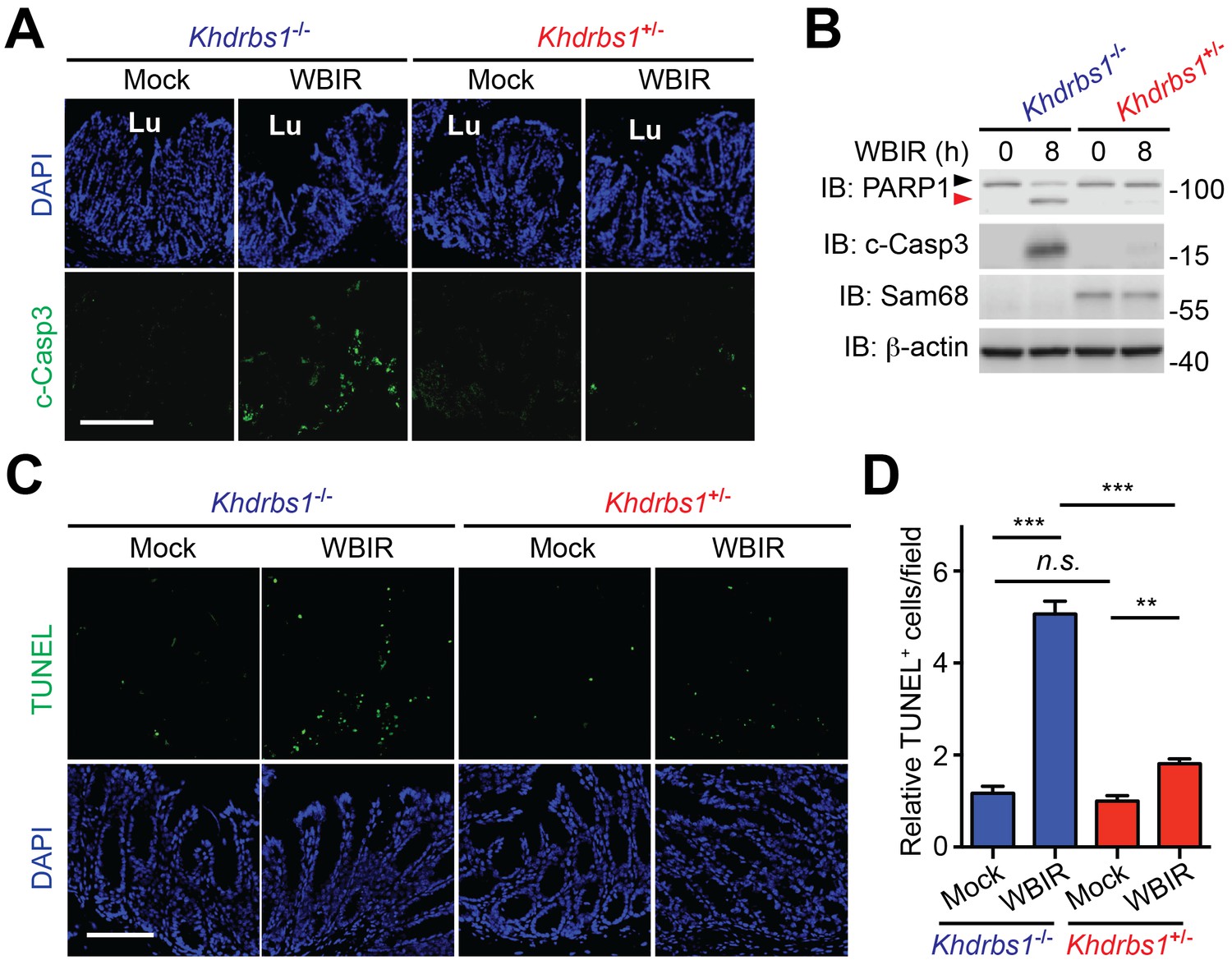
Sam68 deletion sensitizes colon epithelial cells to undergo apoptosis in the γ-irradiated mice.
