Functional dichotomy and distinct nanoscale assemblies of a cell cycle-controlled bipolar zinc-finger regulator
- Institute of Genetics and Genomics in Geneva (iGE3), Faculty of Medicine, University of Geneva, Switzerland
- École Polytechnique Fédérale de Lausanne, Switzerland
- Institute for Cell and Molecular Biosciences, Newcastle University, United Kingdom
Abstract
Protein polarization underlies differentiation in metazoans and in bacteria. How symmetric polarization can instate functional asymmetry remains elusive. Here, we show by super-resolution photo-activated localization microscopy and edgetic mutations that the bitopic zinc-finger protein ZitP implements specialized developmental functions – pilus biogenesis and multifactorial swarming motility – while shaping distinct nanoscale (bi)polar architectures in the asymmetric model bacterium Caulobacter crescentus. Polar assemblage and accumulation of ZitP and its effector protein CpaM are orchestrated in time and space by conserved components of the cell cycle circuitry that coordinate polar morphogenesis with cell cycle progression, and also act on the master cell cycle regulator CtrA. Thus, this novel class of potentially widespread multifunctional polarity regulators is deeply embedded in the cell cycle circuitry.
https://doi.org/10.7554/eLife.18647.001eLife digest
Living cells become asymmetric for many different reasons and how they do so has been a long-standing question in biology. In some cells, the asymmetry arises because a given protein accumulates at one side of the cell. In particular, this process happens before some cells divide to produce two non-identical daughter cells that then go on to develop in very different ways – which is vital for the development of almost all multicellular organisms. The single-celled bacterium Caulobacter crescentus also undergoes this type of asymmetric division. The polarized Caulobacter cell produces two very different offsprings – a stationary cell and a nomadic cell that swims using a propeller-like structure, called a flagellum, and has projections called pili on its surface.
Before it divides asymmetrically, the Caulobacter cell must accumulate specific proteins at its extremities, or poles. Two such proteins are ZitP and CpaM, which appear to have multiple roles and are thought to interact with other factors that regulate cell division. However, little is known about how ZitP and CpaM become organized at the poles at the right time and how they interact with these regulators of cell division.
Mignolet et al. explored how ZitP becomes polarized in Caulobacter crescentus using a combination of approaches including biochemical and genetic analyses and very high-resolution microscopy. This revealed that ZitP accumulated via different pathways at the two poles and that it formed distinct structures at each pole. These structures were associated with different roles for ZitP. While ZitP recruited proteins, including CpaM, required for assembly of pili to one of the poles, it acted differently at the opposite pole.
By mutating regions of ZitP, Mignolet et al. went on to show that different regions of the protein carry out these roles. Further experiments demonstrated that regulators of the cell division cycle influenced how ZitP and CpaM accumulated and behaved in cells, ensuring that the proteins carry out their roles at the correct time during division. These findings provide more evidence that proteins can have different roles at distinct sites within a cell, in this case at opposite poles of a cell. Future studies will be needed to determine whether this is seen in cells other than Caulobacter including more complex, non-bacterial cells.
https://doi.org/10.7554/eLife.18647.002Introduction
Some regulatory proteins that execute important developmental, cytokinetic or morphogenetic functions are localized in monopolar fashion, whereas others are sequestered to both cell poles (Dworkin, 2009; Martin and Goldstein, 2014; Shapiro et al., 2002; St Johnston and Ahringer, 2010). It is unclear if bipolar proteins can confer specialized functions from each polar site, but examples of proteins with a bipolar disposition have been reported for eukaryotes and prokaryotes (Davis et al., 2013; Martin and Berthelot-Grosjean, 2009; Tatebe et al., 2008; Treuner-Lange and Sogaard-Andersen, 2014).
The synchronizable Gram-negative α-proteobacterium Caulobacter crescentus (henceforth Caulobacter) is a simple model system to study pole-specific organization and cell cycle control (Tsokos and Laub, 2012). The Caulobacter predivisional cell is overtly polarized and spawns two morphologically dissimilar and functionally specialized daughter cells, each manifesting characteristic polar appendages (Figure 1A). The swarmer progeny is a motile and non-replicative dispersal cell that samples the environment in search of food. It harbours adhesive pili and a single flagellum at one pole and is microscopically discernible from the stalked cell progeny, a sessile and replicative cell that features a stalk, a cylindrical extension of the cell envelope, on one cell pole. While the stalked cell resides in S-phase, the swarmer cell is in a quiescent G1-like state from which it only exits concomitant with the differentiation into a stalked cell. During this G1→S transition, the polar flagellum and pili of the swarmer cell are eliminated and replaced by the stalk that elaborates from the vacated cell pole. Upon sequential transcriptional activation of developmental factors during the cell cycle (Panis et al., 2015), the nascent stalked cell re-establishes polarization and ultimately gives rise to an asymmetric pre-divisional cell that yield a swarmer and a stalked progeny.
Figure 1
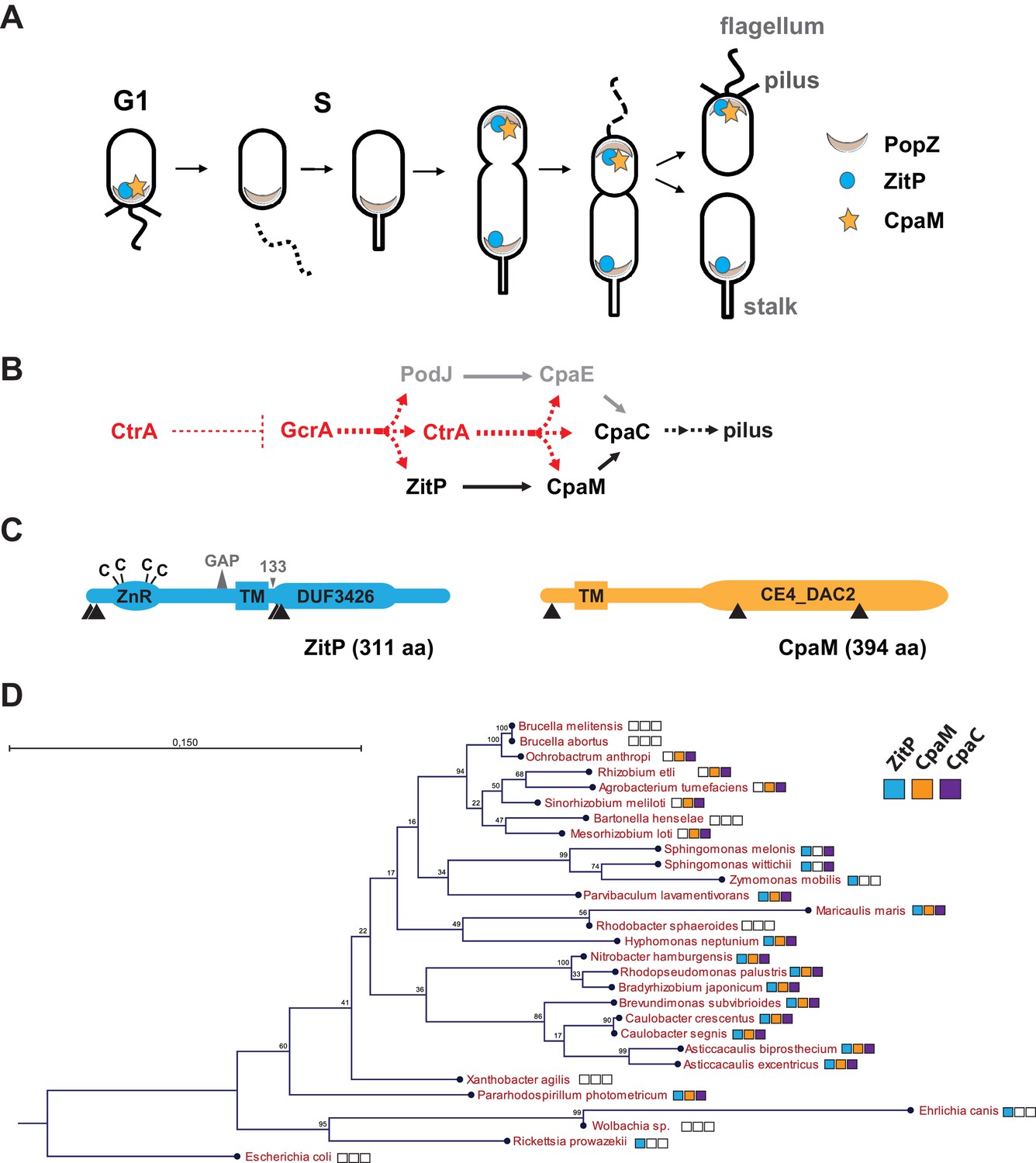
Cell cycle profile and phylogeny of ZitP and CpaM.
(A) Scheme depicting the polarized factors PopZ, ZitP and CpaM during the cell cycle of the dimorphic bacterium C. crescentus. (B) Pilus assembly pathways and global dependencies of the two master cell cycle regulators GcrA and CtrA on the expression of the polar factors PodJ, CpaE, ZitP, CpaM and CpaC that control pilus biogenesis. Red and black dashed lines highlight transcriptional activation and polar recruitment, respectively. (C) Schematic representation (drawn to scale) of ZitP (blue) and CpaM (yellow). ZnR: zinc finger domain; TM: transmembrane domain, C: cysteine. Arrowheads below each protein pinpoint the site of truncation due to transposon insertion in the coding sequence. The large triangle on top of ZitP shows the 2 amino acid residues deleted in the ZitPGAP variant and the small triangle depicts the position of residue 133 where the ZitP coding sequence is truncated in the ZitP1-133 variant. (D) Conservation of ZitP (blue), CpaM (yellow) and CpaC (purple) across the α-proteobacterial clades. The phylogenetic tree was built in CLC Main Workbench (http://www.clcbio.com/products/clc-main-workbench/) from 16S RNA alignments based on the Neighbor Joining method (Juke Cantor substitution model) with 100 bootstrap replicates. Empty boxes mean that no ortholog was found in the genome. Scale bar, 0.15 substitution per site.
