USP5/Leon deubiquitinase confines postsynaptic growth by maintaining ubiquitin homeostasis through Ubiquilin
- Institute of Molecular Biology, Academia Sinica, Taiwan
- National Yang Ming University, Taiwan
- Chang Gung University, Taiwan
Abstract
Synapse formation and growth are tightly controlled processes. How synaptic growth is terminated after reaching proper size remains unclear. Here, we show that Leon, the Drosophila USP5 deubiquitinase, controls postsynaptic growth. In leon mutants, postsynaptic specializations of neuromuscular junctions are dramatically expanded, including the subsynaptic reticulum, the postsynaptic density, and the glutamate receptor cluster. Expansion of these postsynaptic features is caused by a disruption of ubiquitin homeostasis with accumulation of free ubiquitin chains and ubiquitinated substrates in the leon mutant. Accumulation of Ubiquilin (Ubqn), the ubiquitin receptor whose human homolog ubiquilin 2 is associated with familial amyotrophic lateral sclerosis, also contributes to defects in postsynaptic growth and ubiquitin homeostasis. Importantly, accumulations of postsynaptic proteins cause different aspects of postsynaptic overgrowth in leon mutants. Thus, the deubiquitinase Leon maintains ubiquitin homeostasis and proper Ubqn levels, preventing postsynaptic proteins from accumulation to confine postsynaptic growth.
https://doi.org/10.7554/eLife.26886.001Introduction
A synapse is a specialized structure where signals are transmitted from a neuron to another neuron or other target cells such as muscles. Proper synapse formation is prerequisite to building functional synapses and constructing neuronal circuits. Synapse abnormalities are suggested to induce neurological and psychological disorders such as autism spectrum disorders and fragile X syndrome (van Spronsen and Hoogenraad, 2010). Formation of postsynapses requires coordinated formation of several specialized structures. One prominent postsynaptic feature at neuromuscular junctions (NMJs) is the extensively folded muscular membranes. Specialized folding of postjunctional membranes is thought to increase the area exposed to the synaptic cleft and ensure the effectiveness of neuromuscular transmission (Sanes and Lichtman, 1999; Wu et al., 2010). In addition to membrane specializations, the postsynaptic density (PSD) is also a common element whose size requires proper control. The PSD contains scaffolding proteins that recruit signaling protein complexes and neurotransmitter receptors, matching precisely the presynaptic active zones (Feng and Zhang, 2009; Sheng and Hoogenraad, 2007). Formations of postsynaptic membrane and PSD are tightly controlled and coordinated yet these processes remain elusive.
The Drosophila NMJ is a model to study synapse formation and activity-dependent synapse remodeling (Collins and DiAntonio, 2007; Ruiz-Cañada and Budnik, 2006). Synaptic boutons are swollen structures of axonal terminals embedded in highly folded muscular membranes called the subsynaptic reticulum (SSR) and each bouton contains tens of neurotransmitter release sites paired with PSDs. During larval development, the SSR and the PSD concomitantly form and gradually increase their sizes. Two crucial factors, postsynaptic density protein-95/Discs large (Dlg) localized at the SSR and Drosophila p21-activating kinase (dPak) localized at the PSD, regulate SSR formation (Albin and Davis, 2004; Budnik et al., 1996; Lahey et al., 1994). At the PSD, two types of localized glutamate receptors (GluRs), IIA and IIB, appear in distinct GluR clusters (Marrus et al., 2004). The abundance of GluRIIA at the PSD is regulated by PSD-localized dPak and the SSR-localized NF-κB complex, NF-κB/Dorsal (Dl), IκB/Cactus (Cact) and IRAK/Pelle (Pll) (Albin and Davis, 2004; Heckscher et al., 2007; Parnas et al., 2001; Zhou et al., 2015). Thus, the postsynaptic protein could localize at either SSR or PSD, and confer growth regulation on SSR, PSD or both.
Ubiquitination plays essential roles in various cellular processes including synaptic growth (DiAntonio et al., 2001). Ubiquitin species are dynamically balanced among free and substrate-conjugated forms of mono-ubiquitin and ubiquitin chains. Ubiquitin homeostasis, i.e. the maintenance of diverse ubiquitin species in proper proportions and levels, is regulated in cellular growth and differentiation (Hallengren et al., 2013; Kimura and Tanaka, 2010). Deubiquitinases (DUBs), a large superfamily of ubiquitin regulators, participate in the dynamic equilibrium of ubiquitin species. While some DUBs process newly synthesized ubiquitin precursors for ubiquitin supply, others recycle ubiquitin by cleaving ubiquitin chains from protein substrates prior to proteasomal degradation. USP5, the focus of this study, is dedicated to disassembly of free ubiquitin chains for recycling (Hochstrasser, 2009; Komander et al., 2009). Physiologically, heat shock stress in yeast causes a reduction of the mono-ubiquitin level. To compensate for ubiquitin depletion, the level of the DUB Doa4 is elevated, leading to an increase in the mono-ubiquitin level by cleaving free ubiquitin chains (Kimura et al., 2009). The ataxia mice axJ, carrying mutations in the DUB USP14, displayed nerve swelling and abnormal neurotransmission at NMJs. The defects are caused by a reduction in the ubiquitin level as lower ubiquitin levels were detected in the mutant mice and introducing an ubiquitin transgene suppressed the axJ phenotypes (Chen et al., 2011, 2009). Thus, regulation of the ubiquitin level is a critical step in synapse development and for preventing neurological disorders.
Drosophila USP5/Leon is essential to maintain ubiquitin homeostasis during tissue formation and controls activation of apoptosis and the JNK pathway during eye development (Fan et al., 2014; Wang et al., 2014). In this study, we characterized the role of Leon in postsynaptic growth after synapse formation. In leon mutants, while the presynapse maintains normal morphology, the postsynapse overelaborates, displaying expanded SSR, enlarged PSD and excess PSD-localized GluR clusters. Free ubiquitin chains and ubiquitinated substrates accumulate in leon postsynapses, revealing defects in ubiquitin homeostasis. Genetic analysis shows that accumulations of several postsynaptic proteins accounts for overelaborated postsynaptic structures. The ubiquitin receptor Ubqn recognizes and transfers ubiquitinated substrates to the proteasome for degradation (Finley, 2009; Lipinszki et al., 2011). The Ubqn level is elevated in leon postsynapses and reducing the Ubqn level suppresses leon mutant phenotypes. Importantly, co-overexpression of free ubiquitin chains and Ubqn promotes expansion of these postsynaptic features. Thus, ubiquitin homeostasis such as disassembly of free ubiquitin chains, timely degradation of proteins, and normal function of the ubiquitin receptor Ubqn are compromised in leon mutants, leading to postsynaptic overgrowth.
Results
Abnormal NMJ morphology in leon mutants
We examined NMJs in third-instar larvae of leon2/19-2 mutants that died at mid-pupal stages and leon1/19-2 mutants that died at the late-third larval stages. Based on the viability and Western blot analysis, leon2/19-2 is considered a hypomorphic mutant and leon1/19-2 is a close-to-null mutant (Wang et al., 2014). Both NMJ phenotypes were compared to the control w1118 that had been used to backcross all leon alleles. At control NMJs, axonal terminals immunostained for presynaptic horseradish peroxidase (HRP) branched out extensively from initial targeting sites; synaptic boutons revealed by postsynaptic Dlg staining spread evenly along axonal tracks, displaying the beads-on-a-string pattern (Figure 1A). In leon mutants, axonal terminals failed to extend and synaptic boutons aggregated, making them larger in appearance (Figure 1A). To quantify the morphological defects, we scored the number of boutons and branch lengths of NMJs at muscles 6/7. In both leon1/19-2 and leon2/19-2 mutants, the bouton numbers were significantly decreased by about 25% (Figure 1B). Total branch length was also reduced in leon1/19-2, although the reduction was not significant in leon2/19-2 (Figure 1C). In both mutants, the muscle sizes were comparable to controls (Figure 1D). Reductions in both bouton number and branch length in leon1/19-2 were completely restored by GFP-leon-GR, a genomic rescue transgene of leon, suggesting that leon is required for normal NMJ formation (Figure 1A–C).
Figure 1 with 1 supplement see all
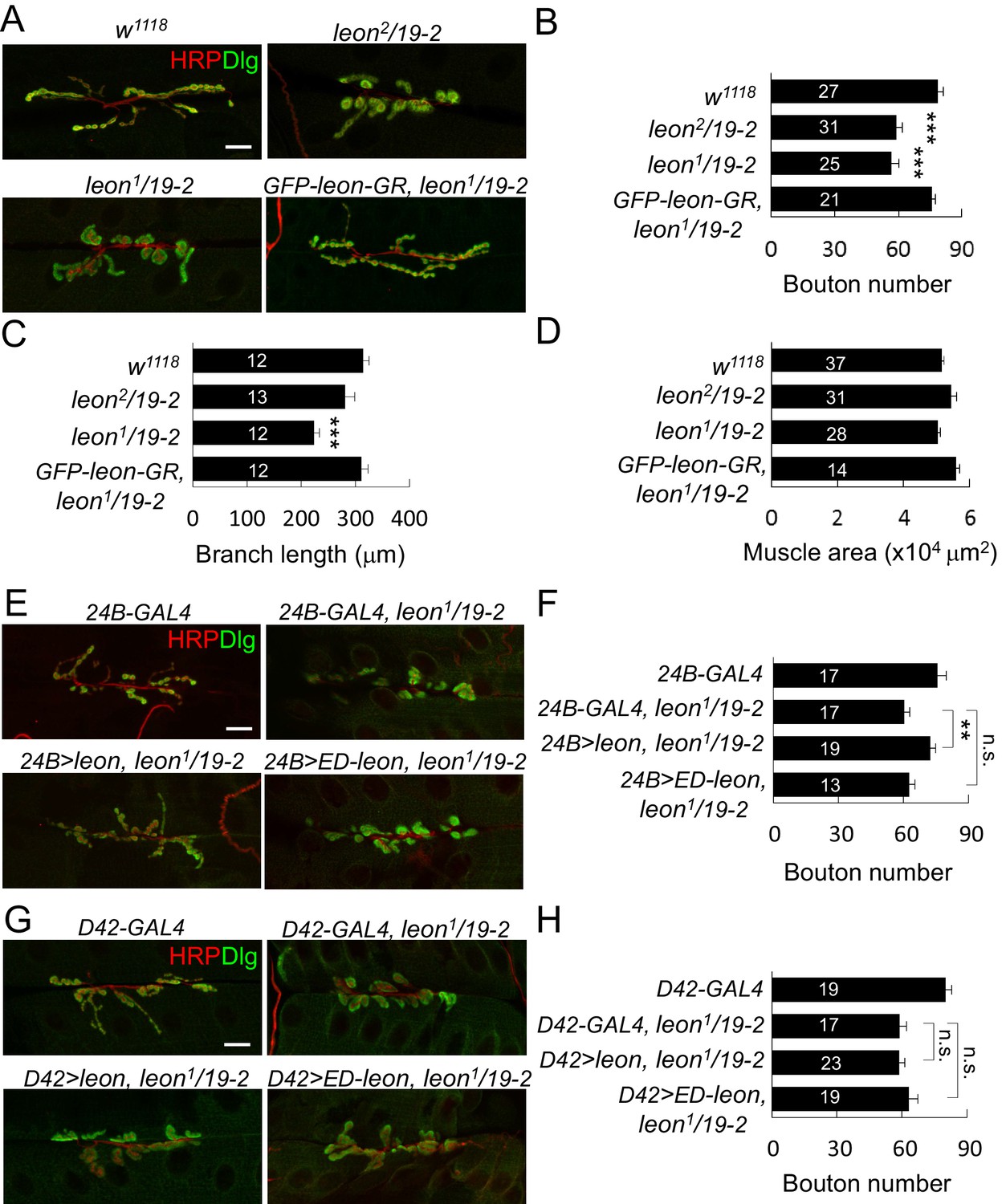
leon mutants display abnormal NMJ morphology.
(A) Immunostaining images show NMJs of w1118, leon2/19-2, leon1/19-2 and GFP-leon-GR, leon1/19-2 for HRP (red) and Dlg (green). (B–D) Bar graphs show means ± SEM (standard error of mean) of bouton numbers (B), branch lengths (C) and muscle areas (D, quantified by phalloidin staining, not shown). (E and G) Immunostaining images co-stained by HRP (red) and Dlg (green) show transgene rescue of leon1/19-2 NMJs by UAS-leon or UAS-ED-leon driven by postsynaptic 24B-GAL4 (E) or presynaptic D42-GAL4 (G). (F and H) Bar graphs show means ± SEM of bouton numbers in postsynaptic or presynaptic rescue. Scale bars, 20 μm. All data were compared to controls unless specifically indicated by brackets, with n.s. indicating no significance, * for p<0.05, and *** for p<0.001 according to Student’s t tests. The detail statistic numbers also see Supplementary file 1.
We then analyzed whether leon is required in pre- or post-synapses by performing tissue-specific rescue of leon mutants. Expression of UAS-leon by 24B-GAL4 in muscles of leon1/19-2 restored NMJ morphology and bouton number to the wild-type level (Figure 1E,F). In contrast, expression by D42-GAL4 in motor neurons failed to rescue leon mutant phenotypes (Figure 1G,H). The postsynaptic requirement of leon was further confirmed by driving the leonRNAi transgene in individual compartments. When driven by 24B-GAL4, the NMJ morphological defect was identical to that of leon mutants, with a significant reduction in the bouton number (Figure 1—figure supplement 1A,C). These phenotypes, however, were not detected by neuronal expression of leonRNAi (Figure 1—figure supplement 1B,C).
To analyze the requirement of Leon deubiquitinating activity for NMJ growth, the UAS-ED-leon transgene that expresses enzyme-dead Leon was introduced into leon mutants. Expression of UAS-ED-leon in muscles or motor neurons failed to rescue any of the leon mutant phenotypes (Figure 1E–H). Thus, in comparison to the effective rescue by wild-type UAS-leon, this result suggests that Leon functions as a DUB in regulating NMJ development.