(A) Immunofluorescence micrographs of cleaved Caspase-3 (c-Casp3) in colon tissue collected from Khdrbs1+/− and Khdrbs1−/− mice at 8 hr post whole body γ-irradiation (WBIR) or mock irradiation, with nuclei counterstained by DAPI. Lu, lumen; Scale bar, 200 μm. (B) Colon epithelial cells were isolated from mice treated as in (A) and whole cell lysates were derived and immunoblotted (IB) for indicated proteins, with β-actin as a loading control. The full-length and cleaved PARP1 are indicated by a black triangle and a red triangle, respectively. (C) Micrographs of TUNEL staining in colon tissue collected from mice treated as in (A), with nuclei counterstained by DAPI. Scale bar, 100 μm. (D) Relative cells with TUNEL staining from four random fields, as in (C), were quantified. Data are representative of at least two independent experiments. Results in (D) are expressed as mean and s.e.m. n.s., non-significant difference and **p<0.01, ***p<0.001 by Student’s t tests.
Sam68 is crucial for the NF-κB-mediated radioprotection in the colon of γ-irradiated animals
Previous studies reveal that the intestine and the colon are hypersensitive to radiotoxicity (Barlow et al., 1996; de Murcia et al., 1997; Gannon et al., 2012) and that NF-κB signaling pathway executes an important protective function in the γ-irradiated colon (Egan et al., 2004). To assess the impact of Sam68 on the radiodamage to the colon tissue, we examined the morphology of the colon from mice, relative to mock-treated controls, by gross dissection and histological staining. Indeed, the colonic morphology, length, and structure of mock-irradiated Khdrbs1+/− and Khdrbs1−/− mice were indistinguishable, suggesting that Sam68 is dispensable for mouse colon development (Figure 4A–E). Fourteen days post WBIR, the colons in Khdrbs1+/− mice were comparable to those from mock-irradiated animals in morphology and length (Figure 4A–C). In contrast, the γ-irradiated Khdrbs1−/− mice, compared