The GcrA transcriptional regulator predominates in early S-phase (Holtzendorff et al., 2004) (Figure 1A–B). It accumulates during the G1→S transition and activates expression of polarity factors that are required for pilus or flagellum biogenesis and cytokinetic components (Davis et al., 2013; Fioravanti et al., 2013; Murray et al., 2013; Quon et al., 1996; Viollier et al., 2002b) (Figure 1A–B). Among GcrA target promoters, is the promoter controlling expression of the PodJ polar organizer that localizes to the pole opposite the stalk and directs assembly of the Caulobacter pilus assembly (cpa) machine at that site. In this cascade, PodJ recruits the cytoplasmic CpaE protein that then promotes the localization and assembly of CpaC secretin localization (Figure 1B) (Viollier, 2002a). Another key promoter controlled by GcrA is the one driving expression of the master cell cycle regulator CtrA that induces the synthesis of a second wave of polar and morphogenesis factors in late S-phase including the cpa operon (Figure 1B). The abundance of CtrA and GcrA is regulated at the level of synthesis and degradation (Collier et al., 2006; Domian et al., 1997) and as a result, cell division spawns a swarmer and stalked cell progeny containing CtrA and GcrA, respectively.
An important polarity determinant in the α-proteobacteria is the conserved matrix protein PopZ (Figure 1A) that organizes poles by forming a molecular lattice that traps polar determinants and effectors (Bowman et al., 2008; Deghelt et al., 2014; Ebersbach et al., 2008; Grangeon et al., 2015; Laloux and Jacobs-Wagner, 2013). PopZ is bipolar in the Caulobacter predivisional cell and it interacts directly with numerous cell cycle kinases, the ParAB chromosome segregation proteins and cell fate determinants (Holmes et al., 2016). Here, we dissect at the genetic and cytological level the polar localization and function of two poorly characterized trans-membrane proteins, the zinc-finger protein ZitP and the CpaM effector protein, that are polarly localized and that execute multiple regulatory functions. We unearthed two separate localization pathways for each cell pole, one PopZ-dependent and another that is PopZ-independent, and we provide evidence by photo-activated localization microscopy (PALM) and by genetic dissection that each polar cluster has a distinctive architecture and a specialized function.
Results
ZitP and CpaM are required for pilus biogenesis.
As pili are necessary for infection by the lytic caulophage CbK (φCbK) (Skerker and Shapiro, 2000), we specifically sought mutants in pilus assembly factors encoded outside of the major pilus assembly cpa gene locus (pilA-cpaA-K) (Christen et al., 2016; Skerker and Shapiro, 2000). To this end, we conducted himar1-transposon (Tn) mutagenesis of wild-type (WT) Caulobacter in the presence of φCbK (see Methods) and recovered mutants with Tninsertions in CCNA_02298, renamed here zitP (zinc-finger targeting the poles) because of the pleiotropic roles detailed below, or in cpaM (CCNA_03552) (Figure 1C) (Marks et al., 2010). While both genes have previously been implicated in polar functions and their transcription is cell cycle-regulated (Christen et al., 2016; Fioravanti et al., 2013; Fumeaux et al., 2014; Hughes et al., 2010; McGrath et al., 2007), they are poorly characterized. The zitP gene is predicted to encode a 311-residue bitopic trans-membrane (TM) protein harbouring a CXXC-(X)19-CXXC motif that binds a zinc ion (zinc_ribbon_5 or PF13719 superfamily, residues 1-37) at the cytoplasmic N-terminus (Bergé et al., 2016) and a conserved domain-of-unknown function (DUF3426, residues 128-245) in the C-terminal region that is predicted to reside in the periplasm (Figure 1C). The cpaM gene codes for a 394-residue protein harbouring a single N-terminal TM domain and a C-terminal CE4_DAC2-like polysaccharide deacetylase domain predicted to be periplasmic (Figure 1C). ZitP and CpaM are not restricted to the Caulobacter lineage as BLASTP searches revealed orthologs in many α-proteobacterial clades (Figure 1D). To confirm the phenoytpes of the Tn insertion mutants, we engineered strains with an in-frame deletion in zitP (ΔzitP) or cpaM (ΔcpaM) and found that the mutants no longer supported plaque formation (lysis) by the pilus-specific bacteriophage φCbK. By contrast, plaques were still formed by the S-layer specific caulophage φCr30 (Edwards and Smit, 1991) (Figure 2A), showing that mutations in cpaM or zitP prevent infection of φCbK, but not all phages. This defect was corrected upon expression of either ZitP or CpaM from an ectopic locus in ΔzitP or ΔcpaM cells, respectively (Figure 2A).
Figure 2 with 2 supplements see all
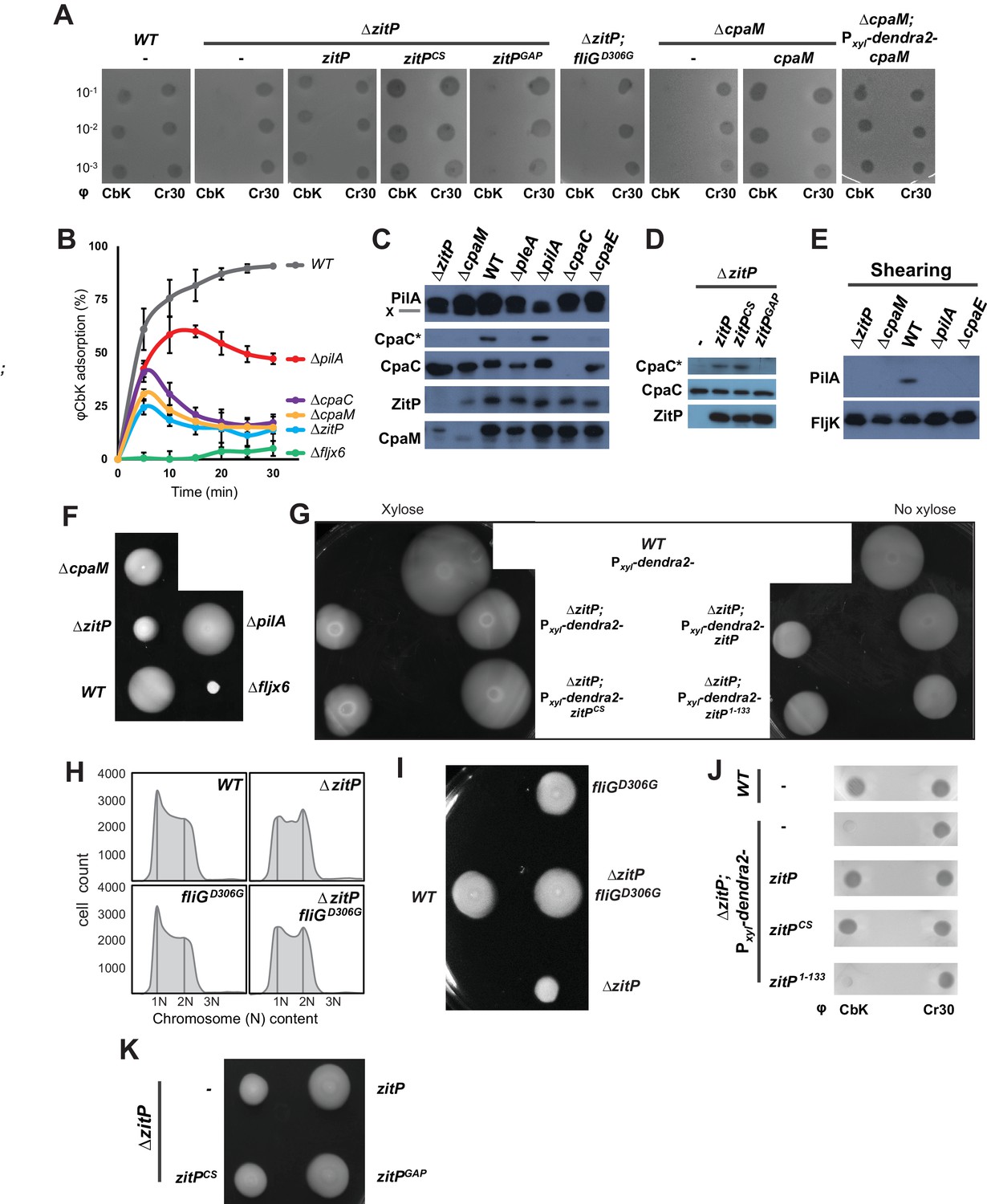
Functional dichotomy in ZitP and effects on polar morphogenesis.
(A) Bacteriophage infection assays of WT, ΔzitP, ΔzitP;fliGD306G and ΔcpaM mutant cells. Cells harbour empty pMT335 or a complementing plasmid (pMT335 backbone) and were grown in the absence of vanillate. No xylose was added to the agar for the phage assay on ΔcpaM; Pxyl-dendra2-cpaM cells. The phages φCbK and φCr30 were spotted with serial dilution on C. crescentus embedded in top agar. Sensitivity to phages is indicated by plaques (lysis). (B) Adsorption kinetics of φCbK to WT and mutant cells. (C) Steady-state levels of ZitP, CpaM, CpaC, modified CpaC (CpaC*) and PilA in WT and mutant cells as determined by immunoblotting. In the PilA immunoblots, the asterisk (*) points to a non-specific band. (D) Immunoblots showing the steady-state levels of monomeric CpaC and CpaC* in ΔzitP cells harbouring pMT335 or derivatives encoding ZitPWT, ZitPCS or ZitPGAP grown in the presence of vanillate (50 µM). (E) Immunoblots showing PilA and FljK abundance in supernatants of WT and various mutant cells. Supernatants were harvested from mid-log cultures after shearing. (F) Swarming motility test performed on soft (0.3%) agar with WT, ΔzitP, ΔcpaM, ΔpilA and Δfljx6 mutant cells. (G) Complementation of the motility defect on swarm (0.3%) agar displayed by the ΔzitP cells expressing Dendra2-ZitP variants from Pxyl at the xylX locus. Xylose was added to the swarm (0.3%) agar as indicated. (H) Flow cytometry of exponential phase WT and ΔzitP cells. N refers to chromosome equivalents. (I) Suppression of the ΔzitP motility phenotype by fliGD306G point mutation as shown on a swarm (0.3%) agar plate. (J) Phage spot tests with φCr30 and φCbK on WT or ΔzitP cells expressing Dendra2-ZitP variants from Pxyl at the xylX locus. Cells were embedded in top agar containing xylose (0.3%). (K) Motility assays of ΔzitP cells expressing WT ZitP (ZitPWT), ZitPCS or ZitPGAP from pMT335. Swarming motility was assessed in absence of vanillate on 0.3% agar.