Immunostaining of larval tissues by anti-Leon antibodies showed that Leon was expressed ubiquitously. At NMJs, Leon was enriched within synaptic boutons (Figure 1—figure supplement 1D, arrowheads). Leon was also expressed in postsynapses with lower levels in the SSR marked by Cact (Figure 1—figure supplement 1E). These Leon expressions were almost diminished in the null 19–2 homozygous larvae, confirming that these signals represent Leon expression (Figure 1—figure supplement 1D, bottom panels). Residual puncta (arrowheads) within presynaptic boutons may represent background signals. The genomic rescue transgene GFP-leon-GR showed a similar expression pattern to endogenous Leon (Figure 1—figure supplement 1F), further confirming Leon expression at the NMJ.
Defective ubiquitin homeostasis in leon postsynapses
To examine whether ubiquitin homeostasis is disrupted in the leon mutant, we first performed Western blots to analyze ubiquitin profiles in dissected body wall muscles. By blotting with the anti-ubiquitin antibodies, we found that the levels of free ubiquitin chains were increased in leon mutants (Figure 2A). Whereas moderate increases were detected in hypomorphic leon2/19-2 (2.33 ± 0.64 folds, see Materials and methods), large increases were found in null 19-2/19-2 and close-to-null leon1/19-2 (4.25 ± 1.38 and 4.62 ± 1.61 folds, respectively). In addition, smearing signals at higher molecular weights representing ubiquitinated substrates were also increased in 19-2/19-2 and leon1/19-2 (1.88 ± 0.31 and 1.76 ± 0.37 folds, respectively). The higher molecular-weight smears, however, were increased slightly in leon2/19-2 (1.29 ± 0.78 folds). In contrast, the level of monoubiquitin was strongly increased in hypomorphic leon2/19-2 (1.9 ± 0.77 folds) but the increases were not prominent in 19-2/19-2 and leon1/19-2 (0.98 ± 0.17 and 1.25 ± 0.2 folds, respectively). Therefore, the severity in defective ubiquitin homeostasis, represented by the increased levels of free ubiquitin chains and ubiquitinated substrates, correlates with increasing leon mutant strengths.
Figure 2
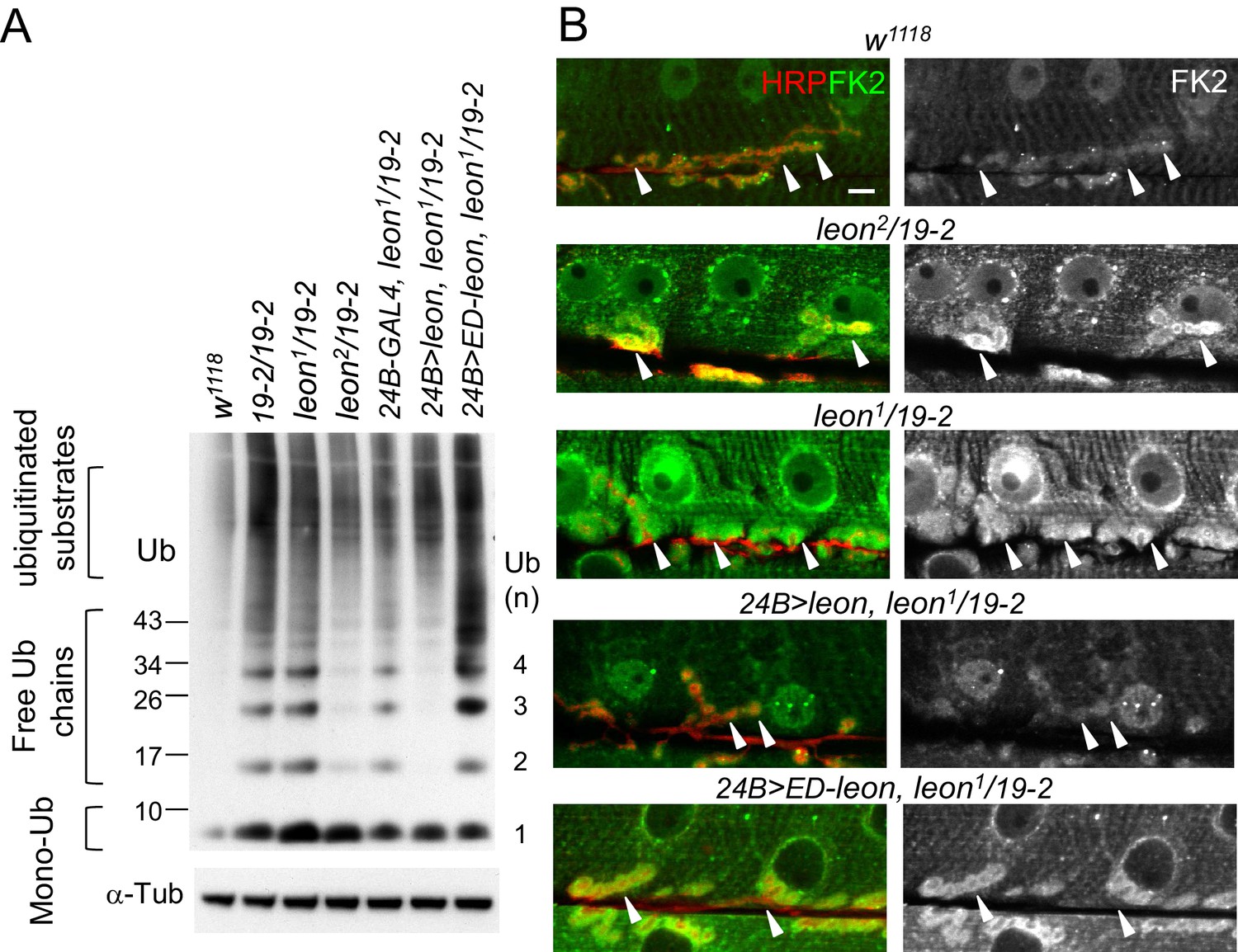
Defective ubiquitin homeostasis in leon mutants.
(A) Western blot probed with ubiquitin (Ub) antibodies shows ubiquitin expression patterns in w1118, 19-2/19-2, leon1/19-2, leon2/19-2, and postsynaptic expression of UAS-leon or UAS-ED-leon driven by 24B-GAL4 in leon1/19-2. α-Tub as control. (B) Images show FK2 (green) and HRP (red) immunostaining of NMJs in w1118, leon2/19-2 and leon1/19-2, and postsynaptic expression of UAS-leon or UAS-ED-leon driven by 24B-GAL4 in leon1/19-2. The single FK2 images (right) are also shown. Arrowheads indicate synaptic regions. Scale bar, 10 μm.
To further confirm disrupted ubiquitin homeostasis in leon mutants, the FK2 antibody that recognizes mono- and poly-ubiquitinated substrates, and free ubiquitin chains was used to immunostain dissected larval body walls. In wild-type controls, FK2 signals were detected at NMJs and in nuclei and Z-bands of muscles (Figure 2B). In leon mutants, overall FK2 signals were enhanced, and the enhancement was very prominent at the postsynaptic sites surrounding presynaptic boutons (indicated by arrowheads, Figure 2B). Consistent with Western blot analysis, the enhancement in the FK2 signal level was more pronounced in leon1/19-2 than in leon2/19-2 (Figure 2B).
We then examined the requirement of postsynaptic Leon and its deubiquitinating activity for ubiquitin homeostasis. In leon1/19-2, muscle expression of UAS-leon, but not UAS-ED-leon, by 24B-GAL4 suppressed the enhanced FK2 signals (Figure 2B). Western blot analysis showed that 24B-GAL4-driven UAS-leon expression suppressed elevated free ubiquitin chains in leon1/19-2 (0.49 ± 0.02 folds in comparison to 24B-GAL4, leon1/19-2) and partially suppressed elevated ubiquitinated substrates (0.91 ± 0.04 folds). The partial suppression could be attributed to the limited muscle expression in the mutant. Interestingly, the expression of UAS-ED-Leon induced higher levels of free ubiquitin chains (2.17 ± 0.16 folds) and ubiquitinated substrates (2.19 ± 0.43 folds). It is possible that the enzymatic activity-deficient ED-Leon can associate with ubiquitin chains but cannot deubiquitinate them, and binding to ED-Leon sequesters ubiquitinated substrates from degradation. Taken together, these results indicate that Leon deubiquitinating activity is required for maintaining ubiquitin homeostasis in postsynapses.
Increases of postsynaptic proteins in leon mutants
Aberrant NMJ morphology and postsynaptic ubiquitin homeostasis defects in leon mutants prompted us to examine constituents of synaptic organization. Several pre- and post-synaptic proteins were analyzed for their expression patterns and levels in leon mutants. Immunostaining for Dlg that is enriched in the SSR revealed a striking phenotype. In controls, SSR-localized Dlg exhibited thin circular rings in bouton sections. In leon mutants, Dlg-localized rings were expanded, showing much greater thickness and higher protein levels (Figure 3A). The thickness of Dlg-positive zones increased by 66% in leon2/19-2 and by almost two folds in leon1/19-2 in comparison to controls (Figure 3C, left panel). We further examined SSR-localized Cact and Dl expressions, which show Dlg-resembling ring patterns (Heckscher et al., 2007). In leon mutants, circular Cact and Dl patterns were also expanded (Figure 3A). As Dlg, Cact and Dl present different aspects of postsynaptic functions, with Dlg promoting SSR formation, and Cact and Dl regulating GluRIIA abundance (Budnik et al., 1996; Heckscher et al., 2007; Lahey et al., 1994), expansions of SSR-localized Dlg, Cact and Dl suggest that the SSR is also likely expanded in leon mutants.
Figure 3 with 4 supplements see all
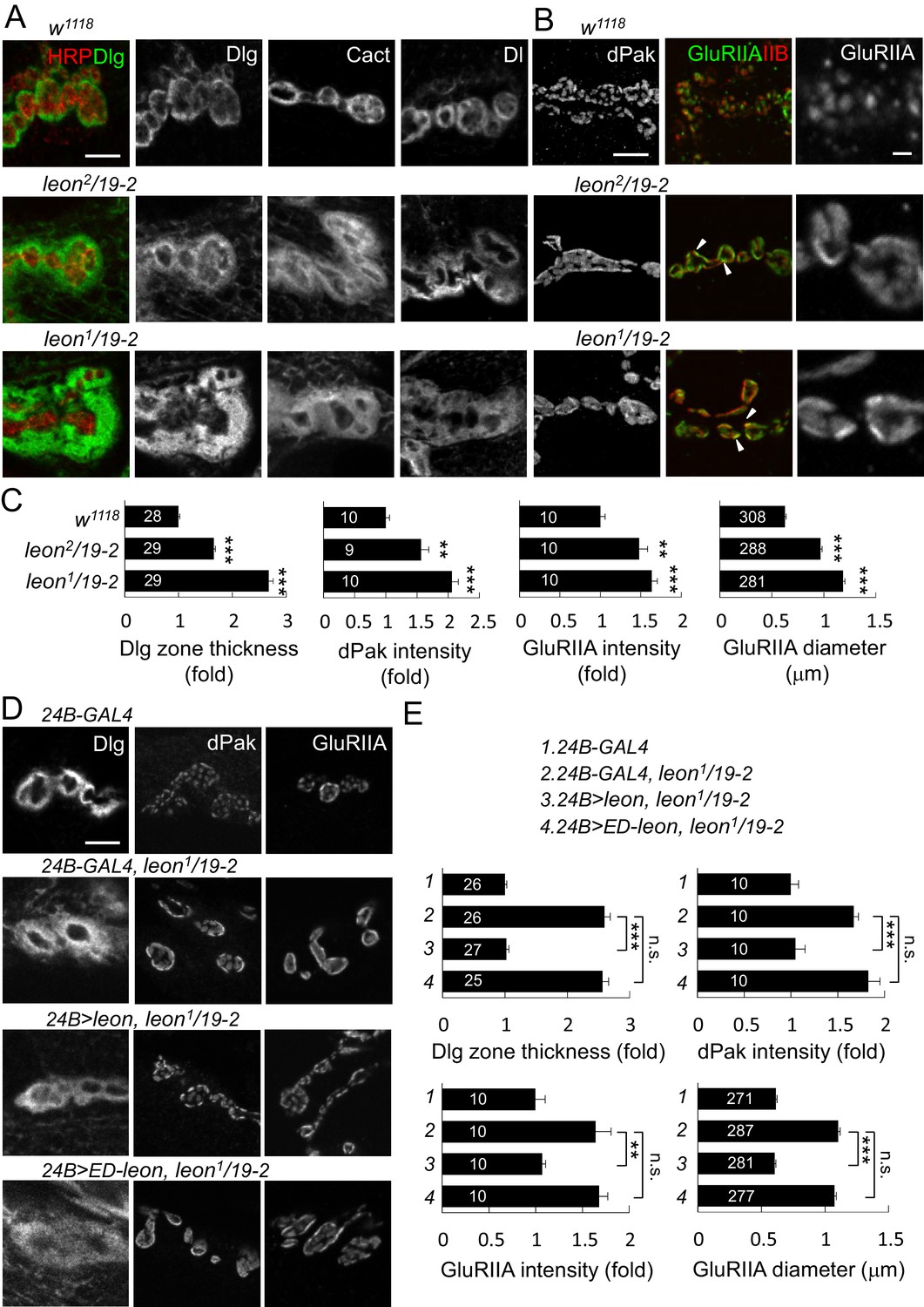
Expansion of SSR- and PSD-localized proteins at leon NMJs.