to Khdrbs1+/− controls, suffered more severe and widespread damage to the colon, with substantially shortened colon lengths (Figure 4A–C). Moreover, histological analyses showed more severe crypt shrinkage, more goblet cell depletion, and less crypt survival in the colon derived from Khdrbs1−/− mice than in those from Khdrbs1+/− controls following WBIR (Figure 4D–E). Consequently, far fewer Khdrbs1−/− mice survived the sublethal dose of WBIR, compared to Khdrbs1+/− controls (Figure 4F), demonstrating that Sam68 deletion promotes the mice to be hypersensitive to radiotoxicity. Consistent with the reported crucial role of NF-κB signaling for providing radioprotection to the colon epithelium (Egan et al., 2004), our results emphasize that Sam68 executes a key function in genotoxic stress-induced NF-κB signaling and transactivation of a panel of anti-apoptotic genes, thus conferring radioprotection to the colon in the whole-body γ-irradiated mice.
Figure 4
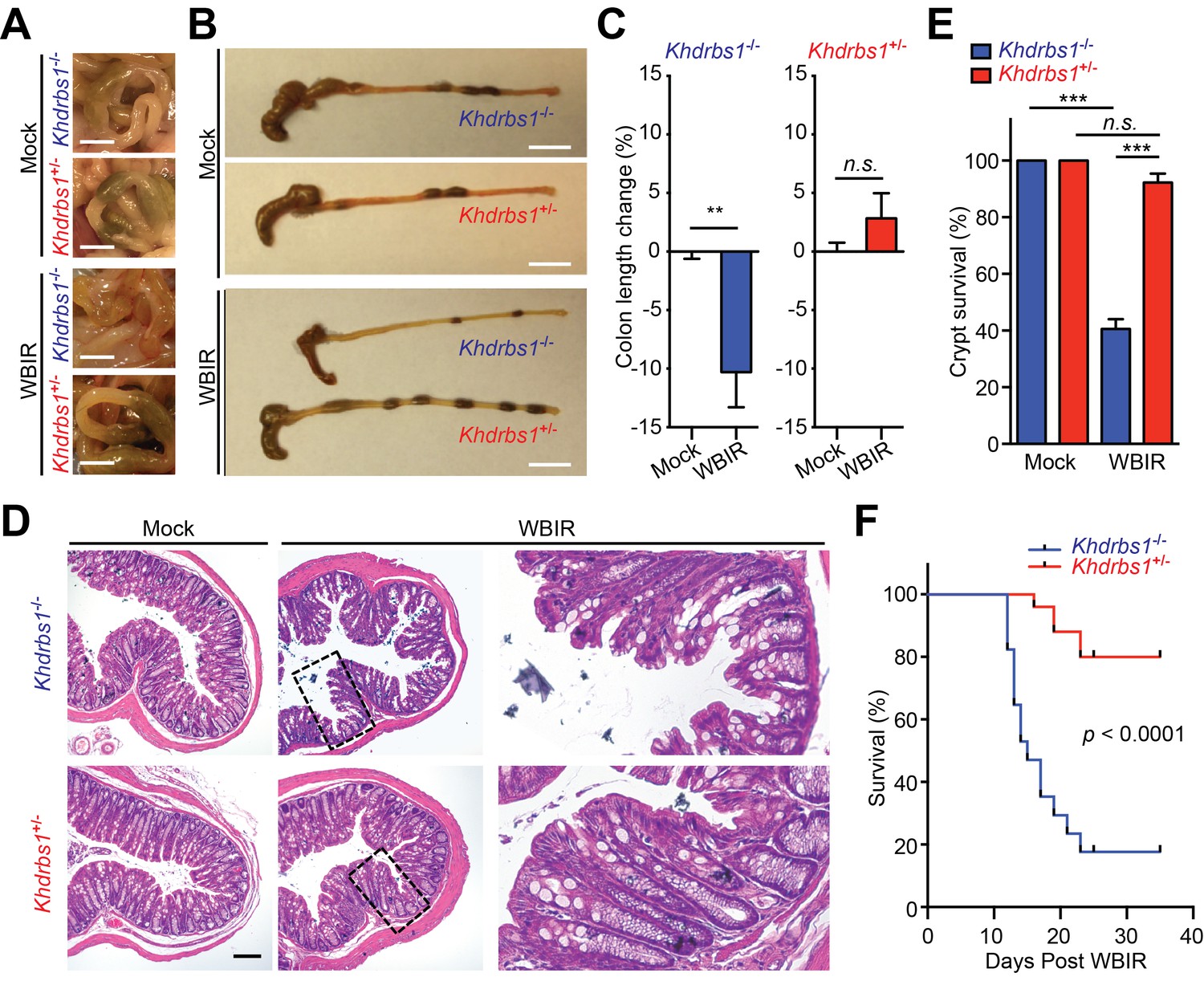
Sam68 is essential for the NF-κB-mediated radioprotection in vivo.
(A and B) Representative photographs of colons in (A) and collected from (B) Khdrbs1+/− and Khdrbs1−/− mice at 14 days post whole body γ-irradiation (WBIR) or mock irradiation. Scale bars, 5 mm (A) and 1 cm (B), respectively. (C) The length changes in the colons derived from Khdrbs1+/− (n = 3) and Khdrbs1−/− (n = 3) mice at 14 days post WBIR or mock-irradiation, normalized to mock-irradiated controls. (D) Hematoxylin and eosin staining of colon tissue sections collected from mice treated as in (A). Scale bar, 100 μm. (E) Percentage of crypt survival in (D) was quantified. (F) Kaplan-Meier analysis of the survival rate in Khdrbs1+/− (n = 25) and Khdrbs1−/− (n = 17) mice following WBIR. p<0.0001 by Gehan-Breslow-Wilcoxon test. Results in (C and E) are expressed as mean and s.e.m. n.s., non-significant difference and **p<0.01, ***p<0.001 by Student’s t tests.