Next, we conducted time-course adsorption assays and found the adsorption kinetics of ΔzitP and ΔcpaM cells to be substantially compromised compared to WT cells (Figure 2B). The φCbK adsorption kinetics of the mutants closely resemble those for ΔcpaC cells that cannot assemble pili because they lack the CpaC secretin (Skerker and Shapiro, 2000). Moreover, immunoblotting revealed that ΔzitP and ΔcpaM cells do not accumulate the modified form of CpaC, CpaC* (Figure 2C–D). A comparable reduction in CpaC* abundance has been previously reported for ΔcpaE, ΔpodJ and ΔpleA cells that no longer assemble a polar CpaC pilus channel in the outer membrane and cannot be infected by φCbK (Viollier and Shapiro, 2003; Viollier et al., 2002b). However, CpaC* accumulates in ΔpilA cells (Figure 2C), suggesting that the CpaC channel forms independently of PilA. To test whether ΔzitP and ΔcpaM cells assemble a pilus filament on the cell surface, we conducted shearing assays followed by immunoblotting using antibodies to the PilA pilin, the subunit of the pilus filament (Figure 2E) (Skerker and Shapiro, 2000). Whereas PilA was efficiently released from WT cells into the supernatant by shearing, no PilA was detectable in the supernatants of ΔcpaE, ΔzitP and ΔcpaM cells after shearing (Figure 2E), even though PilA is clearly expressed in these cells (Figure 2C). As the major subunit of the flagellar filament, the FljK flagellin, accumulates in the supernatants in all samples (Figure 2E), we conclude that ZitP and CpaM are required for the presentation of PilA on the cell surface and, as shown below, that they act in the same pathway (Figure 1B).
Control of motility, G1-phase and the CtrA regulon.
The φCbK adsorption kinetics hinted that motility might be altered in ΔzitP and ΔcpaM cells. This hypothesis is based on the comparison of the φCbK adsorption kinetics to WT, ΔpilA and Δfljx6 (lacking all six flagellin genes: fljJ/K/L/M/N/O) cells to ΔzitP and ΔcpaM cells. While pililess ΔpilA cells assemble a flagellum and are motile (Figure 2F), Δfljx6 cells are flagellumless, but piliated (φCbK sensitive) (Guerrero-Ferreira et al., 2011). The kinetics of adsorption of φCbK to ΔzitP and ΔcpaM cells was strongly reduced compared to WT, fitting halfway between the adsorption curves of φCbK to ΔpilA and Δfljx6 cells (Figure 2B). Since it is known that φCbK first reversibly adsorbs to the flagellar filament rotating counter-clockwise, before the irreversibly attachment to the pilus portal is established for productive infection (Guerrero-Ferreira et al., 2011), we wondered whether there are fewer motile cells in the ΔzitP and ΔcpaM populations than in WT or if motility in these mutants is altered in other ways. In fact, motility tests on swarm (0.3%) agar revealed a mild reduction in motility of ΔcpaM cells and a severe reduction of ΔzitP cells compared to WT (Figure 2F). However, ΔzitP cells still have residual motility that allows them to spread in swarm agar compared to Δfljx6 cells (Figure 2F). Expression of Dendra2-ZitP from an ectopic locus confers near WT motility to ΔzitP cells (Figure 2G), showing that this deficiency in motility is indeed due to the absence of ZitP.
As Caulobacter divides into a motile G1-phase cell and a sessile S-phase cell, mutants accumulating fewer G1-phase cells in the population can exhibit reduced motility on soft agar (Sanselicio et al., 2015; Sanselicio and Viollier, 2015). To test if ZitP controls the G1 cell number, we used flow cytometry to quantify the number of G1 cells and indeed observed fewer G1 cells in the ΔzitP population compared to WT (Figure 2H). Knowing that the master cell cycle transcriptional regulator CtrA retains cells in G1-phase and activates many cell cycle-regulated promoters that fire in G1-phase (Domian et al., 1997; Fumeaux et al., 2014; Quon et al., 1996), we then conducted promoter-probe assays using several CtrA-activated promoters fused to the promoterless lacZ gene and quantified CtrA-dependent promoter activity in WT and ΔzitP cells (Figure 2—figure supplement 1). While all such promoter-probe reporters for the CtrA regulon exhibited a decrease in activity by 30-40% in ΔzitP versus WT cells, promoter-probe reporters for the GcrA regulon or other reporters were unaffected. Thus, ZitP is required for optimal CtrA activity and G1 cell accumulation.
The reduction in CtrA-dependent transcription does not appear to be solely responsible for the motility defect of ΔzitP cells. First, promoter-probe assays revealed that ΔcpaM cells also suffer from reduced CtrA-dependent activation (Figure 2—figure supplement 2A), even though their motility exceeds that of ΔzitP cells (Figure 2F). Second, we were able to mitigate the defect in CtrA-dependent transcription by ectopic expression of the (p)ppGpp alarmone, a signalling molecule that enhances CtrA function and stability via a poorly understood mechanism (Gonzalez and Collier, 2014). We accomplished this by heterologously expressing the truncated version of the E. coli (p)ppGpp-synthase RelA (RelA’) from the xylose-inducible promoter at the xylX locus in WT and ΔzitP cells. LacZ-based promoter-probe assays revealed that ectopic induction of (p)ppGpp restores CtrA-dependent promoter activity to near WT levels (Figure 2—figure supplement 2B). However, the motility of ΔzitP cells ectopically producing (p)ppGpp is still substantially lower than that of WT cells (Figure 2—figure supplement 2C–D), indicating that ZitP also promotes motility through a CtrA- and (p)ppGpp-independent pathway.
To reinforce this conclusion, we isolated a spontaneous motile suppressor of ΔzitP cells (see Materials and Methods, Figure 2I) with a single point mutation in the fliG flagellar gene (fliGD306G) that neither corrects the pilus assembly defect (φCbK-resistance, Figure 2A), nor the reduction in G1 cell number of the ΔzitP mutant (Figure 2H). As FliG encodes a component of the flagellar motor that is associated with the cytoplasmic membrane (Macnab, 2003), we conclude that ZitP controls pilus biogenesis and a multifactorial motility phenotype, with a minor contribution from a CtrA-dependent pathway and a major one from a CtrA-independent pathway(s) that can be bypassed by a mutant variant of FliG.
Distinct polar ZitP assemblies control CpaM localization
To investigate if ZitP also controls its polar functions from the cell pole, we resorted to live-cell fluorescence imaging by epifluorescence microscopy (Figure 3—figure supplement 1A–D) and photo-activated localization microscopy (PALM, Figure 3A–B and D–E) (Betzig et al., 2006) using WT, ΔzitP or ΔcpaM cells expressing functional Dendra2-CpaM or Dendra2-ZitP. We observed Dendra2-ZitP to adopt a bipolar disposition in dividing cells, whereas Dendra2-CpaM is restricted to the pole opposite the stalk where the pilus biogenesis machinery assembles (Figure 3A–B; Figure 3—figure supplement 2A–C). While Dendra2-ZitP localization is not noticeably perturbed in ΔcpaM cells (Figure 3—figure supplement 1B–C), Dendra2-CpaM is dispersed in ΔzitP cells (Figure 3A; Figure 3—figure supplement 1D and 2B). Moreover, biochemical pull-down experiments with ZitP-TAP (Figure 3—figure supplement 3) and reciprocal co-immunoprecipitation experiments using antibodies to ZitP and CpaM (Figure 3C) showed that ZitP and CpaM reside in a complex. Since Dendra2-ZitP and Dendra2-CpaM localization is not affected in ΔpodJ, ΔcpaE or ΔcpaC cells (Figure 3F, Figure 3—figure supplement 1C and D) and since CpaE localization is not noticeably altered in ΔzitP and ΔcpaM cells (Figure 3—figure supplement 4A–B), we conclude that ZitP and CpaM are part of a previously unknown (PodJ/CpaE-independent) polarization pathway for pilus assembly in Caulobacter in which ZitP recruits CpaM (Figure 1B).