(A) Images show SSR-localized proteins Dlg, Cact and Dl in immunostaining of boutons in w1118, leon2/19-2 and leon1/19-2. Co-staining of Dlg (green) and HRP (red) is shown (left panels). (B) Images show PSD-localized dPak, GluRIIA and GluRIIB immunostaining at synapses of w1118, leon2/19-2 and leon1/19-2. Arrowheads indicate overlapping signals of expanded GluRIIA (green) and GluRIIB (red) clusters in middle panels. GluRIIA images are shown with magnification (right panels). (C) Bar graphs show means ± SEM of Dlg-positive zone thickness, dPak intensity and GluRIIA intensity and diameter in w1118, leon2/19-2 and leon1/19-2. (D) Images show Dlg, dPak and GluRIIA immunostaining at NMJs, with postsynaptic 24B-GAL4-driven UAS-leon or UAS-ED-leon expression in leon1/19-2. (E) Bar graphs show means ± SEM of Dlg-positive zone thickness, dPak intensity and GluRIIA intensity and diameter. All scale bars represent 5 μm except in magnified GluRIIA images, which is 1 μm. All data were compared to controls unless specifically indicated by brackets with n.s. indicating no significance, ** for p<0.01 and *** for p<0.001 according to Student’s t tests. The detail statistic numbers also see Supplementary file 1.
At NMJs, each bouton contains multiple release sites paired with discrete receptor clusters that can be revealed by the localizations of presynaptic ELKS/CAST family protein Bruchpilot (Brp), and postsynaptic dPak (Albin and Davis, 2004; Kittel et al., 2006; Wagh et al., 2006). Well-matched pairs of Brp and dPak were evenly distributed along the rim of and within bouton sections (Figure 3—figure supplement 1A). In leon mutants, the size, spacing and density of Brp puncta appeared normal (Figure 3—figure supplement 1A–B). Strikingly, the postsynaptic dPak patches were enormously enlarged, and the spacing among them was often diminished, appearing as a continuous structure in bouton sections, particularly in leon1/19-2 (Figure 3B). Quantification showed that the dPak levels were increased in leon postsynapses (Figure 3C). The normal one-to-one pairing between Brp and dPak could not be resolved in leon mutants because expanded and fused dPak patches could accommodate a few Brp puncta.
We further examined PSD-localized GluRs. GluRs are composed of four subunits, including essential subunits GluRIIC/GluRIII, GluRIID and GluRIIE and one of the two interchangeable subunits GluRIIA and GluRIIB (DiAntonio, 2006; Marrus et al., 2004). The GluRIIA and GluRIIB receptor clusters were distributed evenly in wild-type boutons. In leon mutants, GluRIIA and GluRIIB clusters were enlarged and overlapped, filling most of the space in bouton sections (Figure 3B, arrowheads). The wild-type GluRIIA cluster pattern, shown in the magnified image, was no longer present in leon mutants. Instead, GluRIIA clusters also appeared much like the dPak fusion pattern (Figure 3B, right panels). The average size of GluRIIA clusters was increased by 54% in leon2/19-2% and 89% in leon1/19-2 (Figure 3C). The GluRIIC clusters were also enlarged in leon mutants (Figure 3—figure supplement 1A). Statistically, the intensities of GluRIIA, GluRIIB and GluRIIC immunostaining were all elevated in both leon mutants (Figure 3C and Figure 3—figure supplement 1B). These results indicate that the levels of postsynaptic SSR- and PSD-localized proteins are increased in leon mutants.
GluR clustering appears after axonal terminals innervate muscles in mid embryonic stages and SSR is formed in the first instar stage (Guan et al., 1996; Harris and Littleton, 2015). The increases of SSR- and PSD-localized proteins in leon mutants could be a cumulative process from early larval stages. We compared expressions of SSR-localized Cact and PSD-localized GluRIIA in controls and leon mutants in the same stages. At leon NMJs, the thickness of Cact-positive zones and the intensity and size of GluRIIA clusters were larger than wild-type controls during 72 hr, 96 hr, and 120 hr AEL, with more severe defects in leon1/19-2 than in leon2/19-2 (Figure 3—figure supplement 2A–C). Therefore, the increases of SSR- and PSD-localized postsynaptic proteins in leon mutants are already prominent in early larval stages and progressively enhanced throughout later larval stages.
We then examined whether Leon deubiquitinating activity is required for proper postsynaptic growth. Expression of UAS-leon in muscles suppressed the expanded Dlg rings and the enlarged dPak and GluRIIA clusters in leon1/19-2. In contrast, enzyme-dead UAS-ED-leon failed to suppress these phenotypes (Figure 3D–E). Also, presynaptic expression of either UAS-leon or UAS-ED-leon had no effect on the increased size or intensity of the Dlg-positive zone, the dPak patch and the GluRIIA cluster in leon mutants (Figure 3—figure supplement 3A–B). To further confirm the requirement of postsynaptic leon for proper control of SSR- and PSD-localized protein levels, leonRNAi knockdown was performed in pre- or post-synaptic sites. When the leonRNAi transgene was driven by postsynaptic 24B-GAL4, the Dlg-positive zone, the dPak patch, and the GluRIIA cluster were increased (Figure 3—figure supplement 4A,C). These phenotypes, however, were not detected by presynaptic knockdown in neurons (Figure 3—figure supplement 4B,C). Taken together, these analyses suggest that the deubiquitinating activity of Leon is required in postsynapses to control postsynaptic protein levels.
With the normal distribution, size and intensity of Brp puncta in leon mutants, we examined expression patterns of other presynaptic proteins. Immunostaining for synaptic vesicle-associated Synapsin and cysteine string protein (CSP) that display a pattern of small spreading puncta in boutons (Fuentes-Medel et al., 2009), revealed no abnormality in leon mutants. The signals of microtubule-associated Futsch staining reach the terminal boutons in wild-type larvae, but stopped short without reaching terminals in leon mutants, suggesting a possible defect in the microtubule structure or stability in presynapses (Figure 3—figure supplement 1C).
Enlarged SSRs and synaptic membranes in leon mutants
We further examined the ultrastructures of leon mutant boutons by transmission electron microscopy. Folded SSR surrounding boutons (colored in pink in Figure 4A), electron-dense membranes (within the pairs of red arrows in Figure 4B) and T-bars (indicated by blue arrows) were analyzed in 19-2/19-2 and leon1/19-2. In both mutants, SSR areas were expanded and membrane folds were tightly packed, resulting in compact membrane layers surrounding the sectioned bouton (Figure 4A,A’). While the bouton sizes in both leon mutants were comparable to controls, the SSR areas were dramatically increased by as much as two-fold (Figure 4C). Thus, the ultrastructural analysis confirms the expansion of SSR suggested by analyzing the SSR-localized Dlg, Cact and Dl in leon mutants (Figure 3A).
Figure 4
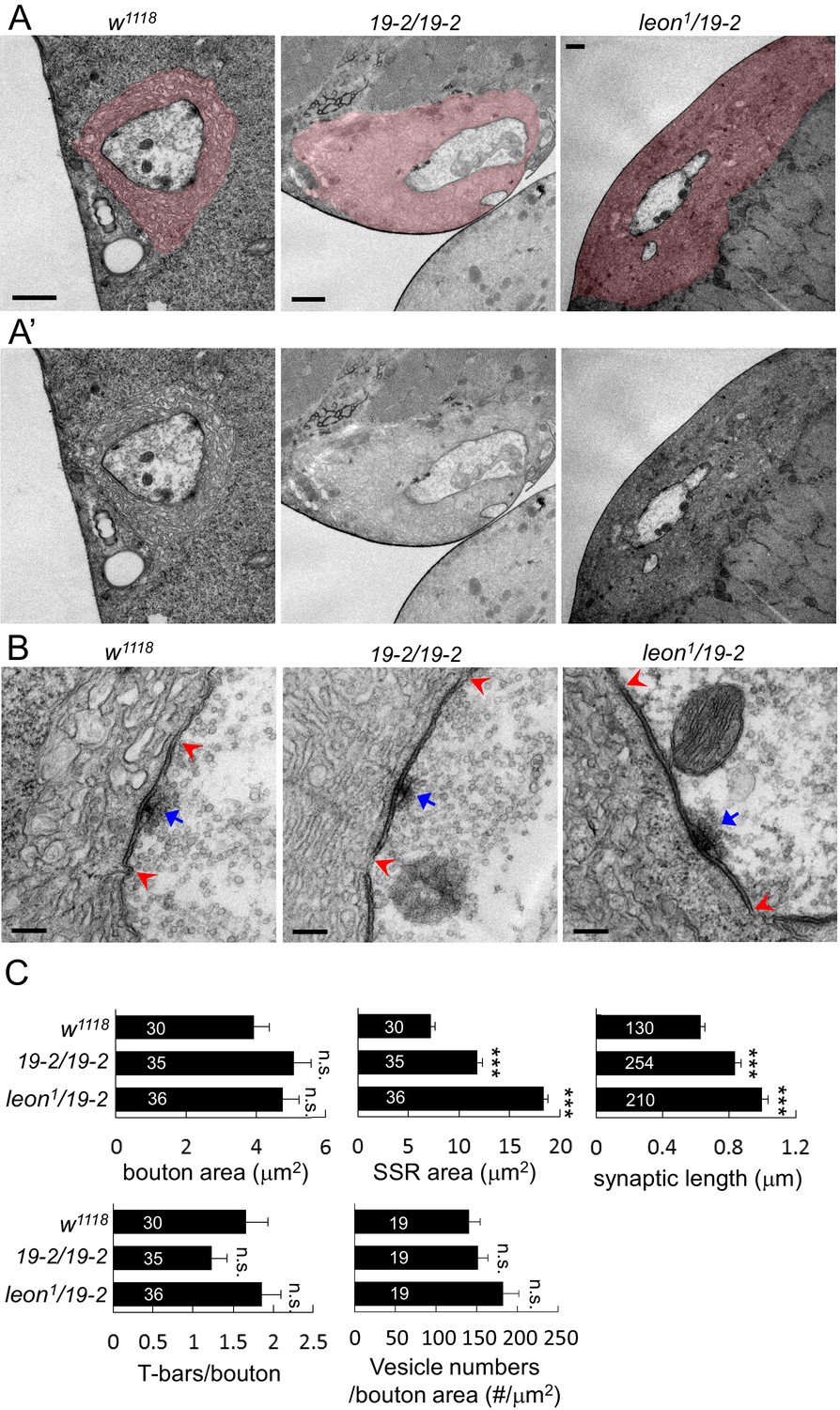
Ultrastructural analysis of leon mutant boutons showed enlarged SSR and longer synaptic membrane.
(A and A’) Electron micrographs of type Ib boutons in w1118, 19-2/19-2 and leon1/19-2. The SSRs are colored in (A). Scale bars, 1 μm. (B) Electron-dense membranes with presynaptic T-bars, postsynaptic SSRs and vesicles are shown in w1118, 19-2/19-2 and leon1/19-2. Each pair of red arrowheads delineates the boundary of synaptic membranes, and arrows (blue) indicate T-bars. Scale bars, 0.2 μm. (C) Bar graphs show means ± SEM of bouton area, SSR area, synaptic length, T-bar/bouton and vesicles/bouton areas in w1118, 19-2/19-2 and leon1/19-2. All data were compared to w1118 with n.s. indicating no significance and *** for p<0.001 by Student’s t tests. The detail statistic numbers also see Supplementary file 1.
The electron-dense synaptic membranes along the bouton circumference were also prominently increased in leon mutants (Figure 4B). The average length of synaptic membranes was significantly increased in both leon mutants (Figure 4C). Therefore, synaptic membranes account for more than 60% of the bouton circumference in leon mutants, as compared to about 35% in controls. This phenotype is consistent with the enlargement of the PSD that is suggested by immunostaining of dPak and GluRs (Figure 3B). Presynaptically, the number of T-bars and vesicle numbers showed no significant difference to controls (Figure 4C). Thus, the ultrastructural analysis confirms the expansion of SSR and PSD in leon mutants.
Impaired electrophysiological properties at leon mutant NMJs
We then examined the electrophysiological properties in leon mutants because of abnormal NMJ morphology in leon mutants. In both 19-2/19-2 and leon1/19-2 mutants, the mEJC frequencies were comparable to the +/19–2 control, showing no significant differences (Figure 5A). mEJC amplitudes in 19-2/19-2 and leon1/19-2 were slightly larger than that in +/19–2 control, although no statistical significance was detected (p=0.07 and p=0.13, respectively). Another independent mEJC amplitudes recording also suggested leon1/19-2 had slightly larger mEJC amplitudes than +/19–2 and w1118 controls (Figure 5—figure supplement 1A). Interestingly, the EJC showed a dramatic reduction in leon mutants compared to the +/19–2 control. The reduction in EJC leads to a reduction in the quantal content, determined by the ratio of the EJC amplitude to the mEJC amplitude (Figure 5A, bottom panels). Failure analysis also suggests that the quantal content was significantly reduced in the leon1/19-2 mutant (Figure 5—figure supplement 1B). Both EJC amplitude and quantal content were restored by introducing GFP-leon-GR into leon mutants (Figure 5A).