Discussion
Herein, we report that Sam68 is critical for γ-irradiation-initiated NF-κB signaling and anti-apoptotic transcription in the colon in vivo and that Sam68-dependent NF-κB activation executes a protective function to the colon epithelium in the whole-body γ-irradiated animals. Sam68 deletion substantially dampens the γ-irradiation-initiated signaling cascade essential for NF-κB activation, which includes PAR synthesis, p65 phosphorylation, and p65 nuclear translocation, in the colon derived from mice at various time periods post WBIR. As a consequence, γ-irradiation-induced expression of NF-κB target genes, in particular Bcl2l1 encoding the anti-apoptotic protein Bcl-XL, is remarkably tempered in the colon epithelium from Khdrbs1−/− mice, compared to Khdrbs1+/− controls. These results are consistent with our prior report that Sam68 deletion diminishes the genotoxic stress-induced NF-κB signaling and NF-κB-mediated anti-apoptotic gene expression in the cultured MEFs and CECs in vitro (Fu et al., 2016). Moreover, WBIR fosters Khdrbs1−/− CECs to undertake apoptosis in situ in the colon from Khdrbs1−/− mice, but not Khdrbs1+/− controls, which also mirrors our prior report that Khdrbs1−/− CECs are hypersensitive to γ-irradiation and other genotoxic stresses in culture (Fu et al., 2016). Our results generated from whole-body γ-irradiated animals, along with our previous reports in the cultured cells, further support the physiological relevance of Sam68 in orchestrating genotoxic stress-initiated NF-κB activation signaling in the colon epithelium in response to genotoxic stresses. As elucidated previously (Fu et al., 2016), knockdown/knockout of Sam68 substantially sensitizes human colon cancer cells to undergo spontaneous apoptosis and retards colon tumor development in Apcmin716/+ mice, which highlights the critical role of Sam68-dependent NF-κB transactivation in the cellular responses to the intrinsic DNA damage that occurs frequently in the rapidly-dividing/proliferating cancer cells. We show here that Khdrbs1−/− mice suffer more severe damage in the colon and succumb rapidly from acute radiotoxicity than their Khdrbs1+/− controls post the extrinsic DNA damage challenge by WBIR. These results, extending additional support to the reported key role of NF-κB in providing radioprotection to the colon epithelium (Egan et al., 2004), highlight the pathophysiological relevance of the Sam68-dependent NF-κB activation in colonic cell survival and recovery from extrinsic/environmental DNA damage.
Elevation in Sam68 protein levels has been proposed as a prognostic marker in multiple cancers (Chen et al., 2012; Liao et al., 2013; Song et al., 2010; Zhang et al., 2009), although the exact function of Sam68 in these cancers remains obscure. We recently revealed that Sam68 plays a crucial role in controlling DNA damage-induced PARP1 activation and PAR production; hence Sam68 deficiency dramatically dampens the PAR-dependent NF-κB signaling and DNA repair pathways initiated by DNA damage (Fu et al., 2016; Sun et al., 2016). As a key early signaling regulator that converges at the proxy of the DNA damage-triggered signaling cascade in the nucleus, Sam68 could provide a novel target for cancer therapeutics. In support of this notion, manipulation of Sam68 sensitizes colon cancer to DNA damage-triggered apoptosis in human colon cancer cell lines and retards colon tumor burden in Apcmin716/+ mice (Fu et al., 2016). Besides its crucial role in cancer cells to overcome the frequently-occurred intrinsic DNA damage, our results here demonstrate that Sam68-dependent NF-κB transactivation is pivotal for normal cells in the colon epithelium by executing an important physiological function to prevent the radiodamage to the colon caused by extrinsic/environmental γ-irradiation. The levels of Sam68 proteins in both normal and cancerous colon tissues could be a potential biomarker to facilitate the optimization of the administered dose of γ-irradiation, when employed as a single therapy or combined with other means for cancer treatment, in order to achieve superior outcomes via an elegant balance between the antitumor effects to tumor tissue and the acute side-effects to normal tissue caused by γ-irradiation.
Materials and methods
Mice and ethics statement
Request a detailed protocolAll animal experiments were performed according to protocol number MO16-H285, approved by the Johns Hopkins University’s Animal Care and Use Committee and in direct accordance with the NIH guidelines for housing and care of laboratory animals. Khdrbs1−/− mice and their gender-matched littermate Khdrbs1+/− mice were produced using heterozygous breeding pairs, as previously described (Fu et al., 2016). Mice were maintained in a specific pathogen-free facility and fed autoclaved food and water ad libitum.