Figure 3 with 8 supplements see all
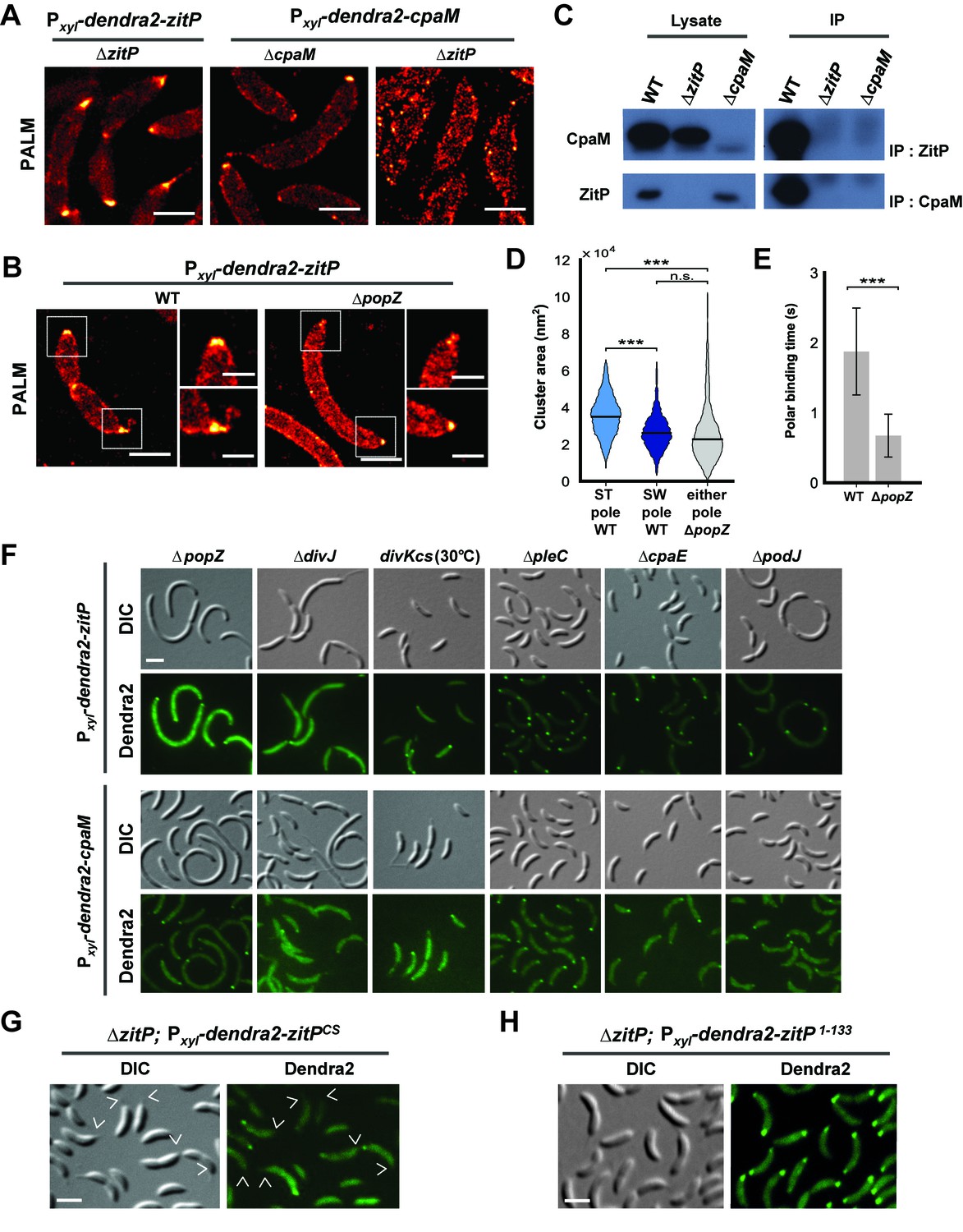
Distinct ZitP nanoscale assemblies and localization determinants.
(A) Photo-activated light microscopy (PALM) imaging of Dendra2-ZitP or Dendra2-CpaM expressed from the xylose-inducible Pxyl promoter on a plasmid integrated at the chromosomal xylX locus in ΔzitP or ΔcpaM cells exposed to xylose 3 hours before imaging. Scale bar: 1 µm. (B) PALM imaging of Dendra2-ZitP in WT or ΔpopZ::Ω cells. We induced expression of Dendra2-ZitP from the xylose-inducible Pxyl promoter on a plasmid integrated at the chromosomal xylX locus by the addition of xylose 3 hours before imaging. Scale bar: 1 µm. Scale bar of zoomed images: 0.5 µm. (C) Co-immunoprecipitation (co-IP) of ZitP or CpaM with polyclonal antibodies to CpaM or ZitP, respectively. Immunoprecipitates and cell lysates from WT, ΔzitP or ΔcpaM cells were probed for the presence of ZitP or CpaM. (D) Projected area of the Dendra2-ZitP polar complex as determined by PALM from Dendra2-ZitP expressed in WT and ΔpopZ::Ω cells. Black lines indicate medians. Statistical significance from Mood’s median test: n.s, p>0.05; ***p<0.001. (E) ZitP polar binding times in WT and ΔpopZ::Ω cells, measured via single particle tracking PALM. Error bars indicate 95% confidence interval of the fit to the data (Figure 3—figure supplement 6D). Statistical significance from a 2 sample t-test: ***p=p<0.001. (F) Epifluorescence (Dendra2) and Nomarski (DIC) images depicting the localization of Dendra2-ZitP or Dendra2-CpaM in ΔpopZ::Ω, ΔdivJ, divKcs, ΔpleC, ΔcpaE or ΔpodJ cells. Expression of Dendra2-ZitP or Dendra2-CpaM was induced from the chromosomal xylX locus with xylose 4 hours before imaging. Scale bars: 1 µm. (G) (H) Epifluorescence (Dendra2) and Nomarski (DIC) images depicting the localization of the motility-deficient and pilus-proficient Dendra2-ZitPCS variant (G) or the motility-proficient and pilus-deficient Dendra2-ZitP1-133 variant (H) in ΔzitP cells. Arrow heads pinpoint stalked poles. We induced expression of Dendra2-fusions from the xylose-inducible Pxyl promoter on a plasmid integrated at the chromosomal xylX locus by the addition of xylose 4 hours before imaging. Scale bars: 1 µm.
PALM analysis disclosed differently shaped and sized complexes of Dendra2-ZitP at each Caulobacter pole. Both Dendra2-ZitP clusters appear extended, suggesting that ZitP multimerization along the polar membrane is spatially restricted (Figure 3A–B; Figure 3—figure supplement 2A and C). Quantification of the 2D area and shape-based analyses (circularity, solidity and eccentricity) showed that ZitP clusters extending into the base of the stalk are significantly larger and differently shaped than the extended fluorescent foci lining the cap of the opposite (swarmer) pole (Figure 3B and D; Figure 3—figure supplement 2A, C and 5A–D). In further support of the existence of two distinct nanostructures of ZitP at each pole, genetic experiments revealed that different pathways govern ZitP polarization: one requiring PopZ and another operating independently of PopZ. Imaging of Dendra2-ZitP in ΔpopZ cells revealed mainly monopolar foci (Figure 3B and F; Figure 3—figure supplement 1C and 2C), resembling those seen at the pole opposite the stalk in WT cells (Figure 3B and D; Figure 3—figure supplement 2C and 5C–D). Quantitative analysis of the polar residence time using stroboscopic single particle tracking PALM (Gebhardt et al., 2013) revealed a strong reduction in polar binding times of Dendra2-ZitP in ΔpopZ compared to that of WT cells (Figure 3E; Figure 3—figure supplement 6A–D). Thus, PopZ promotes the formation of a large polar ZitP assembly at the stalked pole, whereas a small complex of ZitP sequesters independently of PopZ at the opposite pole.
Localization and functional determinants in ZitP
To identify the determinants within ZitP governing the differential polar localization and to test if they support specific functions, we first constructed a mutant variant of ZitP in which all four zinc-coordinating cysteine residues in the zinc-finger domain (Bergé et al., 2016) are replaced by serine residues (henceforth ZitPCS, Figure 1C). The motility of ΔzitP cells expressing ZitPCS or Dendra2-ZitPCS is reduced compared to those expressing the WT version of ZitP (ZitP or Dendra2-ZitP; Figure 2G and K). While Dendra2-ZitPCS exclusively localizes to the pole opposite the stalk in ΔzitP cells (Figure 3G; Figure 3—figure supplement 7A), it still supports lysis by φCbK (Figure 2A) and CpaC* assembly (Figure 2D). ZitPCS supports localization of Dendra2-CpaM to the pole opposite the stalk and co-immunoprecipitation experiments show that it interacts with CpaM (Figure 3—figure supplement 7B–D). ZitPCS also confers (CpaM-dependent) firing of CtrA-activated promoters with similar efficiency as WT ZitP (Figure 3—figure supplement 8A). Since Dendra2-CpaM is also still monopolar in ∆popZ cells, zinc-binding within the zinc_ribbon_5 domain is necessary for the interaction between PopZ and ZitP (Bergé et al., 2016), but not for CpaM localization/interaction. Thus, inactivation of the zinc-coordinating residues in ZitP effectively mimics the monopolar localization of Dendra2-ZitP in ∆popZ cells and functions as unmodified ZitP with respect to the functions that depend on CpaM.
By contrast, the opposite effect was seen when ZitP1-133, a ZitP variant that lacks the periplasmic DUF3426 but retains the cytoplasmic and TM domains (residues 1-133, Figure 1C), is expressed in ∆zitP cells. ZitP1-133 supports efficient motility and is polarly localized, but no longer supports pilus function (i.e. plaque formation by φCbK), CpaM localization and efficient CtrA-activated transcription (Figure 2G and J, Figure 3—figure supplement 8A–B). Thus, the periplasmic DUF3426 plays a critical role in promoting pilus assembly through the polar recruitment of CpaM.
Support for the notion that DUF3426 function is regulated from sequences N-terminal to the DUF3426 came from a forward genetic screen (see Materials and Methods) that led to the identification of ZitPGAP (Figure 1C), a mutant variant in which residues Arg93 and Ala94 preceding the TM domain are deleted. ZitPGAP supports motility (Figure 2K), but neither plaque formation by φCbK, nor CpaC* production (Figure 2A and D). As ZitPGAP still localizes to the cell poles, interacts with CpaM and recruits Dendra2-CpaM (Figure 3—figure supplements 7A–D and 8C), ZitP also acts on pilus biogenesis independently of CpaM localization.
Taken together our experiments indicate that function and localization of ZitP can be genetically uncoupled. The periplasmic DUF3426 region is required for pilus biogenesis and CtrA-dependent transcription and it implements these functions via the recruitment of CpaM to the pole opposite the stalk. The zinc_ribbon_5 domain promotes PopZ-dependent localization of ZitP to the stalked pole and efficient swarming motility by an unknown mechanism. Interestingly, in a related study, we recently found that ZitP controls PopZ localization independently of the DUF3426 (Bergé et al., 2016).