Figure 5 with 1 supplement see all
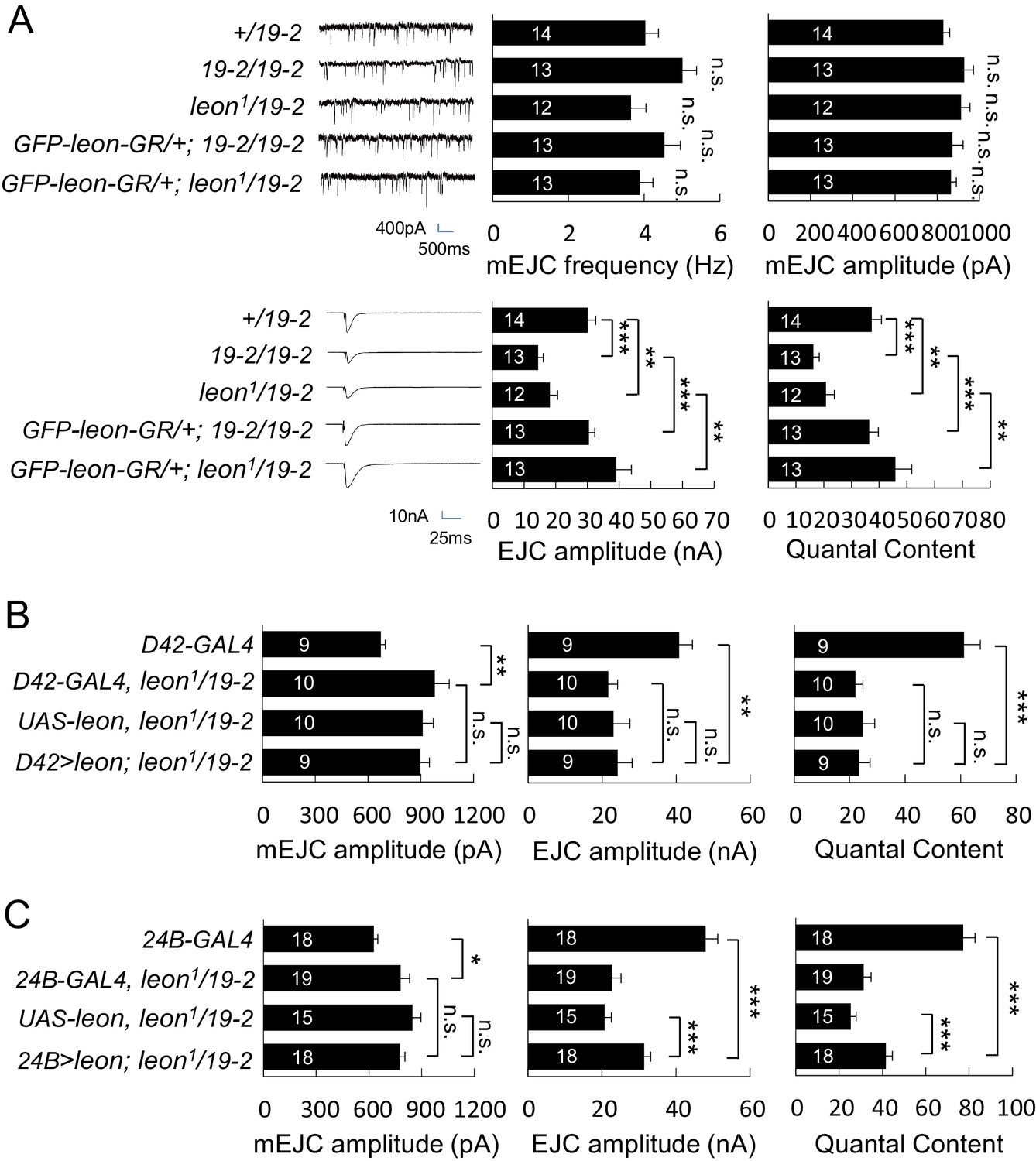
Impaired electrophysiological properties at leon mutant NMJs.
(A) Bar graphs compare frequency, mEJC amplitude, EJC amplitude, and quantal content in 19-2/19-2 and leon1/19-2 to +/19–2 control, and the rescue of 19-2/19-2 and leon1/19-2 by GFP-leon-GR. (B) Bar graphs show that presynaptic D42-GAL4-driven Leon expression failed to restore both EJC amplitude and quantal content in leon1/19-2 mutants. (C) Bar graphs show that 24B-GAL4-driven muscular expression of leon partially restored EJC amplitude and quantal content in leon1/19-2. All data were compared to controls unless specifically indicated by brackets with n.s. indicating no significance, * for p<0.05, ** for p<0.01 and *** for p<0.001 by Student’s t tests. The detail statistic numbers are in Supplementary file 1.
We then examined whether Leon is required in pre- or post-synapses for restoring the EJC and quantal content in leon1/19-2. Presynaptic Leon expression by D42-GAL4 failed to restore both EJC amplitude and quantal content in leon mutants (Figure 5B). Postsynaptic Leon expression by 24B-GAL4 partially restored the EJC amplitude and the quantal content in leon1/19-2 (Figure 5C). The mEJC in leon mutants carrying D42-GAL4 or 24B-GAL4 was significantly larger than that in respective GAL4 driver control, suggesting the mEJC increase in leon mutants might be sensitive to the variation in genetic backgrounds (Figure 5B,C). As postsynaptic expression of Leon also suppressed bouton reduction in the leon mutant, the reduction of the bouton and hence the total release sites in leon mutants could contribute partly to the electrophysiological defects. Indeed, there were about 20% reduction in the bouton number (Figure 1A,B,E and F), and about 15% reduction in the release sites at the leon mutant NMJ (Brp number per NMJ: w1118, 731.6 ± 31.6; leon2/19-2, 631.9 ± 28.1; leon1/19-2, 619.5 ± 31.1; n = 10 for all genotypes). With the 50% or more reduction in the EJC amplitude and the quantal content, other processes are also likely defective in the leon mutants.
Suppression of leon mutant phenotypes by reducing postsynaptic proteins
Accumulations of postsynaptic proteins in the SSR or the PSD of leon mutants might cause some aspects of the mutant phenotypes. To test this idea, we examined whether reductions of the gene dosage for these postsynaptic proteins would alleviate leon mutant phenotypes. We first examined SSR-localized Dlg that is required for SSR formation (Lahey et al., 1994). Replacing the wild-type allele by the null dlgX1-2 allele significantly suppressed the SSR-localized Cact expansion in leon1/19-2 (Figure 6A,B). However, the intensity and size of PSD-localized GluRIIA clusters was unaltered. Thus, this result suggests that the Dlg might mediate SSR but not PSD expansion in the leon mutant.
Figure 6 with 1 supplement see all
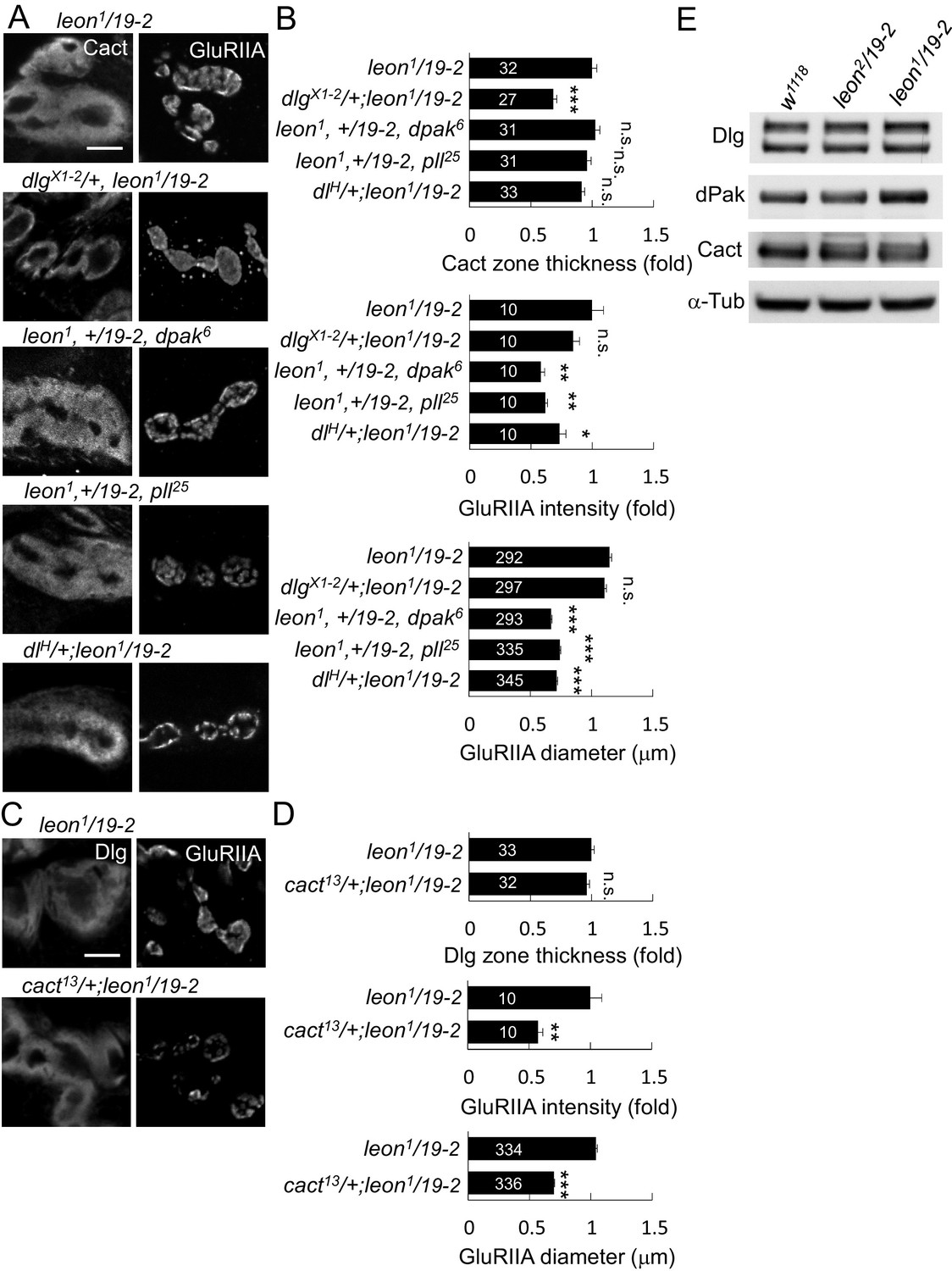
leon mutant phenotypes suppressed by reductions of gene dosages.
(A) Immunostaining images show suppression of Cact expansion in leon1/19-2 by dlgX1-2, and suppression of GluRIIA enlargement by dpak6, pll25 and dlH. (B) Bar graphs show means ± SEM of Cact-positive zone thickness and GluRIIA intensity and diameter. (C) Immunostaining images of NMJs for Dlg and GluRIIA show suppression of GluRIIA enlargement in cact13/+; leon1/19-2 as compared to leon1/19-2. (D) Bar graphs show means ± SEM of Dlg-positive zone thickness and GluRIIA intensity and diameter. Comparisons to leon1/19-2 were assessed by Student’s t tests with n.s. indicating no significance, * for p<0.05, ** for p<0.01 and *** for p<0.001. Scale bar, 5 μm. The detail statistic numbers are in Supplementary file 1. (E) Western blots show enhanced signals of Dlg, dPak and Cact in leon1/19-2 in comparison to w1118. α-Tub as control.
The PSD-localized dPak is required for GluRIIA cluster localization and SSR formation (Albin and Davis, 2004). Replacing the wild-type copy of dpak by the null dpak6 allele in leon1/19-2 suppressed the GluRIIA cluster size and intensity. The Cact-positive zone, however, was not affected (Figure 6A,B). We also examined the SSR-localized complex of Pll, Dl and Cact that regulate GluRIIA abundance at the PSD (Heckscher et al., 2007). Replacing the wild-type allele with respective pll25, dlH, or cact13 in leon1/19-2 suppressed the intensity and size of GluRIIA clusters but had no effect on the thickness of Cact- or Dlg-positive zones (Figure 6A–D). These results suggest that dPak and the Dl/Pll/Cact complex may mediate more specifically the expansion of GlRIIA clusters in the leon mutant.
To further confirm that reduced expressions of these postsynaptic proteins would suppress SSR or PSD enlargement, dlgRNAi, dpakRNAi, dlRNAi, cactRNAi, and pllRNAi that have been used previously (Dent et al., 2015; Sun and Irvine, 2011; Zhou et al., 2015) or shown in this study (Figure 6—figure supplement 1A) to effectively knockdown respective gene expression were introduced into leon1/19-2. To show the effect on the membranous SSR, muscle-expressed mCD8GFP that is enriched in the SSR membrane was used (Figure 6—figure supplement 1B). Expansion of mCD8GFP areas and enhancement of the signals were also detected in leon1/19-2, consistent with the increase of SSR (Figure 6—figure supplement 1C,D). Similarly, dlg knockdown suppressed mCD8GFP-enriched areas in leon1/19-2. However, the expanded PSD-localized GulRIIA clusters remained the same. Instead, pll, dl or cact knockdown suppressed the intensity and size of GluRIIA clusters in leon1/19-2 but had no effect on the expanded mCD8GFP area (Figure 6—figure supplement 1C,D). Interestingly, dpak knockdown in leon1/19-2 suppressed both mCD8GFP areas and GluRIIA clusters (Figure 6—figure supplement 1C,D), consistent with its role in the formation of both SSR and GluRIIA clusters (Albin and Davis, 2004). Thus, these genetic suppressions support that accumulations of postsynaptic proteins at the SSR or PSD could mediate the expansion of postsynaptic specializations in leon mutants.
We then addressed whether the accumulations of postsynaptic proteins at the SSR or PSD are accompanied with increases in the total protein levels. Western blots for examining Dlg, dPak and Cact protein levels were performed in isolated body-wall muscles of wild-type and leon mutants. Quantification of the protein levels showed that Dlg, dPak and Cact were increased in leon1/19-2 (1.19 ± 0.01, 1.3 ± 0.06, and 1.57 ± 0.28 folds, respectively), which were not significantly altered in leon2/19-2 (0.97 ± 0.06, 0.93 ± 0.09, and 1.13 ± 0.13 folds, respectively). Thus, the increases in the protein levels, as well as other mechanisms, could contribute to postsynaptic accumulations of Dlg, dPak and Cact in leon mutants.