Whole-body γ-irradiation
Request a detailed protocolWhole-body γ-irradiation (WBIR) in mice was performed as previously described (Sun et al., 2016). The γ-irradiated mice were sacrificed at indicated time points post WBIR for the indicated analyses, and the mortality and survival of mice were also monitored post γ-irradiation.
Antibodies and reagents
Request a detailed protocolAntibodies used were: Sam68 (RRID: AB_631869) and p65 (RRID: AB_632037) from Santa Cruz Biotechnology (Dallas, TX); β-actin (RRID: AB_476744) from Sigma-Aldrich (St. Louis, MO); PAR (RRID: AB_2572318) from Trevigen (Gaithersburg, MD); PARP1 (RRID: AB_2160739), phospho-p65 (RRID: AB_330570), Bcl-2 (RRID: AB_1903907), and cleaved Caspase-3 (RRID: AB_2341188) from Cell Signaling Technology (Danvers, MA); Bcl-XL (RRID: AB_1949733) from GeneTex (Irvine, CA). 4',6-diamidino-2-phenylindole (DAPI) was obtained from Sigma-Aldrich.
Immunofluorescence staining
Request a detailed protocolImmunofluorescence staining on colon tissue sections was performed as we did previous (Fu et al., 2016). Briefly, after euthanizing mice, the entire colons were excised under aseptic conditions and frozen in optimal cutting temperature (O.C.T.) media (Tissue-Tek, Elkhart, IN) or embedded in paraffin (Sigma-Aldrich). Tissue sections (5-micron) were cut, collected on coated slides, fixed in paraformaldehyde, washed with PBS, and blocked with appropriate sera in PBS. After incubating with appropriate antibodies, sections were washed and incubated with fluorescence dye-conjugated second antibodies and 1 µg/ml of DAPI (Sigma-Aldrich). Stained sections were washed and mounted under a coverslip using Fluoro-gel with Tris Buffer (Electron Microscopy Sciences, Hatfield, PA) and examined using an Axio Observer fluorescence microscope (Zeiss, Oberkochen, Germany).
Isolation of primary colonic epithelial cells
Request a detailed protocolColonic epithelial cells (CECs) were isolated from mice as previously described (Fu et al., 2016; Hodgson et al., 2015).
Subcellular fractionation
Request a detailed protocolSubcellular fractionation was performed by differential centrifugation as previously described (Wan et al., 2007; Wier et al., 2012).
Immunoblot
Request a detailed protocolImmunoblot assays were conducted as previously described (Fu et al., 2013; Hodgson et al., 2015). In brief, cells were harvested and lysed on ice by 0.4 ml of lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40 and 0.5% sodium deoxycholate, 1 × complete protease inhibitor cocktail [Roche Applied Science, Indianapolis, IN]) for 30 min. The lysates were centrifuged at 10,000 × g at 4°C for 10 min. The protein-normalized lysates were separated by SDS-PAGE under reduced and denaturing conditions. The resolved protein bands were transferred onto nitrocellulose membranes and probed by the Super Signaling system (Thermo Scientific) according to the manufacturer's instructions, and imaged using a FluorChem E System (Protein Simple, Santa Clara, CA).