Cell cycle control of ZitP and CpaM assemblies
Synchronization studies and genetic experiments with cell cycle mutants showed that ZitP and CpaM polarization is temporally and functionally coordinated with cell cycle progression. Immunoblotting revealed the steady-state levels of ZitP and CpaM to fluctuate during the cell cycle (Figure 4A), exhibiting a trough during the G1→S transition and concomitant loss of polar fluorescence at this time (Figure 4B–C). Consistent with the genetic and cytological hierarchy, ChIP-Seq data shows that the early S-phase regulator GcrA directly promotes ZitP and CtrA expression, while the late S-phase regulator CtrA activates expression of CpaM (Fiebig et al., 2014; Fioravanti et al., 2013; Fumeaux et al., 2014; Murray et al., 2013). Moreover, ZitP, CtrA and CpaM abundance is reduced when GcrA is depleted or inactivated (Figure 4D). ZitP expression is also strongly reduced in the absence of the CcrM adenine methyltransferase that methylates adenines at the N6-position in the context of 5’-GANTC-3’ sequences. GANTC methylation is required for efficient recruitment of GcrA to its target promoters (Fioravanti et al., 2013; Murray et al., 2013).
Figure 4
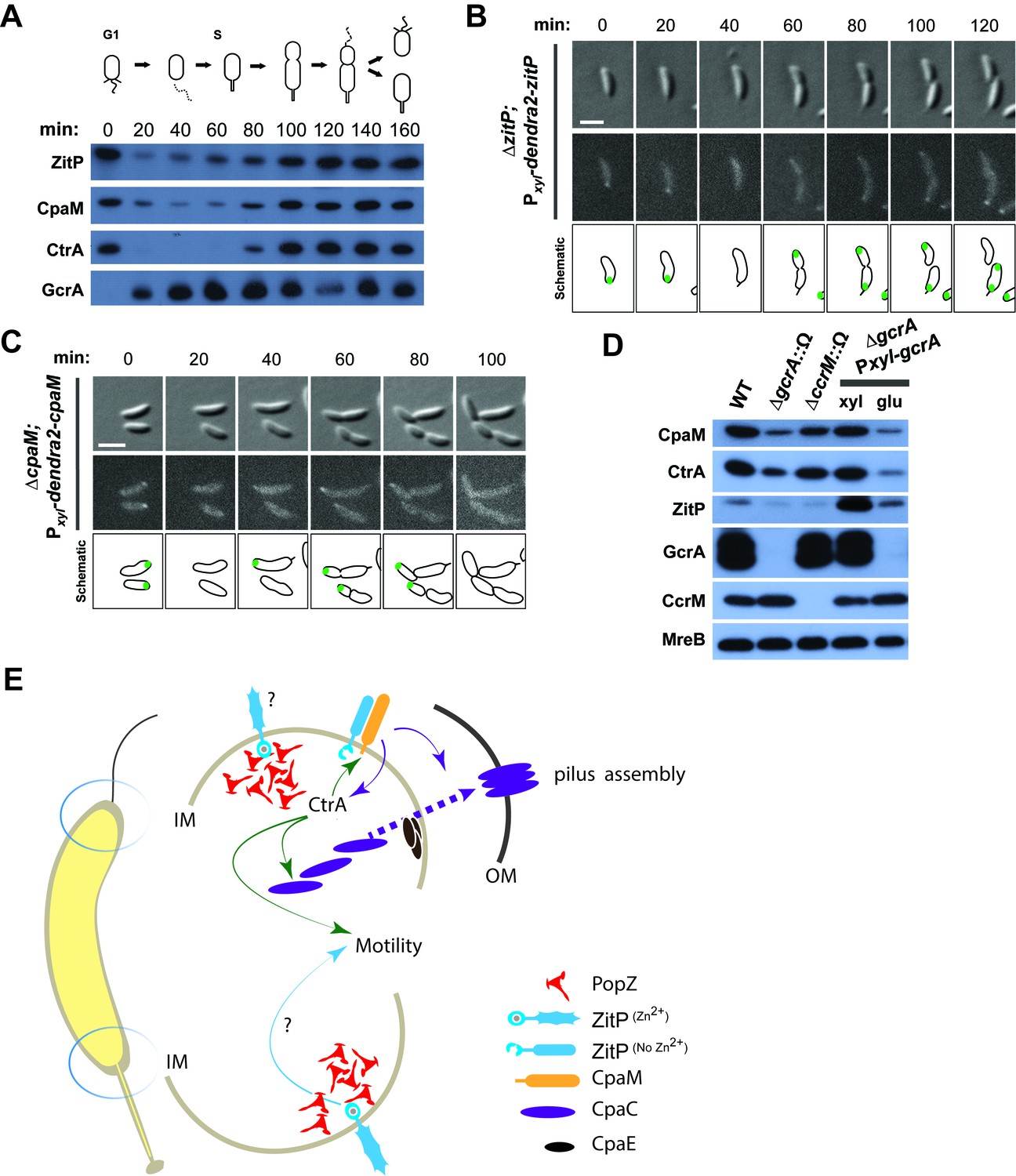
Cell cycle regulation of ZitP and CpaM localization.
(A) Immunoblots showing the levels of ZitP, CpaM and master cell cycle regulators along the C. crescentus cell cycle in a synchronized WT population. The upper scheme depicts C. crescentus cell cycle stages. (B) (C) Epifluorescence (Dendra2) and Nomarski (DIC) images depicting the localization of Dendra2-ZitP (B) and Dendra2-CpaM (C) in synchronized ΔzitP or ΔcpaM cells, respectively. We induced expression of Dendra2 fusions expressed from the xylose-inducible Pxyl promoter on a plasmid integrated at the chromosomal xylX locus. Schematic drawings highlight Dendra2 localizations. After synchronization, cells were resuspended in M2G and imaged every 20 minutes. Scale bars: 1 µm. (D) Steady-state levels of ZitP, CpaM, CtrA, GcrA, CcrM and MreB (control) in WT, gcrA and ccrM mutant cells. Xylose (0.3%, xyl) or glucose (0.2%, glu) were supplemented to the medium in order to induce/deplete GcrA in ΔgcrA xylX::Pxyl-gcrA cells. (E) Schematic representation of the two Caulobacter cell poles. At the stalked pole, the PopZ matrix promotes the recruitment of ZitP. The Zn2+-bound zinc-finger domain of ZitP prevents ZitP/CpaM association and influences CtrA activity and swarming motility. At the opposite pole, the inactive Zn2+-unbound zinc-finger domain allows the formation of the ZitP/CpaM complex and the export and assemblage of CpaC in the outer membrane (OM)independently of PopZ.
Additionally, we found that the DivJ-PleC-DivK (kinase-phosphatase-substrate) system that regulates cell cycle progression and polar development influences the appearance of polar Dendra2-ZitP and Dendra2-CpaM (Figure 3F, Figure 3—figure supplement 1C–D). Specifically examining the localization in mutants where the phosphoflux is shifted towards the accumulation of the phosphorylated form of the DivK cell fate determinant (Tsokos and Laub, 2012), we found that such a mutation (inactivation of the PleC phosphatase, ∆pleC) promotes ZitP/CpaM polarization as indicated by the bipolar localization of Dendra2-CpaM. By contrast, mutations that have the opposite effect on DivK activity or DivK phosphorylation (caused by the divKCS or ∆divJ mutation), disfavour Dendra2-ZitP (but not Dendra2-CpaM) polarization (Figure 3F, Figure 3—figure supplement 1C–D). Thus, polar reprogramming of ZitP and CpaM is deeply integrated into the Caulobacter cell cycle through conserved components of the α-proteobacterial cell cycle (Brilli et al., 2010).
Discussion
The pole-specific and distinctly shaped assemblies of ZitP are governed via independent localization pathways and linked with functional specialization (Figure 4E). While ZitP acts on pilus assembly by recruiting CpaM and, subsequently, the CpaC pilus channel to the pole opposite the stalk (1B and 4E), CpaM is also required for efficient activation of CtrA-dependent promoters by an unknown mechanism. A similar reduction in CtrA-dependent transcription occurs in ∆zitP cells that are unable to localize CpaM. While diminished CtrA activity can undermine motility by reducing the number of motile G1-phase cells in the population (Sanselicio et al., 2015; Sanselicio and Viollier, 2015), ZitP affects motility in another way, since ∆zitP cells are diminished in motility compared to ∆cpaM cells. Moreover, ectopic induction of the alarmone (p)ppGpp reinforces CtrA abundance and activity (Boutte et al., 2012; Gonzalez and Collier, 2014; Lesley and Shapiro, 2008; Ronneau et al., 2016; Sanselicio and Viollier, 2015), but only modestly improves the motility of ∆zitP cells.
Such a motility defect also manifests when ZitPCS, a variant that no longer localizes to the stalked pole, is expressed in ∆zitP cells. How ZitP promotes swarming motility from the stalked pole is unclear, but there is precedence of other regulators (SpmX/Y and CpdR) that localize exclusively to the stalked pole and affect Caulobacter motility indirectly by regulating cell cycle factors (Janakiraman et al., 2016; McGrath et al., 2006; Radhakrishnan et al., 2008). Moreover, SpmX and CpdR interact with PopZ directly and their localization is compromised in the absence of PopZ (Bowman et al., 2010; Holmes et al., 2016). It is therefore conceivable that ZitP also affects motility indirectly from the stalked pole, possibly via cell cycle regulation, flagellar performance and/or polarity. The fact that the motility defect of ∆zitP cells can be restored by compensatory mutations in a switch component (FliG) of the flagellar motor (Kojima and Blair, 2004), suggests that flagellar performance, reversals or timing (i.e. the length of flagellation in the cell cycle) could be altered by the ∆zitP mutation.