Suppression of leon mutant phenotypes by reducing Ubqn levels
In leon mutants, ubiquitinated substrates accumulate (Figure 2A) while the enzymatic activities of the proteasome remain intact (Wang et al., 2014), suggesting that ubiquitinated substrates fail to be transported for proteasomal degradation. We examined the involvement of the ubiquitin-like (UBL) and ubiquitin-association (UBA) domain proteins (UBL-UBA) that function as ubiquitin receptors to bind and shuttle ubiquitinated substrates to the proteasome for degradation. The Drosophila genome encodes three UBA-UBL ubiquitin receptors, Rad23, Ddi1 and Ubqn/Dsk2. We then tested whether any of the UBL-UBA proteins contribute to postsynaptic defects in leon mutants. RNAi transgenes for knocking down Ubqn, Rad23 or Ddi1 were effective in suppressing respective gene expression (Figure 7—figure supplement 1A), and were introduced into leon mutants to test their suppression of leon mutant phenotypes. Interestingly, postsynaptic Ubqn knockdown suppressed the size or intensity of Dlg-positive zones, dPak patches and GluRIIA clusters in both leon2/19-2 and leon1/19-2 (Figure 7A,B and Figure 7—figure supplement 1B,C). In contrast, reductions in Rad23 or Ddi1 expression showed no obvious alteration of leon mutant phenotypes, except for a slight enhancement of Dlg-positive zones in leon2/19-2 by Rad23RNAi (Figure 7—figure supplement 1B,C). These results suggest that Ubqn plays a prominent role in mediating postsynaptic phenotypes in leon mutants. We then investigated whether Ubqn knockdown has any effect on the defective ubiquitin homeostasis in leon mutant postsynapses. The FK2 immunostaining intensity, while increased in leon mutants, was dramatically reduced in muscles and synapses in Ubqn knockdown (Figure 7A,B). Therefore, the UBL-UBA protein Ubqn mediates ubiquitin homeostasis defects and postsynaptic defects in leon mutants.
Figure 7 with 1 supplement see all
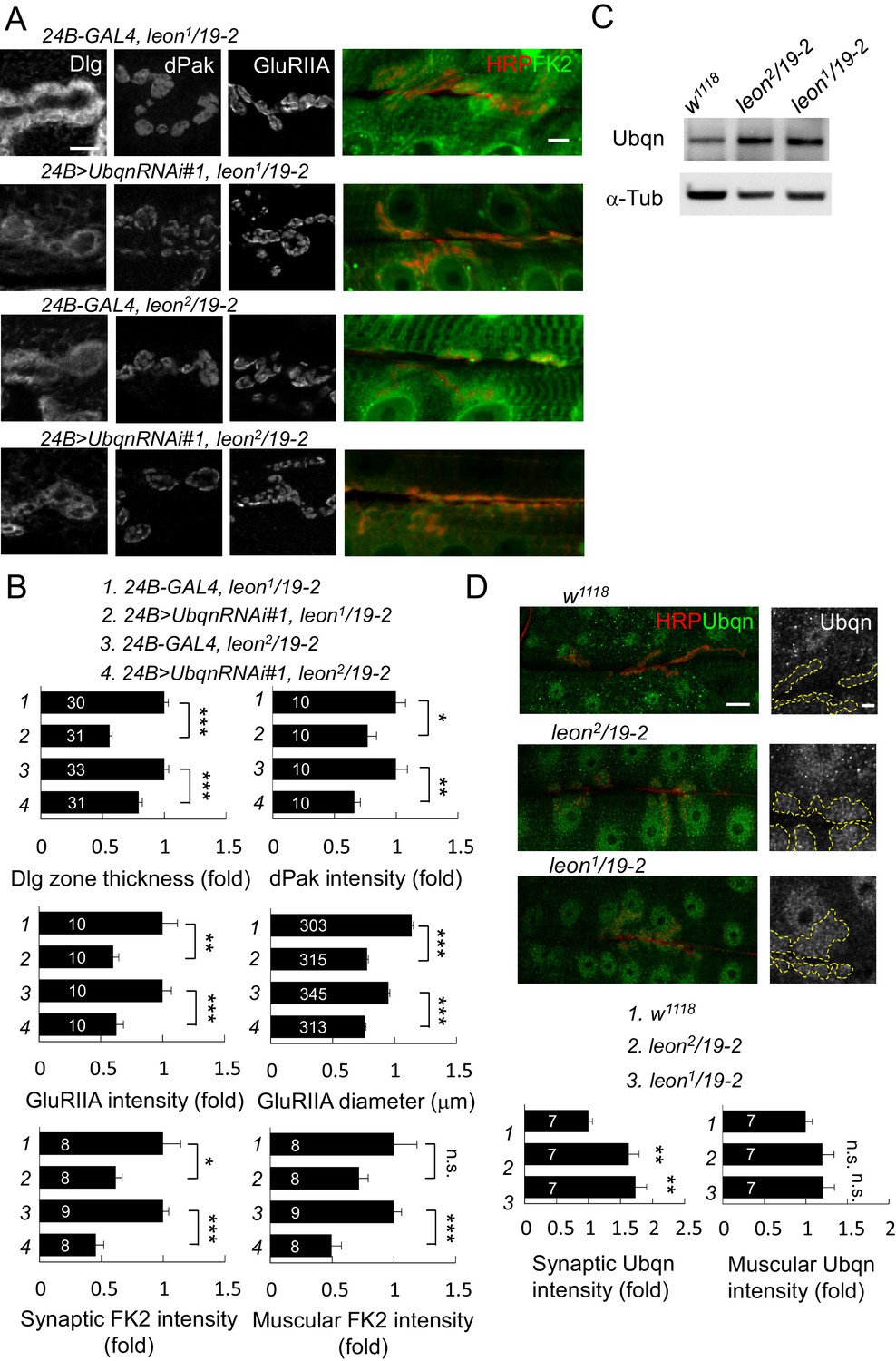
Ubqn-dependent postsynaptic expansion and ubiquitin homeostasis defects in leon mutants.
(A) Immunostaining images show suppression of Dlg, dPak and GluRIIA expansions, and reduction of FK2 intensity in leon1/19-2 and leon2/19-2 by 24B-GAL4-driven postsynaptic expression of UbqnRNAi#1. Scale bars: left, 5 μm, and right, 10 μm. (B) Bar graphs show means ± SEM of Dlg-positive zone thickness, dPak intensity, GluRIIA intensity and diameter, and FK2 intensities on synapses and muscles. (C) Western blot shows increases of Ubqn levels in leon2/19-2 and leon1/19-2 as compared to w1118. α-Tub as control. (D) Images show enhanced immunostaining signal of Ubqn (green, co-stained HRP in red) in leon2/19-2 and leon1/19-2 compared to w1118. The single Ubqn channel is also shown and yellow dashed lines delineate Cact-positive areas (Cact staining is not shown). Scale bars: left, 20 μm and right, 5 μm. Bar graphs show means ± SEM of Ubqn intensity at synaptic (left) or muscle (right) areas. All data were compared to controls unless specifically indicated by brackets with n.s. indicating no significance, * for p<0.05, ** for p<0.01 and *** for p<0.001 by Student’s t tests. The detail statistic numbers are in Supplementary file 1.
Given the reduction in Ubqn suppressed leon mutant phenotypes, we examined whether Ubqn expression is altered in leon mutants. As expected, Ubqn protein levels on Western blots showed increases to more than two folds in both leon mutants (Figure 7C, leon2/19-2: 2.2 ± 0.54 folds and leon1/19-2: 2.03 ± 0.35 folds). Immunostaining for Ubqn revealed ubiquitous expression at NMJs and in muscles, with enrichment in nuclei (Figure 7D). In leon mutants, however, postsynaptic Ubqn immunostaining signals were highly elevated at the SSR (>1.5 folds) and only weakly in other regions (1.2 folds in muscle). These results are consistent with that the increase in the Ubqn level in leon mutants could mediate expansions of Dlg-positive zones, dPak patches and GluRIIA clusters in leon mutants.
Ubqn induces and associates with ubiquitinated postsynaptic proteins
We then examined the role of Ubqn in postsynaptic development. The Flag-Ubqn transgene expressed by 24B-GAL4 in postsynapses caused expansions of Dlg-positive zones and GluRIIA clusters (Figure 8A,B), suggesting that elevated Ubqn levels could promote postsynaptic protein accumulation. However, Ubqn depletion by UbqnRNAi knockdown had no effect on these postsynaptic proteins (Figure 8A,B). As a ubiquitin receptor, Ubqn could associate with free ubiquitin chains, ubiquitinated substrates, or both in leon mutants. Immunoprecipitates of Flag-Ubqn probed by ubiquitin antibodies in Western blots displayed only smearing ubiquitin signals in high molecular weights, which were more prominent in leon mutants (Figure 8C, left panels). The ubiquitin signals represent Ubqn-associated, ubiquitinated substrates rather than ubiquitinated Ubqn, as SDS treatment of the Flag-Ubqn immunoprecipitates largely depleted the associated signals (Figure 8C, right panels). Taken together, these results indicate that Ubqn associates with ubiquitinated substrates but not free ubiquitin chains.
Figure 8
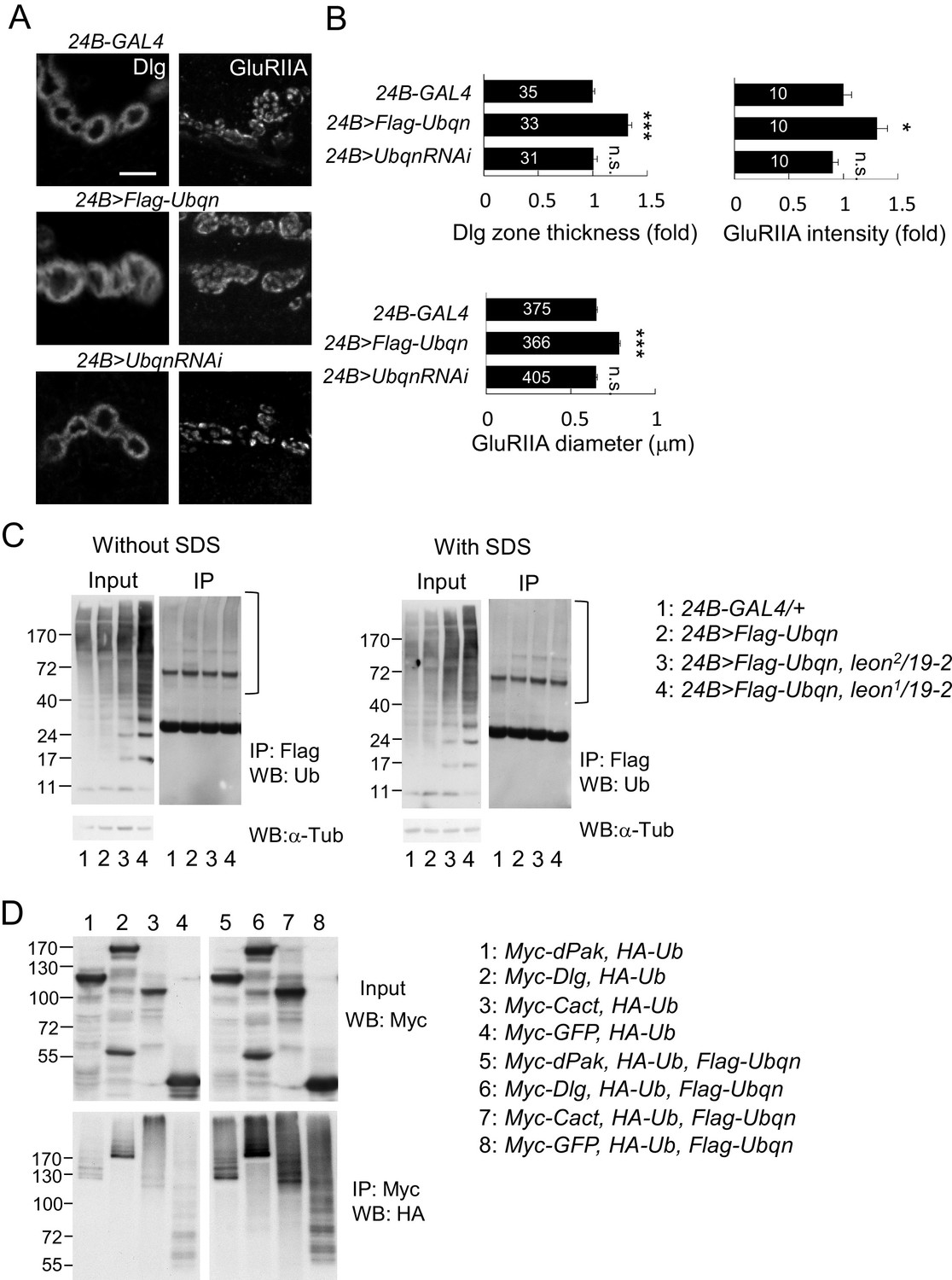
Ubqn promotes postsynaptic expansion and associates with ubiquitinated substrates.
(A) Images show 24B-GAL4-driven postsynaptic expression of Flag-Ubqn and UbqnRNAi, with immunostaining for Dlg or GluRIIA. Scale bar, 5 μm. (B) Bar graphs show means ± SEM of Dlg-positive zone thickness and GluRIIA intensity and diameter. All data were compared to 24B-GAL4 control with n.s. indicating no significance, and *** for p<0.001 according to Student’s t tests. The detail statistic numbers are in Supplementary file 1. (C) Western blots show input and immunoprecipitation of Flag-Ubqn from 24B-GAL4 control and 24B-GAL4-driven Flag-Ubqn expression in wild type, leon2/19-2 and leon1/19-2. The immunoprecipitates were probed with ubiquitin antibody. Left panels were performed in normal lysis buffer and right panels in lysis buffer containing SDS to disrupt protei association. Brackets indicate the smearing ubiquitin signals. α-Tub as control. (D) Western blots show input probed by Myc antibody (top panel) and immunoprecipitation of Myc-proteins probed by HA antibody. S2 cell transfected with plasmids for expressing HA-Ub and either Myc-dPak, Myc-Dlg, Myc-Cact, or Myc-GFP (lanes 1–4) or further co-transfected with Flag-Ubqn (lanes 5–8).