mRNA in situ hybridization
Request a detailed protocolDigoxigenin (DIG)-labeled probes were employed to visualize Bcl2l1 mRNA encoding Bcl-XL in colon tissues, as previously described (Hobbs et al., 2015). Briefly, Bcl2l1 gene specific sequence was first ligated to the pCRII-TOPO Vector (Life Technologies). The antisense and sense complementary RNA probes specific for Bcl2l1 mRNA were transcribed using a Lig'n Scribe Kit (Life Technologies), and then labeled with DIG using a DIG RNA labeling kits (Roche Applied Science) according to the manufacturer’s instructions. The mRNA in situ hybridization on frozen colon tissue sections was performed using adapted methods from Gu and Coulombe (2007). Briefly, colon tissues were post-fixed in 4% paraformaldehyde/PBS for 20 min, followed by proteinase K digestion at 37°C for 6 min and re-fixed in 4% paraformaldehyde/PBS, then acetylated by 0.25% acetic anhydride in 0.1 M triethanolamine (10 min). Hybridization solution containing 3 μg of each denatured DIG-labeled probe was mixed with the samples for overnight incubation at 65°C. The next day, slides were rinsed and incubated in the HSW solution (50% formamide, 0.5 × standard sodium citrate, 0.1% Tween-20) for 30 min at 65°C. The slides were then washed in the HSW solution (2 × 20 min) at 65°C, 2 × standard sodium citrate, 0.1 × standard sodium citrate at 37°, respectively. The slides were switched to blocking solution (10% normal goat serum [NGS] in PBST) for 1 hr, followed by an incubation in alkaline phosphatase (AP)-conjugated sheep anti-DIG-antibody (Roche Applied Science), 1:2000 diluted in PBST/1% NGS overnight at 4°C in the dark. To visualize the mRNA in situ hybridization signal, tissues were washed with PBST (3 × 2 hr) and NTMT (0.1 M NaCl, 0.1 M Tris-HCl [pH7.9], 50 mM MgCl2, 0.1% Tween-20) for 10 min, and incubated in BM purple AP-substrate (Roche Applied Science) containing 0.5 mg/ml levamizole overnight, then stopped the reaction by washing in PBS. Hybridized tissues are mounted in crystal/mount media in preparation for microscopy.
TUNEL assays
Request a detailed protocolTerminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) in situ on colon tissue sections were carried out using a DNA fragmentation Image Kit (Roche Applied Science), according to the manufacturer's instructions.
Histology
Request a detailed protocolHistological analyses were carried out as we did previously (Fu et al., 2016). In brief, the excised entire colons were embedded in paraffin. Tissue sections (5-micron) were cut, deparaffinised, rehydrated, and stained with hematoxylin and Eosin (H and E) staining and stained sections were washed and mounted under a coverslip and examined under light microscopy (Zeiss). The crypt survival assays (Lai and Egan, 2013) were employed to evaluate the radio-sensitivity of the colon post whole-body γ-irradiation in mice.
Statistical analyses
Request a detailed protocolAll statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA). Standard errors of means (s.e.m.) were plotted in graphs. Significant differences were considered: ns, non-significant difference; * at p<0.05; ** at p<0.01; *** at p<0.001; **** at p<0.0001 by unpaired Student’s t-test.
References
-
Overexpression and cytoplasmic localization of Sam68 correlate with tumour progression and poor prognosis in patients with clinically N0 oral tongue cancerHead & Neck Oncology 4:61.
-
Keratin expression provides novel insight into the morphogenesis and function of the companion layer in hair folliclesThe Journal of Investigative Dermatology 127:1061–1073.https://doi.org/10.1038/sj.jid.5700673
-
Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signalGenes & Development 19:1951–1967.https://doi.org/10.1101/gad.1331805
-
ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaksJournal of Biological Chemistry 276:8898–8903.https://doi.org/10.1074/jbc.M009809200
-
Sam68, the KH domain-containing superSTARBiochimica et Biophysica Acta (BBA) - Reviews on Cancer 1653:73–86.https://doi.org/10.1016/j.bbcan.2003.09.001
-
DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside outImmunological Reviews 246:311–326.https://doi.org/10.1111/j.1600-065X.2012.01101.x
-
Reaching for the stars: Linking RNA binding proteins to diseasesAdvances in Experimental Medicine and Biology 693:142–157.
-
Regulation of nuclear factor-κB in autoimmunityTrends in Immunology 34:282–289.https://doi.org/10.1016/j.it.2013.01.004
-
Regulation and function of NF-kappaB transcription factors in the immune systemAnnual Review of Immunology 27:693–733.https://doi.org/10.1146/annurev.immunol.021908.132641
-
Identification of an N-terminal truncation of the NF-κB p65 subunit that specifically modulates ribosomal protein S3-dependent NF-κB gene expressionJournal of Biological Chemistry 287:43019–43029.https://doi.org/10.1074/jbc.M112.388694
-
Many faces of NF-kappaB signaling induced by genotoxic stressJournal of Molecular Medicine 85:1187–1202.https://doi.org/10.1007/s00109-007-0227-9
-
Expression and cytoplasmic localization of SAM68 is a significant and independent prognostic marker for renal cell carcinomaCancer Epidemiology Biomarkers & Prevention 18:2685–2693.https://doi.org/10.1158/1055-9965.EPI-09-0097
Article and author information
Author details
Funding
National Institute of General Medical Sciences (R01GM111682)
- Fengyi Wan
American Cancer Society (RSG-13-052-01-MPC)
- Fengyi Wan
National Cancer Institute (T32CA009110)
- Eric M Wier
- Ryan P Hobbs
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Xin Guo for help with histological analyses.