Zinc-finger domain proteins other than ZitP may be implicated in linking motility and polarity. The gliding motility protein AgmX confers a flagellum- and pilus-independent form of surface motility in Myxococcus xanthus (Nan et al., 2010), a δ-proteobacterium that periodically reverses the polarity of movement. Since AgmX also harbors a related N-terminal Zinc-finger domain, at least two related zinc-finger domains control different types of motility. This is intriguing and hints at a potentially important and conserved role of such zinc-finger domain proteins in developmental processes that rely on protein polarization in bacteria and polar matrix proteins such as PopZ to interact with them. In a complementary study, we additionally show in vitro and in vivo that zinc-bound ZitP binds PopZ directly and regulates PopZ localization without the periplasmic DUF3426 domain (Bergé et al., 2016), suggesting that this activity in ZitP may underlie the aforementioned CtrA-independent role in motility.
The conservation of ZitP, CpaM (Figure 1D) and PopZ orthologs (Bowman et al., 2010) in distant α-proteobacterial lineages that reside in different ecological niches hints that the functions that these proteins control are not unique to the Caulobacter branch. Indeed, we describe an interaction between ZitP and PopZ in several distinct α-proteobacterial lineages (Bergé et al., 2016). On a more general scale, our work suggests that pole-specific functions conferred by bipolar regulators may be commonly used in bacteria and possibly eukaryotes. Such mechanisms could be relevant for toggle proteins, moonlighting/trigger enzymes (Commichau and Stulke, 2015) and other bifunctional regulators (Radhakrishnan and Viollier, 2012) that have more than one biochemical activity and function, for example kinase-phosphatases or synthase-hydrolases of cyclic-di-GMP sequestered to both cell poles (Boyd, 2000; Kazmierczak et al., 2006; Tsokos and Laub, 2012).
In sum, the functional and topological versatility of ZitP illustrates how a conserved regulator is used to coordinate multiple functions from different locations and structures in the same cell, relying on distinct protein domains and partners to control localization or to implement function. As these functions and polar remodelling events are coordinated with cell cycle progression in Caulobacter via conserved cell cycle proteins, it is likely that superimposed temporal layers similarly act on ZitP and CpaM orthologs in other α-proteobacterial cell cycles.
Materials and methods
Strains and growth conditions
Request a detailed protocolCaulobacter crescentus NA1000 and derivatives were grown at 30°C in PYE or in M2 salts plus 0.2% glucose (M2G) supplemented with 0.4% liquid PYE (Ely, 1991). Escherichia coli S17-1, S17-1 λpir and EC100D (Epicentre Technologies, Madison, WI) were cultivated at 37°C in LB. We added 1.5% agar to PYE plates, and motility was assayed on PYE plates containing 0.3% agar. We added D-xylose (0.3% except if otherwise stated), glucose (0.2%), sucrose (3%), kanamycin (solid, 20 µg/ml; liquid, 5 µg/mL), tetracycline (1 µg/mL), spectinomycin (liquid, 25 µg/mL), spectinomycin/streptomycin (solid, 30 and 5 µg/mL, respectively), apramycin (10 µg/mL), gentamycin (1 µg/mL) and nalidixic acid (20 µg/mL), as required. Swarmer cell isolation, electroporation, biparental mating, and bacteriophage φCr30-mediated generalized transduction were performed as described before (Chen et al., 2005; Ely, 1991; Simon et al., 1983; Viollier and Shapiro, 2003).
Bacterial strains, plasmids, and oligonucleotides
Request a detailed protocolBacterial strains, plasmids, and oligonucleotides used in this study are listed and described in supplementary tables.
β-Galactosidase assays
Request a detailed protocolβ-Galactosidase assays were performed at 30°C as described previously (Huitema et al., 2006; Viollier and Shapiro, 2003). Experimental values represent the averages (standard error of the mean, SEM) of at least three independent experiments.
PALM imaging conditions
Request a detailed protocolTo image C. crescentus, overnight cultures were diluted in fresh PYE, xylose was added (0.3% final concentration), and the cells were grown for 3 hours to mid-exponential phase (OD (660) ~ 0.4). Two uL of culture was placed on a agarose pad containing PYE. The agarose pad was mounted in a silicone gasket (Grace Biolabs 103280) sandwiched between two microscope coverslips to minimize shrinkage of the agarose. The temperature of the microscope enclosure during experiments was 24°C. Images were acquired using a previously described custom built PALM microscope (Holden et al., 2014). Fluorescent proteins were excited at 560 nm, and photoactivation was induced at 405 nm at ~ 0–16 W/cm2. For PALM images of Dendra2-ZitP in C. crescentus, cells were imaged at an exposure time of 10 milliseconds for 10,000 frames, and an excitation intensity of ~4 kW/cm2. For stroboscopic single particle tracking PALM measurement of ZitP binding time, cells were imaged at an exposure time of 30 milliseconds, with a variable interval between each frame, at an excitation intensity of ~1 kW/cm2. PALM localizations were accumulated in a 2D histogram; the resulting image was blurred with a 2D Gaussian of radius 15 nm to reflect the localization uncertainty of the measurement. The image was gamma adjusted to 0.5 to compensate for the large dynamic range of the image, and the ‘Red Hot’ ImageJ colormap was applied.
Measurement of ZitP binding time by PALM
Request a detailed protocolBinding time, τoff, of ZitP to the C. crescentus poles was determined via stroboscopic single particle tracking PALM (Gebhardt et al., 2013; Manley et al., 2008). Under these conditions, Dendra2 bleached under continuous illumination with a photobleaching lifetime, τb, on the order of 50 milliseconds. Since rapid diffusion means that Dendra2-ZitP is only visible when bound to the membrane, and since photobleaching will shorten the observed binding time, the effective on-time of a single Dendra2-ZitP molecule, τeff, will be the convolution of the photobleaching lifetime, τb, and the binding lifetime τoff,
(1)
Effective on-time was measured by combining individual Dendra2-ZitP localizations in adjacent frames into tracks (Crocker and Grier, 1996), and fitting a single exponential model to the observed the track length distribution (Figure 3—figure supplement 6A). In order to measure binding times longer than the photobleaching lifetime, the photobleaching lifetime of the fluorescent protein may be artificially extended by using stroboscopic illumination, introducing large gaps between short periods of illumination. This increases the effective bleaching lifetime to:
(2)
where τtl is duration of the gap (time lapse/strobe interval), τint is camera integration time. By measuring the effective on-time for multiple different stroboscopic illumination times, τtl, and performing a fit of:
(3)
to the data, both the binding time and photobleaching lifetime may be calculated (Gebhardt et al., 2013) (Figure 3—figure supplement 6B and C Model 1). We performed non-linear least squares fitting of the raw τeff data directly to Eq. 3, instead of calculating the quantity τtl/τeff and performing a linear fit as proposed by Gebhardt and coworkers (Gebhardt et al., 2013), since the inverse transform proposed results in a non-linear transformation of the sample error distribution incompatible with least squares fitting. We observed that for stroboscopic illumination times significantly greater than the binding time, the data appeared to transition from the hyperbolic relationship predicted by Eq. 3 to a zero-gradient plateau (Figure 3—figure supplement 6B), giving very poor fits between Eq 3 and the data, especially for the ΔpopZ strain which appeared to have a shorter Dendra2-ZitP binding lifetime (Figure 3—figure supplement 6B). We hypothesized that this was due to an inability to accurately estimate effective on-time when molecules bind and unbind in a time significantly less than the duration of a single strobing interval (since the observed track length will almost always equal 1 frame). We confirmed this hypothesis by performing the stroboscopic tracking analysis on simulated data (Figure 3—figure supplement 6C). We simulated timetraces of molecules binding/unbinding with finite bleaching lifetimes, and measured the observed on-time for each simulated molecule by fitting a single exponential to the on-time histogram as above. We observed as hypothesized that the observed off-times showed a sharp plateau for long-strobe intervals due to the finite integration time of the measurement, giving a poor fit of Eq 3 to the data (Figure 3—figure supplement 6C). In order to correct for this, we modified the fitting model to include a minimum measurable on-time plateau:
(4)
Use of the modified model allowed us to obtain accurate fits to the entire simulated dataset (Figure 3—figure supplement 6C; Eq 4).
We therefore used our updated model to fit the experimental data (Figure 3E and Figure 3—figure supplement 6B) and to calculate the observed binding times (Figure 3—figure supplement 6D). This gave a much better fit to the data, both at late and early strobe intervals. Notably, independent fits to the WT and ΔpopZ datasets gave similar observed of ~0.4 frames, supporting the use of the updated model.
Measurement of ZitP cluster area and shape by PALM
Request a detailed protocolIn order to estimate the area of Dendra2-ZitP polar complexes, observed localizations were clustered based on local density using DBSCAN (Endesfelder et al., 2013; Ester et al., 1996). Identified clusters were converted to PALM images binarized, and morphologically closed (Figure 3—figure supplement 5Bi-iii). By performing morphological closing on the binary image, we obtained segmented clusters (Figure 3—figure supplement 5Biii) which were less sensitive to noise and better reflected the visually estimated extent of the non-segmented cluster. For each identified cluster, the area of the segmented cluster was calculated.
For the NA1000 xylX::Pxyl-dendra2-zitP strain, clusters were visually identified as belonging to the stalked or flagellar poles based on the PALM and phase contrast images of the region. For the ΔpopZ::Ω xylX::Pxyl-dendra2-zitP strain, there was no clear difference in pole morphology, so the cluster area for cells was calculated without discriminating poles. Measurement noise means that the measured area of even a zero-area cluster will be larger than zero (and approximately proportional to the localization uncertainty). To test whether Dendra2-ZitP formed an extended polar complex, we compared the area of ZitP clusters to the measured area of simulated zero-area clusters by generating simulated datasets containing localizations coming from a point source, with photon count, background noise and total number of localizations equal to the median values of either the WT or ΔpopZ::Ω datasets (Figure 3—figure supplement 5D). The cluster area of the simulated datasets was then calculated as above.
We also calculated the following shape based metrics to further quantify the differences in pole shape: circularity, solidity and eccentricity (Figure 3—figure supplement 5C).
Circularity measures similarity of a shape to a circle, where A is shape area and p is perimeter. Solidity measures the extent to which a shape is convex or concave, where A is shape area and H is the convex hull area of the shape. Eccentricity measures how elongated a shape is, where a is the length of the minor axis and b is the length of the major axis.