We then tested whether elevation of Ubqn could associate and stabilize ubiquitinated postsynaptic proteins. Myc-tagged dPak, Dlg, Cact and GFP were separately co-transfected with HA-Ub into Drosophila S2 cells. Myc-immunoprecipitates probed by HA antibodies in Western blots displayed smearing signals (Figure 8D, lanes 1–4), indicating they are ubiquitinated proteins. Interestingly, upon further co-transfection of Flag-Ubqn, the Myc-precipitates present stronger ubiquitination signals, suggesting that Ubqn enhances ubiquitination of postsynaptic proteins (Figure 8D, lanes 5–8). Ubiquitination of GFP was also enhanced, indicating that Ubqn likely recognizes the conjugated ubiquitin chains for binding, rather than protein substrates. In summary, we propose that Ubqn is elevated in leon postsynaptic sites to bind and stabilize ubiquitinated proteins, a mechanism that could account for the accumulation of postsynaptic proteins in leon mutants.
Induction of leon mutant phenotypes by free ubiquitin chains
In the absence of Leon deubiquitinating activity, free ubiquitin chains also accumulated in the postsynaptic area (Figure 2C). Thus, we tested whether free ubiquitin chains could induce phenotypes observed in leon mutants. To increase the level of free ubiquitin chains in vivo, the conserved C-terminal residues Gly75 and Gly76 that are essential for substrate conjugation were mutated to Ala. UbAA can be conjugated by the endogenous ubiquitin C-terminus onto the Lys48 residue, forming free ubiquitin chains, but is unable to conjugate onto substrates including endogenous ubiquitin due to the C-terminal mutations. Also, UbAA-induced free ubiquitin chains are resistant to Leon enzymatic activity as the C-terminal AA motif would prevent recognition by USP5 (Dayal et al., 2009). As controls, we also generated wild-type UAS-HA-UbGG that can conjugate to substrates, and UAS-HA-UbAA-K48R in which Lys48 was replaced by Arg, preventing ubiquitin chain formation on Lys48 of UbAA (Figure 9A). Larval lysates of UAS-HA-UbGG, UAS-HA-UbAA and UAS-HA-UbAA-K48R driven by 24B-GAL4 were probed with anti-HA antibodies in Western blot analyses (Figure 9B). As expected, UbGG produced smearing signals at higher-molecular weights, representing substrate-conjugated ubiquitin chains (lane 2). As expected, expression of UbAA formed ladders of free ubiquitin chains (lane 3). Finally, expression of UbAA-K48R failed to induce higher-molecular weight, smearing signals and free ubiquitin chains signals, except ubiquitin dimers, likely through conjugation of non-K48 residues (lane 4).
Figure 9
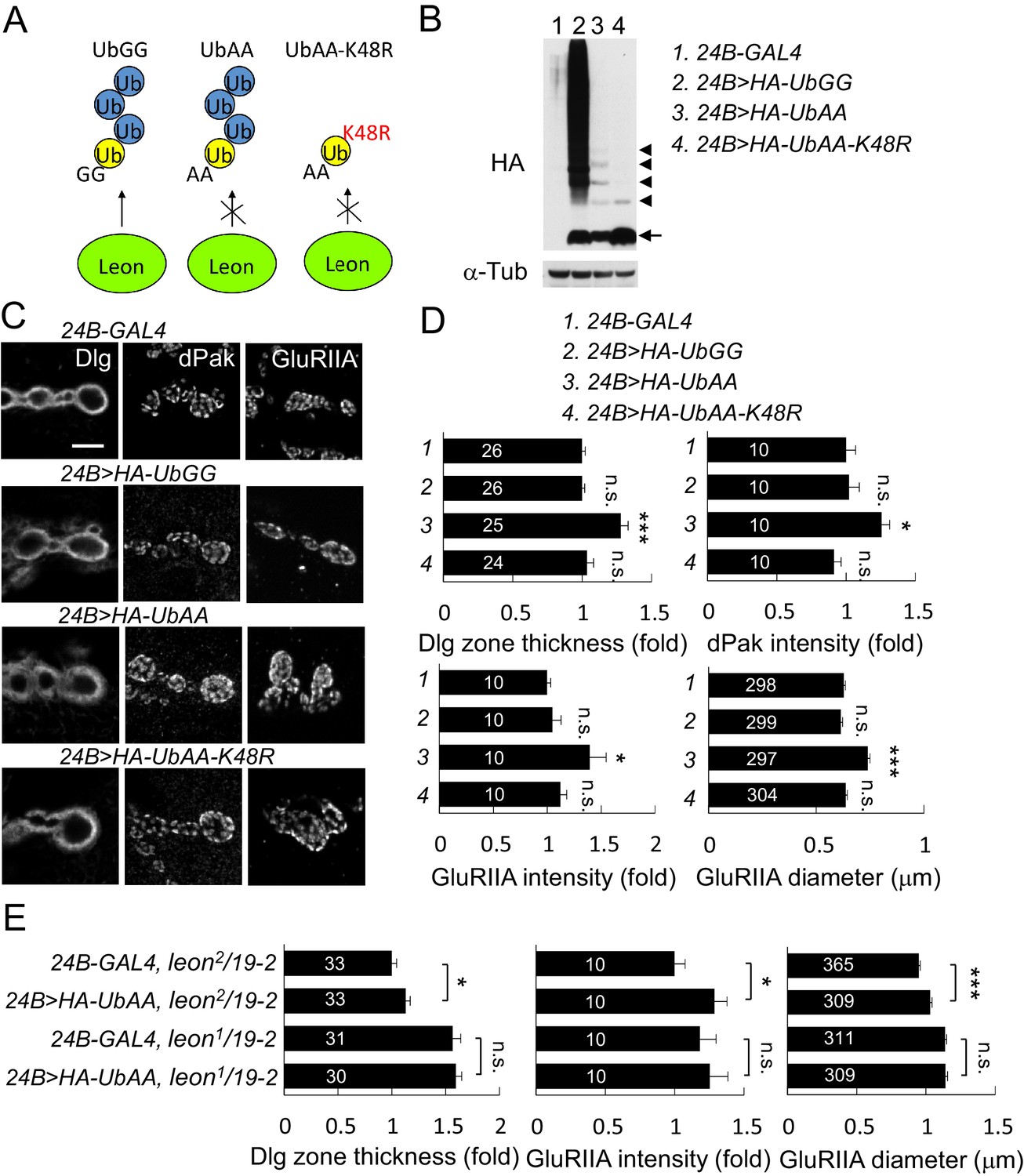
Postsynaptic defects induced by K48-linked free ubiquitin chains.
(A) Diagram shows formation of K48-linked free ubiquitin chains between ectopically expressed UbGG or UbAA (yellow), and endogenous Ub (blue). UbAA-K48R, however, cannot form K48-linked free ubiquitin chains. The free ubiquitin chains initiated by ectopic UbGG, but not UbAA, could be deconjugated by Leon. In addition, UbGG also conjugates to substrates. (B) Western blot probed with HA antibody shows HA expression patterns in 24B-GAL4 control (lane 1), 24B>HA-UbGG (lane 2), 24B>HA-UbAA (lane 3) and 24B>HA-UbAA-K48R (lane 4). α-Tub as control. Arrow indicates ubiquitin monomer and arrowheads the dimer, trimer, tetramer and pentamer. (C) Images show 24B-GAL4-driven postsynaptic expression of UbGG, UbAA and UbAA-K48R, with immunostaining for Dlg, dPak or GluRIIA. Scale bar, 5 μm. (D) Bar graphs show means ± SEM of Dlg-positive zone thickness, dPak intensity, GluRIIA intensity and diameter in 24B-GAL4-driven expression of UbGG, UbAA and UbAA-K48R. (E) Bar graphs show means ± SEM of Dlg-positive zone thickness and GluRIIA intensity and diameter in 24B-GAL4-driven UbAA expression in leon2/19-2 or leon1/19-2. All data were compared to controls unless specifically indicated by brackets with n.s. indicating no significance, * for p<0.05, and *** for p<0.001 according to Student’s t tests. The detail statistic numbers are in Supplementary file 1.
To test which types of ubiquitins have impact on postsynaptic proteins, we performed immunostaining for Dlg, dPak and GluRIIA when these Ub transgenes were expressed in muscles. Quantitative analyses showed that postsynaptic expression of UbAA induced the expansion of Dlg-positive zones and GluRIIA clusters, and enhanced the intensity of dPak and GluRIIA clusters at postsynaptic sites (Figure 9C,D). These phenotypes, however, were not detected in expression of UbGG or UbAA-K48R. Given that UbAA, but not UbGG or UbAA-K48R, induced the formation of free ubiquitin chains (Figure 9B). Thus, these results are consistent with that K48-linked free ubiquitin chains induce higher-levels of postsynaptic proteins at postsynaptic sites.
However, UbAA overexpression failed to fully recapitulate the extreme severity of leon phenotypes, partly due to the difficulty of expressing high levels of free ubiquitin chains in vivo. To further correlate the level of free ubiquitin chains and the phenotypic severity, UbAA was overexpressed in leon2/19-2 and leon1/19-2 in which lower or higher levels of free ubiquitin chains accumulated (Figure 2A). Quantification of Dlg-positive zones and GluRIIA cluster intensity and size indicated that overexpression of UbAA further enhanced the hypomorphic leon2/19-2 phenotypes, but was unable to exacerbate leon1/19-2 defects (Figure 9E). These analyses suggest that the amounts of free ubiquitin chains correlate to the protein levels at postsynaptic sites.
Recapitulation of leon mutant phenotypes by coexpression of free ubiquitin chains and Ubqn
Both levels of free ubiquitin chains and Ubqn were highly elevated in leon mutants. Also, when overexpressed, both were able to induce protein accumulation at postsynaptic sites. We then investigated the relationship between free ubiquitin chains and Ubqn in these processes. Postsynaptic expression of UbAA, but not UbGG or UbAA-K48R, slightly induced the Ubqn levels, including synaptic sites and muscles (Figure 10A,B). Conversely, overexpression of Ubqn could not induce free ubiquitin chains (Figure 10C). As UbAA only induced mild postsynaptic phenotypes accompanying with slight Ubqn elevation, we therefore tested whether co-expression of UbAA and Ubqn could further enhance these phenotypes. This experiment was performed in a leon heterozygous background, +/19–2, in which postsynapses are normal but more sensitive to the induction of phenotypes. Expression of either UbAA or Ubqn in the sensitive background induced mild enhancement of FK2 intensity in postsynaptic sites. However, combined overexpression of UbAA and Ubqn in the same background caused a large increase in FK2 immunostaining intensity (Figure 10D,E). This co-expression also resulted in larger expansion of Cact-positive zones and GluRIIA clusters than expression of either one alone (Figure 10D,E). Thus, these data suggest that Ubqn and free ubiquitin chains in leon mutants could function together to induce defective ubiquitin homeostasis, and protein accumulations at the postsynaptic site.
Figure 10
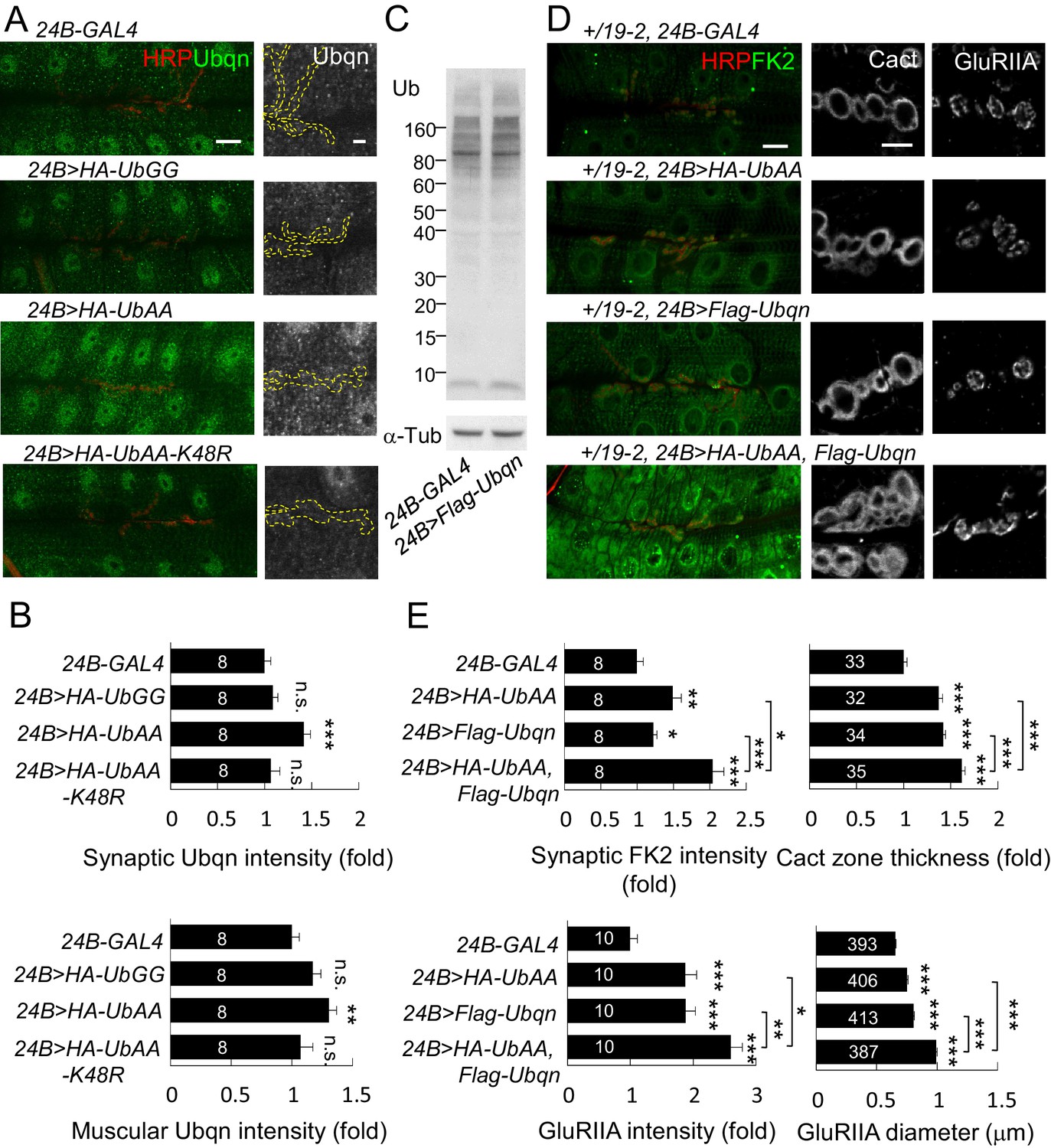
Free ubiquitin chains induce Ubqn levels and enhance postsynaptic defects when co-expressed with Ubqn.