Ethics
Animal experimentation: All animal experiments were performed according to protocol number MO16-H285, approved by the Johns Hopkins University's Animal Care and Use Committee and in direct accordance with the NIH guidelines for housing and care of laboratory animals. Khdrbs1-/- mice and their gender-matched littermate Khdrbs1+/- mice were produced using heterozygous breeding pairs. Mice were maintained in a specific pathogen-free facility and fed autoclaved food and water ad libitum.
Version history
- Received: September 29, 2016
- Accepted: December 19, 2016
- Accepted Manuscript published: December 20, 2016 (version 1)
- Version of Record published: January 4, 2017 (version 2)
Copyright
© 2016, Fu et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 926
- views
-
- 196
- downloads
-
- 13
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Sam68/KHDRBS1-dependent NF-κB activation confers radioprotection to the colon epithelium in γ-irradiated mice
eLife 5:e21957.
https://doi.org/10.7554/eLife.21957
Further reading
-
- Cancer Biology
Tyrosine kinase inhibitors (TKI) directed against MET have been recently approved to treat advanced non-small cell lung cancer (NSCLC) harbouring activating MET mutations. This success is the consequence of a long characterization of MET mutations in cancers, which we propose to outline in this review. MET, a receptor tyrosine kinase (RTK), displays in a broad panel of cancers many deregulations liable to promote tumour progression. The first MET mutation was discovered in 1997, in hereditary papillary renal cancer (HPRC), providing the first direct link between MET mutations and cancer development. As in other RTKs, these mutations are located in the kinase domain, leading in most cases to ligand-independent MET activation. In 2014, novel MET mutations were identified in several advanced cancers, including lung cancers. These mutations alter splice sites of exon 14, causing in-frame exon 14 skipping and deletion of a regulatory domain. Because these mutations are not located in the kinase domain, they are original and their mode of action has yet to be fully elucidated. Less than five years after the discovery of such mutations, the efficacy of a MET TKI was evidenced in NSCLC patients displaying MET exon 14 skipping. Yet its use led to a resistance mechanism involving acquisition of novel and already characterized MET mutations. Furthermore, novel somatic MET mutations are constantly being discovered. The challenge is no longer to identify them but to characterize them in order to predict their transforming activity and their sensitivity or resistance to MET TKIs, in order to adapt treatment.
-
- Cancer Biology
- Genetics and Genomics
Relapse of acute myeloid leukemia (AML) is highly aggressive and often treatment refractory. We analyzed previously published AML relapse cohorts and found that 40% of relapses occur without changes in driver mutations, suggesting that non-genetic mechanisms drive relapse in a large proportion of cases. We therefore characterized epigenetic patterns of AML relapse using 26 matched diagnosis-relapse samples with ATAC-seq. This analysis identified a relapse-specific chromatin accessibility signature for mutationally stable AML, suggesting that AML undergoes epigenetic evolution at relapse independent of mutational changes. Analysis of leukemia stem cell (LSC) chromatin changes at relapse indicated that this leukemic compartment underwent significantly less epigenetic evolution than non-LSCs, while epigenetic changes in non-LSCs reflected overall evolution of the bulk leukemia. Finally, we used single-cell ATAC-seq paired with mitochondrial sequencing (mtscATAC) to map clones from diagnosis into relapse along with their epigenetic features. We found that distinct mitochondrially-defined clones exhibit more similar chromatin accessibility at relapse relative to diagnosis, demonstrating convergent epigenetic evolution in relapsed AML. These results demonstrate that epigenetic evolution is a feature of relapsed AML and that convergent epigenetic evolution can occur following treatment with induction chemotherapy.




{kind=link}
{kind=link}
{kind=link}
{kind=link}