Since the observed distributions showed significant non-normality, statistical significance was assessed by the non-parametric test, Mood’s median test. Stars on Figure 3D and Figure 3—figure supplement 5C indicate statistical significance: n.s, p>0.05; *p<0.05; **p<0.01; ***p<0.001.
The stalked and the other (swamer) pole foci in WT showed statistically significant differences (p<0.001) in area, circularity and solidity, supporting the conclusion that ZitP forms distinct polar assemblies.
The WT stalked pole showed statistically significant differences (p<0.001) to the ΔpopZ::Ω mutant foci for area, circularity, solidity and eccentricity, supporting the conclusion that PopZ specifically promotes the formation of large polar assemblies at the stalked pole.
Isolation of φCbK resistant mutants
Request a detailed protocolA himar1-based transposon mutagenesis of the NA1000 (wild-type, WT) strain was done using the E. coli S17-1 λpir strain containing the himar1-delivery plasmid pHPV414 (Viollier et al., 2004). The mutant library was selected on plates containing nalidixic acid and kanamycin embedded in top agar containing φCbK. Colonies emerging from this selection were pooled. We then created generalized transducing lysate from this pool using phage φCr30 and transduced it into strain PV14 ΔpilA-cpaF::Ωaac3 (conferring resistance to aparamycin), selecting for apramycin and kanamycin resistant transductants to eliminate himar1 insertions in the pilA-cpaF locus. The transductants were pooled and a generalized transducing lysate was prepared from this pool using φCr30. This new lysate was then used to transduce NA1000 to kanamycin resistance and the resulting clones were individually tested for resistance to φCbK. The himar1 insertion site mapping of φCbK–resistant himar1 mutants was done as described before (Viollier et al., 2004).
To isolate the zitPGAP mutation, we generated a mutant library of zitP alleles by electroporating pMT335-zitP into the mutator E. coli XL1-Red strain. We collected and pooled over 20,000 clones for plasmid extraction and we electroporated the plasmid library into the ΔzitP mutant. We incubated the electroporated cells during two hours for regeneration and next added φCbK for one hour in order to eradicate clones that bear a mutated zitP allele restoring effective phage infection. Finally, we plated cells on soft (0.3% swarming) agar to evaluate the motility properties. We picked and streaked out motile clones for amplification and plasmid extraction and introduced the plasmids back into a ΔzitP background in the perspective to confirm the motility-proficient and φCbK resistant phenotypes. We isolated a unique plasmid, pMT335-zitPGAP, which bears the zitPGAP allele (deletion of the Arg93 and Ala94 in the ZitP protein).
Immunoblotting
Request a detailed protocolProtein samples were separated by SDS-PAGE and blotted on PVDF (polyvinylidenfluoride) membranes (Merck Millipore). Membranes were blocked for 1 hour with Tris-buffered saline, 0.05% Tween 20 (TBST), and 5% dry milk and then incubated for an additional 1 hour with the primary antibodies diluted in TBST, 5% dry milk. The membranes were washed 4 times for 5 minutes in TBST and incubated for 1 hour with the secondary antibody diluted in TBST, and 5% dry milk. The membranes were finally washed again 4 times for 5 minutes in TBST and revealed with Immobilon Western Blotting Chemoluminescence HRP substrate (Merck Millipore) and Super RX-film (Fujifilm). Rabbit antisera were used at the following dilutions: anti-CtrA (1:10,000), anti-PilA (1:10,000), anti-FljK (1:50,000), anti-CpaC (1:5000), anti-ZitP (1:5000), anti-CpaM (1:5000) and anti-GcrA (1:2000). HRP-conjugated Anti-rabbit secondary antibody was used at 1:20,000 dilution (Jackson ImmunoResearch, USA).
Epi-fluorescence microscopy
Request a detailed protocolPYE or M2G cultivated cells in exponential growth phase were immobilized using a thin layer of 1% agarose. Fluorescence and DIC images were taken with an Alpha Plan-Apochromatic 100x/1.46 DIC(UV) VIS-IR oil objective on an Axio Imager M2 microscope (Zeiss) with 488 nm laser (Visitron Systems GmbH, Puchheim, Germany) and a CoolSnap HQ (Boutte et al., 2012) camera (Photometrics) controlled through Metamorph V7.5 (Universal Imaging). Images were processed using Image J software. Quantifications were done by manually numbering cells in the diffuse, monopolar or bipolar state.
Protein purification and production of antibodies
Request a detailed protocolThe PCR-amplified zitPCterm and cpaMΔTM genes were cloned into the pET28a vector (Novagen). The His6-ZitPCterm and His6-CpaMΔTM recombinant proteins were overexpressed in E. coli strain Rosetta and purified in standard native conditions on Ni2+-NTA agarose as described previously to raise rabbit polyclonal IgGs in New Zealand White rabbits (Josman LLC, Napa, CA).
Tandem affinity purification (TAP) and mass spectrometry
Request a detailed protocolWe followed the TAP procedure as was previously described (Puig et al., 2001). When a 1 L-culture reached OD660 between 0.4 and 0.6 in the presence of 50 mM vanillate, cells were harvested by centrifugation (6000xg, 10 min). We washed the pellet in 50 mL of buffer I (50mM sodium phosphate pH 7.4, 50 mM NaCl, 1 mM EDTA) and lysed for 15 minutes at room temperature in 10 mL of buffer II (buffer I + 0.5% n-dodecyl-β-D-maltoside, 10mM MgCl2, two protease inhibitor tablets [Complete EDTA-free, Roche] per 50 mL of buffer II, 1x Ready-Lyse lysozyme [Epicentre], 500U of DNase I [Roche]). Cellular debris was removed by centrifugation (7000xg, 20 minutes, 4°C). The supernatant was incubated for 2 hours at 4°C with IgG Sepharose beads (GE Healthcare Biosciences) that had been washed once with IPP150 buffer (10 mM Tris-HCl pH 8, 150 mM NaCl, 0.1% NP40). After incubation, the beads were washed at 4°C three times with 10 mL of IPP150 buffer and once with 10 mL of TEV cleavage buffer (10 mM Tris-HCl pH 8, 150 mM NaCl, 0.1% NP40, 0.5 mM EDTA, 1 mM DTT). The beads were then incubated overnight at 4°C with 1 mL of TEV solution (TEV cleavage buffer with 100 U of TEV protease per ml [Promega]) to release the tagged complex. 3 mM CaCl2 was then added to the solution. The sample with 3 mL of calmodulin-binding buffer (10 mM β-mercaptoethanol, 10 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, 0.1% NP40) was incubated for 1 hour at 4°C with calmodulin beads (GE Healthcare Biosciences) that previously had been washed once with calmodulin-binding buffer. After incubation, the beads were washed three times with 10 mL of calmodulin-binding buffer and eluted five times with 200 µL IPP150 calmodulin elution buffer (calmodulin-binding buffer substituted with 2 mM EGTA instead of CaCl2). Amicon Ultra-4 spin columns (Ambion) were used to concentrate eluates. Eluates were analyzed by SDS-PAGE and stained with silver using the Biorad Silver Stain Plus kit (Biorad, USA). We then cut specific bands and directly sent the gel slices to the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, USA) for mass spectrometric analyses.
Co-immunoprecipitation
Request a detailed protocolCells were harvested from a 50 mL-culture (OD (660 nm) between 0.4–0.6) by centrifugation at 5000xg for 10 minutes. We washed the cell pellet in 10 mL of buffer I (50mM Tris-HCl (pH 7.5); 50 mM NaCl; 1mM EDTA), centrifuged the cell again and resuspended in 1 mL of buffer II (buffer I plus 0.5% n-dodecy-β-D-maltoside; 10 mM MgCl2; EDTA-free protease inhibitors). We incubated the mixture for 15 minutes with 5000 units of Ready-Lyse lysozyme (Epicentre), and 30 units of DNase I (Roche). Cellular debris were removed by centrifugation at 10,000xg for 3 minutes at 4°C. We cleared the supernatant by incubation for 1 hour at 4°C with Protein-A agarose beads (Roche) previously washed three times with 500 µL of buffer II. We removed agarose beads by centrifugation and added to the pre-cleared solution polyclonal IgG rabbit serum for 90 min at 4°C (dilution 1:500). Next, we trapped for 1 hour at 4°C the antibodies-proteins complexes with the addition of Protein-A agarose beads (Roche) previously washed three times with 500 µL of buffer II. The samples were then centrifuged at 3000xg for 1 minute at 4°C and the supernatant was removed. The beads were washed once with buffer I plus 0.5% n-dodecy-β-D-maltoside, three times with 500 µL of wash buffer (10 mM Tris-HCl at pH 7.5; 150 mM NaCl; 0.1% n-dodecy-β-D-maltoside) and finally resuspended in 70 µl SDS sample buffer (50 mM Tris–HCl at pH 6.8), 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 12.5 mM EDTA, 0.02% Bromophenol Blue), heated to 95°C for 10 minutes and stored at −20°C.
Motility assays and phage infectivity tests
Request a detailed protocolSwarming properties were assessed with 5 µl-drops of overnight culture spotted on PYE soft agar plates (0.3% agar) and grown for 24 hours. Phage susceptibility assays were conducted by mixing 500 µL of overnight culture in 6 mL soft PYE agar and overlaid on a PYE agar plate. Upon solidification of the soft (top) agar, we spotted 5 µL-drops of serial dilution of phages (φCbK or φCr30) and scored for plaques after one day incubation at 4°C.
Shearing experiments
Request a detailed protocolWe centrifuged 5 mL mid-log phase cultures of WT or mutant strains and resuspended them in 700 µl of PYE. Then, we pumped in and out (10x) the cells into a syringe endowed with a thin diameter needle. We centrifuged the shear-stressed cells to remove cells debris and collected 200 µL of each supernatant. We added SDS-blue straining and loaded samples on SDS-PAGE gels.