(A) Immunostaining images show NMJs for Ubqn (green) and HRP (red) in 24B-GAL4-driven postsynaptic expression of UbGG, UbAA and UbAA-K48R. The single Ubqn channel is also shown and yellow dashed lines delineate HRP-positive areas. Scale bars: left, 20 μm and right, 5 μm. (B) Bar graphs show means ± SEM of synaptic and muscular Ubqn intensities when UbGG, UbAA or UbAA-K48R was expressed by 24B-GAL4. (C) Western blot probed with Ub antibody shows ubiquitin expression patterns in 24B-GAL4 control and 24B>Flag-Ubqn. α-Tub as control. (D) Images show NMJ immunostaining for FK2 (green, co-stained with HRP in red), Cact or GluRIIA in expression of HA-UbAA, Flag-Ubqn, or both in +/19–2 background. Scale bars: left, 20 μm and right, 5 μm. (E) Bar graphs show means ± SEM of synaptic FK2 intensity, Cact-positive zone thickness and GluRIIA intensity and diameter. All data were compared to controls unless specifically indicated by brackets with n.s. indicating no significance, * for p<0.05, ** for p<0.01, and *** for p<0.001 by Student’s t tests. The detail statistic numbers are in Supplementary file 1.
Discussion
At NMJs, postsynaptic SSR membranes are highly convoluted, showing many layers of folded membranes that surround presynaptic boutons. Each bouton contains multiple release sites where neurotransmitters are released from synaptic vesicles and received by receptors localized at the PSD (Broadie and Richmond, 2002; Collins and DiAntonio, 2007). Leon negatively regulates these unique postsynaptic specializations through control of postsynaptic protein levels. The Leon function in postsynapses is specific as presynaptic defects were not obvious, and leon mutant defects could be rescued mostly by postsynaptic Leon expression. Leon tightly controls postsynaptic specializations throughout larval development. As such dramatic and concomitant expansion of SSRs and PSDs is rarely detected in other reported mutants, our study suggests that Leon likely executes a coordinated program in confining postsynaptic specializations.
The requirement of Leon deubiquitinating activity indicates that Leon functions as a conserved member of the USP5 family to disassemble free ubiquitin chains, thereby maintaining ubiquitin homeostasis in vivo. In leon mutants, accumulations of free ubiquitin chains and ubiquitinated substrates were detected (Figure 2A). The immunostaining signals by FK2 antibodies were also highly intensified, in particular, at the postsynaptic site (Figure 2B). Thus, free ubiquitin chains and ubiquitinated substrates are two major targets that are suppressed by USP5/Leon activity in postsynapses. Postsynaptic expression of UbAA that forms free ubiquitin chains mildly enhanced FK2 intensity and induced higher-levels of SSR- and PSD-localized proteins, similar to but not as dramatic as what was observed in leon mutants. In addition to free ubiquitin chains, ubiquitinated substrates also contribute to SSR and PSD expansion. In leon mutants, accumulation of ubiquitinated substrates could be attributed to disruption of proteasomal degradation, although the enzymatic activity of the proteasome remains active (Wang et al., 2014). We therefore hypothesized that the delivery of ubiquitinated substrates to proteasomal degradation is hindered, leading to their accumulation. In supporting of this idea, we showed that reducing the level of the UBA-UBL ubiquitin shuttling protein Ubqn could suppress SSR- and PSD-localized protein levels in leon mutants. Overexpression of Ubqn also caused accumulations of ubiquitinated substrates in postsynapses. The Ubqn protein itself also accumulated at the SSR region, and bound ubiquitinated substrates. Thus, in leon mutant postsynapses, higher levels of Ubqn could stall proteasomal degradation and cause accumulation of ubiquitinated substrates in postsynapses (Figure 11). Taken together, we propose that accumulated free ubiquitin chains and Ubqn-bound ubiquitinated substrates are two primary causes of leon mutant defects.
Figure 11

Model for Leon in maintaining postsynaptic ubiquitin homeostasis and protein degradation.
Schematics show postsynaptic distributions of free and substrate-conjugated ubiquitin chains in wild-type (upper left) and leon mutant (upper right) postsynaptic sites. (bottom) The proposed pathway for Leon/Usp5 in postsynapses: Leon downregulates the levels of free ubiquitin chains through the deubiquitinating activity. Accumulation of free ubiquitin chains promotes Ubqn elevation, and Leon may suppress Ubqn levels through alternative pathways (dotted lines). Accumulated Ubqn could bind and stabilize ubiquitinated substrates. However, free ubiquitin chains and Ubqn when both are increased, collaborate to induce more accumulations of ubiquitinated substrates. Finally, accumulated substrates contribute to postsynaptic protein accumulation and SSR and PSD expansions (Solid lines: supported by experiments in this study; dash lines: proposed links).
Interestingly, Ubqn level was also upregulated in UbAA overexpression. Conversely, free ubiquitin chains maintained constant levels when Ubqn was overexpressed. These experiments would suggest that accumulation of free ubiquitin chains directly induces higher levels of Ubqn in leon mutants, which could be mediated through stabilization of Ubqn or other pathways. The effect of postsynaptic overexpression of UbAA or Ubqn only induced partial leon mutant phenotypes, such as in the levels of FK2 and postsynaptic proteins, implying tight control on ubiquitin homeostasis and protein substrate levels. Co-overexpression of UbAA and Ubqn together induced much higher FK2 intensity, and higher postsynaptic protein levels than overexpression of either one alone. Thus, Ubqn and UbAA are unlikely to function in a simple linear pathway, as the overexpression of UbAA would have suggested. Other factors such as ubiquitinated substrates have to be taken into account for the full expressivity of leon mutant phenotypes.
With the dramatic expansions of SSR and PSD in leon mutants, we assumed many postsynaptic proteins would be elevated in leon postsynapses. By examining some representative proteins, we showed that those proteins contribute to leon postsynaptic expansions. The SSR-localized Dlg was elevated and its reduction specifically alleviated SSR expansion. PSD-localized dPak was highly elevated near postsynaptic membranes, and dPak accumulation contributes SSR and PSD expansion in leon mutants. Two of SSR-localized NF-κB complex, Cact and Dl, were found to accumulate in leon mutant SSR. Eliminating one wild-type allele or RNAi knockdown of cact, dl, or pll in leon mutants also suppressed the enlargement of GluRIIA clusters, consistent with their roles in regulating GluRIIA cluster abundance. As dramatic increases of protein levels were not detected in Western blots for leon mutants, as would be expected from immunostaining of postsynaptic proteins, other mechanisms like subcellular recruitment to postsynapses could be also involved.
How these postsynaptic proteins accumulated at leon mutant postsynapses? Ubqn could be the key factor in this process. Ubqn associated with ubiquitinated substrates in leon mutants, and could stabilize ubiquitinated dPak, Dlg and Cact (Figure 8D). These results suggest that Ubqn could associate with and stabilize ubiquitinated postsynaptic proteins, which contribute to leon mutant phenotypes. In particular, Ubqn localized at postsynaptic sites and was enriched locally when leon was inactivated, providing the specificity of Leon regulation to postsynaptic specializations (Figure 11). Interestingly, while reduction of Ubqn suppressed SSR- and PSD-localized protein levels in leon mutants, it enhanced leon mutant lethality, indicating the distinct role of Ubqn in postsynapses. Our current model suggests sequential events in leon mutant postsynapses, in which elevated free ubiquitin chains induced Ubqn upregulation, Ubqn further stabilized postsynaptic proteins, and accumulated postsynaptic proteins promote SSR and PSD expansion (Figure 11). The inability to fully recapitulate leon mutant phenotypes could be due to limitation in overexpression or involvement of other factors. Thus, Leon maintains ubiquitin homeostasis through regulation of free ubiquitin chains, Ubqn levels and ubiquitinated substrates for proper development of postsynaptic specializations.
Our model that hinges on postsynaptic Ubqn has resemblances to the pathogenesis of ubiquilin 2-associated ALS (Deng et al., 2011; Ferraiuolo et al., 2011). Ubiquitin and ubiquilin 2 accumulations are common features in ALS patients, reminiscent of Ubqn and free ubiquitin chain accumulations in leon mutants. In addition to ubiquitin homeostatic imbalance and postsynaptic differentiation in leon mutants, overexpression of Ubqn causes pupal lethality and modifies TDP-43 toxicity in an ALS model (Hanson et al., 2010; Lipinszki et al., 2011). Whether ubiquilin 2 accumulation could recruit free ubiquitin chains or ubiquitinated substrates to ubiquitin inclusions and enhance disease progression will be pivotal to understand the pathological mechanisms. Thus, Leon/USP5 holds Ubqn in check and prevents ubiquitin homeostatic imbalance, making USP5 as a potential candidate disease gene in ubiquitin homeostasis-related diseases.
Materials and methods
Fly stocks
Request a detailed protocolleon1, leon2, 19–2 mutant alleles, UAS-Flag-leon and UAS-Flag-ED-leon are described in our previous study (Wang et al., 2014). All flies were reared at 25°C.GFP-leon-GR was constructed by fusing GFP to the ATG codon of leon cDNA driven by the genomic sequence between BtbVII ATG and leon ATG. Transgenic flies carrying UAS-HA-UbGG, UAS-HA-UbAA, UAS-HA-UbAA-K48R and UAS-Flag-Ubqn were generated in this study. dlgX1-2 (Zhang et al., 2007), dlH, cact13and pll25 (Heckscher et al., 2007) and UAS-UbqnRNAi#2 (Ganguly et al., 2008) have been described in respective studies. 24B-GAL4 (RRID:BDSC_1767), D42-GAL4 (RRID:BDSC_8816), da-GAL4 (RRID:BDSC_55851), UAS-mCD8GFP (RRID:BDSC_5137) and dpak6 (RRID:BDSC_8809) were obtained from Bloomington Drosophila Stock Center, Bloomington, IN. UAS-leonRNAi (ID 17567), UAS-UbqnRNAi#1 (ID 47447), UAS-Rad23RNAi (ID 30497), UAS-Ddi1RNAi (ID 40512), UAS-pllRNAi (ID 103774), UAS-dlgRNAi (ID 41136) and UAS-dpakRNAi (ID 108937) were from Vienna Drosophila Resource Center, Austria. UAS-cactRNAi (5848 R-3) was from NIG-FLY, Japan. UAS-dl-BRNAi was from Steven A. Wasserman (Zhou et al., 2015).
Immunostaining
Request a detailed protocolNMJ 6/7 phenotypes were analyzed at the A3 segment of late third instar larvae as previously described (Tsai et al., 2012a, 2012b). Larvae were dissected in cold calcium free HL3 saline (70 mM NaCl, 5 mM KCl, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, and 5 mM HEPES, pH 7.2) and larval body fillets were fixed in 4% paraformaldehyde for 20 min and washed in PBT (0.01% triton-X-100) for 15 min three times. Fixed fillets were incubated with primary antibodies overnight at 4°C, rinsed in PBT three times, and incubated with secondary antibodies for 2 hr at room temperature. Primary antibodies: mouse anti-Dlg (4F3, 1:100; Developmental Studies Hybridoma Bank, DSHB, Iowa City, IA; RRID:AB_528203), mouse anti-Brp (nc82, 1:100; DSHB; RRID:AB_2314868), mouse anti-Synapsin (3C11/SYNORF1, 1:50; DSHB; RRID:AB_528479), mouse anti-CSP (ab49, 1:100; DSHB; RRID:AB_2307345), mouse anti-Futsch (22C10, 1:200; DSHB; RRID:AB_528403), rabbit anti-Cact (1:500; RRID:AB_2314056; [Reach et al., 1996]), rabbit anti-Dl rabbit (1:500; RRID:AB_2570310), anti-dPak (1:1000; RRID:AB_2567913; [Vlachos and Harden, 2011]), rabbit anti-GluRIIB (1:1000; RRID:AB_2568753; [Marrus et al., 2004]), rabbit anti-GluRIIC (1:2500; RRID:AB_2568754; [Marrus et al., 2004]), mouse anti-FK2 (1:500; RRID:AB_10541840; Enzo Life Sciences, Farmingdale, NY.) chicken anti-GFP (1:500; RRID:AB_300798; Abcam, UK.), mouse anti-UBQLN2 (1:100; RRID:AB_565683; Abnova, Taiwan), goat anti-HRP conjugated FITC, rabbit anti-HRP conjugated TRITC or Cy5 (RRID:AB_2314647; RRID:AB_2340257; Jackson ImmunoResearch, West Grove, PA) and mouse anti-Leon (1:100) generated against GST-Leon fusion protein (LTK BioLaboratories, Taiwan). Muscles were visualized by FITC-conjugated phalloidin (1:2000; Sigma-Aldrich, St. Louis, Mo), which are not shown in figures. For GluRIIA (mouse, 1:100, RRID:AB_528269; DSHB) immunocytochemistry, larval fillets were fixed in Bouins fixative (Sigma-Aldrich.) for 5 min, followed by the protocol described above. Larval fillets were mounted onto slides with PBS containing 87.5% glycerol and 0.22M 1, 4-diaza-byciclo (2.2.2) octane (Dabco, Sigma-Aldrich.). NMJ images were acquired by confocal Z-stack scanning (Zeiss LSM510, Germany) using 40x water and 100x oil objectives and processed by LSM 5 image examiner and Adobe Photoshop.
Western blots, Immunoprecipitation and S2 cell transfection
Request a detailed protocolThird instar larva fillets were homogenized in lysis buffer (150 mM NaCl, 5 mM EDTA, 0.5% Triton-X-100, 1% NP-40 and 50 mM Tris-HCl, pH, 7.4) supplemented with protease inhibitor cocktails (Roche, Swiss), 1 mM PMSF, 1 mM DTT and 2 mM Na3VO4 and separated in Nupage 4% ~ 12% Bis-Tris gel (Thermo Fisher Scientific, Waltham, MA) for Western blots. For immunoprecipitation, larvae were lysed in lysis buffer or lysis buffer with 0.5% SDS. Lysates with 0.5% SDS buffer were further diluted to final 0.2% SDS for immunoprecipitation. Lysates after incubation with Flag M2 beads (AB_2637089; Sigma-Aldrich) overnight were washed by lysis buffer three times and separated in Nupage 4% ~ 12% Bis-Tris gel for Western blots. Drosophila S2 cell line was purchased from Invitrogen (RRID:CVCL_Z232; Invitrogen, Thermo Fisher Scientific) and Drosophila S2 cells were cultured at 25°C in Schneider’s Drosophila medium with 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific). S2 cells were transfected with plasmids by Effectene Transfection Reagent (QIAGEN, Germany). Plasmids include Ub-GAL4, Myc-dPak, Myc-Dlg, Myc-Cact, Myc-GFP, Flag-Ubqn, and HA-Ub. After 72 hr transfection, S2 cells were lysed in lysis buffer for immunoprecipitation by Myc agarose beads (9E10; Santa Cruz Biotechnology, Dallas, TX). Immunoprecipitated lysates were washed by lysis buffer three times and separated in 8% acrylamide gels. Antibodies against Dlg (1:1000), dPak (1:5000), Cact (1:5000), ubiquitin (P4D1, 1:1000; RRID; AB_628423; Santa Cruz Biotechnology), mouse anti-UBQLN2 (1:100; Abnova), α-Tubulin (1:10000; Sigma-Aldrich), anti-Myc (9E10, 1:1000; RRID:AB_627268; Santa Cruz Biotechnology) and HA-HRP (1:2000; Sigma-Aldrich) were used in Western blots.
Electron microscopy
Request a detailed protocolProcessing and analysis of ultrastructures of synaptic boutons by electron microscope were as described previously (Tsai et al., 2012b), with some modification. Larval fillets were dissected in cold calcium-free HL3 saline and fixed in modified Trump’s fixative (4% paraformaldehyde/1% glutaraldehyde/0.1 M sodium cacodylate buffer, pH 7.2) and postfixed with 1% aqueous osmium tetroxide/0.1M sodium cacodylate buffer (pH7.2). The dissected muscles 6/7 of A3 segments were stained with 2% uranyl acetate, dehydrated in a graded ethanol series and infiltrated with graded Spurr’s series in a microwave (PELCO BioWave Laboratory Microwave System, Ted Pella, Redding, CA). Thin sections (100 nm) were sectioned by ultramicrotome (Leica, Germany.) and further stained with uranyl acetate and lead citrate. Images were acquired by Tecnai G2 Spirit TWIN (FEI Co, Hillsboro, OR.) and a Gatan CCD Camera (794.10 .BP2 MultiScan). TEM data were quantified by MetaMorph V6.3r7 (Molecular Devices, Sunnyvale, CA).
Quantifications
Request a detailed protocolThe thickness of Dlg-, Cact-, or mCD8GFP-positive zone was analyzed in the original confocal Z-stack images and averaged for lengths across the zone defined by eight straight lines radiating from the bouton center. GluRIIA cluster diameters were analyzed in the original confocal Z-stack images, which were scanned with 0.5 μm intervals and covered overall boutons. Isolated GluRIIA cluster with circular shapes in different Z sections were selected to avoid choosing super-imposed ones for scoring their diameters. When elliptic cluster were chosen, the long and short diameters were averaged to reach the average diameters. Brp number and HRP-labeled NMJ areas were analyzed in the original confocal Z-stack images and Brp density was the ratio of Brp number to HRP-labeled NMJ area. Branch length was calculated from HRP labeling. GluRIIA/IIB/IIC and dPak intensities were the summation from all pixels of NMJs, which was normalized to the total HRP intensity at NMJs by Image J. Synaptic FK2 and Ubqn intensities were calculated by mean pixels in the synaptic area normalized to mean HRP pixels in the synaptic area by Image J. Muscular FK2 and Ubqn intensities were averaged from mean pixels of four muscle areas and normalized to mean HRP pixels in the postsynaptic sites by Image J. The intensities were normalized again to the control genotype for presentation in bar graphs.
To estimate expression levels from Western blots, each set of experiments was performed three times independently, and intensities for bands or areas of interest were processed by Image J with normalization to the internal control α-Tub. The areas of interest for mono-ubiquitin, free ubiquitin chains and ubiquitinated substrates are indicated in Figure 2A.
RT-PCR
Request a detailed protocolLarvae for knockdowns of UAS-UbqnRNAi (#1 and #2), UAS-Rad23RNAi, UAS-Ddi1RNAi, UAS-SyndRNAi and UAS-pllRNAi driven by da-GAL4 were reared at 25°C. mRNA of indicated genotypes was extracted from third-instar larvae by RNAzol RT (Molecular Research Center, Cincinnati, OH), followed by the reverse transcription of cDNAs by ImProm-II Reverse Transcription System through oligo-dT (Promega, Madison, WI). The mRNA expressions were amplified by PCR using Ubqn, Rad23, Ddi1, pll and Rpl19 specific primers.
Electrophysiological recordings
Request a detailed protocolFor sample preparation, larvae were dissected with the segmental nerve cut near the ventral ganglion in cold modified Ca2+-free HL3.1 saline (70 mM NaCl, 5 mM KCl, 10 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, 5 mM HEPES, pH 7.2). Preparations were then incubated in modified HL3.1 saline containing 0.6 mM CaCl2 for stimulation, and recordings were taken at room temperature. The two-electrode voltage-clamp was filled with 3 M KCl and impaled in muscle 6 of the A3 segment. One microelectrode (15 ~ 20 MΩ) monitored the muscle membrane potential while the other (5 ~ 8 MΩ) passed electric currents. The muscle membrane potential was clamped at a command value of −60 mV. mEJCs occurring in the background within 100 s were obtained without any stimulation on the segmental nerve. To record an EJC, the segmental nerve was stimulated every 10 s from the cut end that was connected by a suction electrode with 0.1 msec of pulse duration at the voltage two times that of the threshold. Signals were digitized at 50 kHz by a DigiData 1440 interface (Molecular Devices), low-pass filtered at 10 kHz, and saved on an IBM-compatible PC for analysis. For failure analysis, EJC was evoked in 0.2 mM [Ca2+], and the failure rate was calculated by ln(n/N), with n the number of failure events, and N the total number of stimuli.
References
-
Establishing and sculpting the synapse in Drosophila and C. elegansCurrent Opinion in Neurobiology 12:491–498.https://doi.org/10.1016/S0959-4388(02)00359-8
-
Ubiquitin homeostasis is critical for synaptic development and functionJournal of Neuroscience 31:17505–17513.https://doi.org/10.1523/JNEUROSCI.2922-11.2011
-
Synaptic development: insights from DrosophilaCurrent Opinion in Neurobiology 17:35–42.https://doi.org/10.1016/j.conb.2007.01.001
-
Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53Journal of Biological Chemistry 284:5030–5041.https://doi.org/10.1074/jbc.M805871200
-
Glutamate receptors at the Drosophila neuromuscular junctionInternational Review of Neurobiology 75:165–179.https://doi.org/10.1016/S0074-7742(06)75008-5
-
Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic densityNature Reviews Neuroscience 10:87–99.https://doi.org/10.1038/nrn2540
-
Molecular pathways of motor neuron injury in amyotrophic lateral sclerosisNature Reviews Neurology 7:616–630.https://doi.org/10.1038/nrneurol.2011.152
-
Recognition and processing of ubiquitin-protein conjugates by the proteasomeAnnual Review of Biochemistry 78:477–513.https://doi.org/10.1146/annurev.biochem.78.081507.101607
-
Ubiquilin antagonizes presenilin and promotes neurodegeneration in DrosophilaHuman Molecular Genetics 17:293–302.https://doi.org/10.1093/hmg/ddm305
-
Neuronal ubiquitin homeostasisCell Biochemistry and Biophysics 67:67–73.https://doi.org/10.1007/s12013-013-9634-4
-
Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS)Journal of Biological Chemistry 285:11068–11072.https://doi.org/10.1074/jbc.C109.078527
-
Regulatory mechanisms involved in the control of ubiquitin homeostasisJournal of Biochemistry 147:793–798.https://doi.org/10.1093/jb/mvq044
-
Breaking the chains: structure and function of the deubiquitinasesNature Reviews Molecular Cell Biology 10:550–563.https://doi.org/10.1038/nrm2731
-
Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junctionJournal of Neuroscience 24:1406–1415.https://doi.org/10.1523/JNEUROSCI.1575-03.2004
-
Development of the vertebrate neuromuscular junctionAnnual Review of Neuroscience 22:389–442.https://doi.org/10.1146/annurev.neuro.22.1.389
-
The postsynaptic architecture of excitatory synapses: a more quantitative viewAnnual Review of Biochemistry 76:823–847.https://doi.org/10.1146/annurev.biochem.76.060805.160029
-
Neurofibromin mediates FAK signaling in confining synapse growth at Drosophila neuromuscular junctionsJournal of Neuroscience 32:16971–16981.https://doi.org/10.1523/JNEUROSCI.1756-12.2012
-
Synapse pathology in psychiatric and neurologic diseaseCurrent Neurology and Neuroscience Reports 10:207–214.https://doi.org/10.1007/s11910-010-0104-8
-
The deubiquitinase Leon/USP5 regulates ubiquitin homeostasis during Drosophila developmentBiochemical and Biophysical Research Communications 452:369–375.https://doi.org/10.1016/j.bbrc.2014.08.069
Article and author information
Author details
Funding
Academia Sinica
- Chien-Hsiang Wang
- Yi-Chun Huang
- Pei-Yi Chen
- Ying-Ju Cheng
- Cheng-Ting Chien
Ministry of Science and Technology, Taiwan
- Chien-Hsiang Wang
- Yi-Chun Huang
- Pei-Yi Chen
- Ying-Ju Cheng
- Haiwei Pi
- Cheng-Ting Chien
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank JT Wu, Aaron DiAntonio, Steven A Wasserman, Bloomington Drosophila Stock Center, DGRC, Vienna Drosophila RNAi Center, and DSHB for providing reagents, and SP Lee and SP Tsai for technical support. CTC is supported by grants from Ministry of Science and Technology and Academia Sinica of Taiwan.
Version history
- Received: March 16, 2017
- Accepted: May 9, 2017
- Accepted Manuscript published: May 10, 2017 (version 1)
- Version of Record published: May 19, 2017 (version 2)
Copyright
© 2017, Wang et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,339
- views
-
- 300
- downloads
-
- 18
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
USP5/Leon deubiquitinase confines postsynaptic growth by maintaining ubiquitin homeostasis through Ubiquilin
eLife 6:e26886.
https://doi.org/10.7554/eLife.26886
Further reading
-
- Computational and Systems Biology
- Developmental Biology
Organisms utilize gene regulatory networks (GRN) to make fate decisions, but the regulatory mechanisms of transcription factors (TF) in GRNs are exceedingly intricate. A longstanding question in this field is how these tangled interactions synergistically contribute to decision-making procedures. To comprehensively understand the role of regulatory logic in cell fate decisions, we constructed a logic-incorporated GRN model and examined its behavior under two distinct driving forces (noise-driven and signal-driven). Under the noise-driven mode, we distilled the relationship among fate bias, regulatory logic, and noise profile. Under the signal-driven mode, we bridged regulatory logic and progression-accuracy trade-off, and uncovered distinctive trajectories of reprogramming influenced by logic motifs. In differentiation, we characterized a special logic-dependent priming stage by the solution landscape. Finally, we applied our findings to decipher three biological instances: hematopoiesis, embryogenesis, and trans-differentiation. Orthogonal to the classical analysis of expression profile, we harnessed noise patterns to construct the GRN corresponding to fate transition. Our work presents a generalizable framework for top-down fate-decision studies and a practical approach to the taxonomy of cell fate decisions.
-
- Developmental Biology
- Evolutionary Biology
Despite rapid evolution across eutherian mammals, the X-linked MIR-506 family miRNAs are located in a region flanked by two highly conserved protein-coding genes (SLITRK2 and FMR1) on the X chromosome. Intriguingly, these miRNAs are predominantly expressed in the testis, suggesting a potential role in spermatogenesis and male fertility. Here, we report that the X-linked MIR-506 family miRNAs were derived from the MER91C DNA transposons. Selective inactivation of individual miRNAs or clusters caused no discernible defects, but simultaneous ablation of five clusters containing 19 members of the MIR-506 family led to reduced male fertility in mice. Despite normal sperm counts, motility, and morphology, the KO sperm were less competitive than wild-type sperm when subjected to a polyandrous mating scheme. Transcriptomic and bioinformatic analyses revealed that these X-linked MIR-506 family miRNAs, in addition to targeting a set of conserved genes, have more targets that are critical for spermatogenesis and embryonic development during evolution. Our data suggest that the MIR-506 family miRNAs function to enhance sperm competitiveness and reproductive fitness of the male by finetuning gene expression during spermatogenesis.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}