φCbK adsorption assay
Request a detailed protocolTo determine the adsorption rate of φCbK, Caulobacter crescentus NA1000 and derivatives were first grown overnight in M2G medium at 30°C and then re-started in fresh M2G at 30°C with shaking until the bacterial cell culture reached an OD660 value of 0.4 (0.4 × 108 CFU/ml). Then cell cultures were infected by 0.02 multiplicity of φCbK infection (MOI: ratio of phage to bacteria number). The mixtures were incubated at 30°C without shaking for phage adsorption, followed by separation of unbound phages by centrifugation at 13,000 rpm in specified time points up to 30 minutes. Supernatants were immediately supplemented by the addition of chloroform (1/20 of cell culture volume) and mixed vigorously to kill remaining bacterial cells. A control tube containing only φCbK (equivalent to 0.02 MOI) was maintained in parallel for the duration of the experiment and used as reference to control the initial phage plaque-forming units (pfu) titer. A 50 µL of the phage supernatant from each tube was mixed with 200 µL of Caulobacter crescentus NA1000 culture at log phase and incubated without shaking at room temperature for 10 minutes to allow adsorption. Infected cells were added to 6 mL of soft PYE agar (0.5%) and immediately overlaid on 1.5% PYE agar plates. Plates were incubated at 30°C for 24 hours, when pfu were visible. The φCbK adsorption value (in% of the initial phage pfu titer) was calculated. Values are the mean of three biological replicates; error bars represent data ranges.
Flow cytometry (Fluorescence-activated cell sorting, FACS)
Request a detailed protocolCells in exponential growth phase (OD660nm=0.3–0.6) cultivated in PYE, were fixed in ice-cold 77% ethanol solution. Fixed cells were re-suspended in FACS staining buffer, pH 7.2 (10 mM Tris-HCl, 1 mM EDTA, 50 mM NaCitrate, 0.01% Triton X-100) and then treated with RNase A (Roche) at 0.1 mg mL−1 for 30 minutes at room temperature. Cells were stained in FACS staining buffer containing 0.5 μM of SYTOX Green nucleic acid stain solution (Invitrogen) and then analysed using a BD Accuri C6 flow cytometer instrument (BD Biosciences). Flow cytometry data were acquired and analysed using the CFlow Plus V1.0.264.15 software (Accuri Cytometers Inc.). 20,000 cells were analysed from each biological sample. Experimental values represent the averages of three independent experiments.
fliGD306G swarming pseudo-revertant isolation and backcrossing
Request a detailed protocolWe spotted several 5 µL-drops of ∆zitP overnight culture on soft agar plates and waited for flares spreading out the bulk of cells. Flares were peaked out and streaked on fresh agar plates for amplification and subsequently challenged for motility in comparison to WT and ∆zitP strains. Motility-proficient clones were sent for Illumina HiSeQ 2000 sequencing (Fasteris, www.fasteris.com/). Genomes were compared to NA1000 genome and we identified a single mutation in the fliG gene (D306G).
In order to backcross the fliGD306G allele in different backgrounds, the suppressor strain was electrotransformed with the suicide vector pNTPS138-hook and selected on kanamycin-supplemented plates for single crossing-over in close vicinity of the fliG locus. We prepared lysate of this strain, transduced the fliGD306G-linked pNTPS138 into WT and ∆zitP cells and screen by sequencing for clones harbouring the fliGD306G allele. Finally, we grew up the strain without any antibiotic and selected for plasmid excision by plating an overnight culture on sucrose.
References
-
Ppgpp and polyphosphate modulate cell cycle progression in Caulobacter crescentusJournal of Bacteriology 194:28–35.https://doi.org/10.1128/JB.05932-11
-
Methods of Digital Video Microscopy for Colloidal StudiesJournal of Colloid and Interface Science 179:298–310.https://doi.org/10.1006/jcis.1996.0217
-
De- and repolarization mechanism of flagellar morphogenesis during a bacterial cell cycleGenes & Development 27:2049–2062.https://doi.org/10.1101/gad.222679.113
-
Cellular polarity in prokaryotic organismsCold Spring Harbor Perspectives in Biology 1:a003368.https://doi.org/10.1101/cshperspect.a003368
-
A transducing bacteriophage for Caulobacter crescentus uses the paracrystalline surface layer protein as a receptorJournal of Bacteriology 173:5568–5572.
-
Methods in Enzymology, Vol. 204372–384, Genetics of Caulobacter crescentus, Methods in Enzymology, Vol. 204, 10.1016/0076-6879(91)04019-K.
-
Multiscale spatial organization of RNA polymerase in Escherichia coliBiophysical Journal 105:172–181.https://doi.org/10.1016/j.bpj.2013.05.048
-
Effects of (p)ppGpp on the Progression of the Cell Cycle of Caulobacter crescentusJournal of Bacteriology 196:2514–2525.https://doi.org/10.1128/JB.01575-14
-
The bacterial flagellar motor: structure and function of a complex molecular machineInternational Review of Cytology 233:93–134.https://doi.org/10.1016/S0074-7696(04)33003-2
-
Spatiotemporal control of PopZ localization through cell cycle–coupled multimerizationThe Journal of Cell Biology 201:827–841.https://doi.org/10.1083/jcb.201303036
-
Spot regulates DnaA stability and initiation of DNA replication in carbon-starved Caulobacter crescentusJournal of Bacteriology 190:6867–6880.https://doi.org/10.1128/JB.00700-08
-
How Bacteria assemble FlagellaAnnual Review of Microbiology 57:77–100.https://doi.org/10.1146/annurev.micro.57.030502.090832
-
The genetic basis of laboratory adaptation in Caulobacter crescentusJournal of Bacteriology 192:3678–3688.https://doi.org/10.1128/JB.00255-10
-
A multi-protein complex from Myxococcus xanthus required for bacterial gliding motilityMolecular Microbiology 76:1539–1554.https://doi.org/10.1111/j.1365-2958.2010.07184.x
-
Two-in-one: bifunctional regulators synchronizing developmental events in bacteriaTrends in Cell Biology 22:14–21.https://doi.org/10.1016/j.tcb.2011.09.004
-
Generating and exploiting polarity in bacteriaScience 298:1942–1946.https://doi.org/10.1126/science.1072163
-
Regulation of cell polarity in bacteriaThe Journal of Cell Biology 206:7–17.https://doi.org/10.1083/jcb.201403136
-
Polarity and cell fate asymmetry in Caulobacter crescentusCurrent Opinion in Microbiology 15:744–750.https://doi.org/10.1016/j.mib.2012.10.011
Article and author information
Author details
Patrick H Viollier

Funding
Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (31003A_162716)
- Patrick H Viollier
European Research Council (243016)
- Suliana Manley
Marie Curie Intra-European Fellowship (PIEF-GA-2011-297918)
- Seamus Holden
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Funding support was from Swiss National Science Foundation grants to PHV, European Research Council Starting Grant 243016 to SM and Marie Curie Intra-European Fellowship PIEF-GA-2011-297918 to SH. We are grateful to Florian Gays for technical support with phage adsorption assays.
Version history
- Received: June 9, 2016
- Accepted: November 1, 2016
- Version of Record published: December 23, 2016 (version 1)
Copyright
© 2016, Mignolet et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,187
- Page views
-
- 200
- Downloads
-
- 15
- Citations
Article citation count generated by polling the highest count across the following sources: Crossref, PubMed Central, Scopus.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Functional dichotomy and distinct nanoscale assemblies of a cell cycle-controlled bipolar zinc-finger regulator
eLife 5:e18647.
https://doi.org/10.7554/eLife.18647
Further reading
-
- Cell Biology
- Chromosomes and Gene Expression
Heat stress is a major threat to global crop production, and understanding its impact on plant fertility is crucial for developing climate-resilient crops. Despite the known negative effects of heat stress on plant reproduction, the underlying molecular mechanisms remain poorly understood. Here, we investigated the impact of elevated temperature on centromere structure and chromosome segregation during meiosis in Arabidopsis thaliana. Consistent with previous studies, heat stress leads to a decline in fertility and micronuclei formation in pollen mother cells. Our results reveal that elevated temperature causes a decrease in the amount of centromeric histone and the kinetochore protein BMF1 at meiotic centromeres with increasing temperature. Furthermore, we show that heat stress increases the duration of meiotic divisions and prolongs the activity of the spindle assembly checkpoint during meiosis I, indicating an impaired efficiency of the kinetochore attachments to spindle microtubules. Our analysis of mutants with reduced levels of centromeric histone suggests that weakened centromeres sensitize plants to elevated temperature, resulting in meiotic defects and reduced fertility even at moderate temperatures. These results indicate that the structure and functionality of meiotic centromeres in Arabidopsis are highly sensitive to heat stress, and suggest that centromeres and kinetochores may represent a critical bottleneck in plant adaptation to increasing temperatures.
-
- Cell Biology
High-altitude polycythemia (HAPC) affects individuals living at high altitudes, characterized by increased red blood cells (RBCs) production in response to hypoxic conditions. The exact mechanisms behind HAPC are not fully understood. We utilized a mouse model exposed to hypobaric hypoxia (HH), replicating the environmental conditions experienced at 6000 m above sea level, coupled with in vitro analysis of primary splenic macrophages under 1% O2 to investigate these mechanisms. Our findings indicate that HH significantly boosts erythropoiesis, leading to erythrocytosis and splenic changes, including initial contraction to splenomegaly over 14 days. A notable decrease in red pulp macrophages (RPMs) in the spleen, essential for RBCs processing, was observed, correlating with increased iron release and signs of ferroptosis. Prolonged exposure to hypoxia further exacerbated these effects, mirrored in human peripheral blood mononuclear cells. Single-cell sequencing showed a marked reduction in macrophage populations, affecting the spleen’s ability to clear RBCs and contributing to splenomegaly. Our findings suggest splenic ferroptosis contributes to decreased RPMs, affecting erythrophagocytosis and potentially fostering continuous RBCs production in HAPC. These insights could guide the development of targeted therapies for HAPC, emphasizing the importance of splenic macrophages in disease pathology.




{kind=link}
{kind=link}
{kind=link}
{kind=link}