MYBL2 is a sub-haploinsufficient tumor suppressor gene in myeloid malignancy
- University Hospital Essen, Germany
- Dana-Farber Cancer Institute, United States
- MD Anderson Cancer Center, United States
- University of Helsinki, Finland
- University of California, San Francisco, United States
- Children’s Hospital Oakland Research Institute, United States
- Brigham and Women’s Hospital, United States
Abstract
A common deleted region (CDR) in both myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN) affects the long arm of chromosome 20 and has been predicted to harbor a tumor suppressor gene. Here we show that MYBL2, a gene within the 20q CDR, is expressed at sharply reduced levels in CD34+ cells from most MDS cases (65%; n = 26), whether or not they harbor 20q abnormalities. In a murine competitive reconstitution model, Mybl2 knockdown by RNAi to 20–30% of normal levels in multipotent hematopoietic progenitors resulted in clonal dominance of these ‘sub-haploinsufficient’ cells, which was reflected in all blood cell lineages. By 6 months post-transplantation, the reconstituted mice had developed a clonal myeloproliferative/myelodysplastic disorder originating from the cells with aberrantly reduced Mybl2 expression. We conclude that downregulation of MYBL2 activity below levels predicted by classical haploinsufficiency underlies the clonal expansion of hematopoietic progenitors in a large fraction of human myeloid malignancies.
https://doi.org/10.7554/eLife.00825.001eLife digest
Blood cells are produced within bone marrow by specialized stem cells and progenitor cells. Abnormalities in this process lead to a group of diseases known as myeloid malignancies, which include acute myeloid leukaemia—in which the bone marrow produces abnormal white blood cells—and myelodysplastic syndromes, which are caused by too few mature blood cells being produced.
Many individuals affected by these disorders possess a shortened form of chromosome 20 that lacks a number of genes. This deletion is only ever seen in one of their two copies of the chromosome—suggesting that at least some of these genes are essential for survival—but the identity of the gene(s) that are associated with the increased risk of myeloid malignancies is unknown.
Now, Heinrichs et al. have uncovered a key tumor suppressor among those genes frequently lost on chromosome 20. The gene, which is called MYBL2, encodes a transcription factor that helps to control the cell division cycle. Myeloid malignancy patients lacking one copy of this gene showed levels of MYBL2 expression that were less than 50% of those in healthy individuals. This suggests that additional mechanisms must be acting to reduce expression of their remaining copy of the gene. Surprisingly, MYBL2 levels were also reduced in myeloid malignancy patients who possessed two intact copies of chromosome 20, indicating that loss of a single copy represents only one mechanism to reduce MYBL2 expression, i.e., the ‘tip-of-the-iceberg’. Hence, this finding reveals a more general role for MYBL2 as it indicates that more patients are likely to be affected by altered expression of this gene.
To confirm their findings from studies in patients, Heinrichs et al. used gene silencing techniques to reduce the expression of MYBL2 in mice and showed that this induced symptoms of myeloid malignancies in the animals. Moreover, injection of modified cells from these animals into healthy mice also induced symptoms in the recipients. The modified cells are able to expand more robustly than normal cells, and this dominance induced by downregulation of the tumor suppressor increases the risk of malignancy.
In addition to revealing a new tumor suppressor gene and its contribution to myeloid malignancies, the study by Heinrichs et al. highlights the importance of gene dosage in mediating the effects of tumor suppressors.
https://doi.org/10.7554/eLife.00825.002Introduction
The molecular changes underlying human myeloid malignancies remain difficult to unravel, posing major obstacles to the development of effective countermeasures. Although the silencing of tumor suppressor genes by chromosomal deletions, point mutations, or other mechanisms is an acknowledged factor in myeloid cell transformation, the specific involvement of gene dosage is not well understood. In broadest terms, single-copy loss of a suppressor gene can be sufficient to modify gene function and promote tumorigenesis (classical haploinsufficiency), while in other tumors, the loss of two alleles is required (two-hit paradigm of Knudson) (Knudson, 1971). Recent evidence indicates that more subtle reductions in suppressor gene function may contribute importantly to myeloid malignancy (Rosenbauer et al., 2004; Liu et al., 2007), leading to the need for faithful animal models to establish that such changes are truly involved in tumorigenesis.
Loss of an interstitial segment of chromosome 20q (20q CDR) is detected in about 4% of myelodysplastic syndromes (MDS) (Haase et al., 2007), and this region is variably affected in different types of myeloproliferative neoplasms (MPN), including polycythemia vera (10%) and primary myelofibrosis (12%), and less commonly in acute myeloid leukemia (AML; 1%) (Bench et al., 2000). Notably, only heterozygous deletions have been found in studies of myeloid malignancies with loss of chromosome 20q, without any evidence of homozygous deletion or mutations of a gene within the affected region (Heinrichs et al., 2009; Huh et al., 2010). These findings implicate a gene within the 20q CDR that is essential for cell viability, but whose tumor suppressor function is strongly dose-dependent and does not follow the classical Knudson model (Knudson, 1971), which predicts biallelic gene inactivation. Instead, monoallelic loss, with or without additional epigenetic or microRNA (miRNA)-mediated downregulation of the remaining allele, may reduce gene expression levels sufficiently to promote myeloid cell transformation. Thus, we sought to identify candidate tumor suppressor genes within the 20q CDR on the basis of their reduced expression in malignant myeloid progenitor cells, as we have reported previously for CTNNA1 in myeloid malignancies with deletions of chromosome 5q (Liu et al., 2007; Ye et al., 2009).
Here we identify MYBL2, which encodes a transcription factor with functions in checkpoint control of the G2 cell cycle phase, as a key tumor suppressor gene in over one half of all MDS cases, whether characterized by a 20q deletion or a normal karyotype. Reductions of MYBL2 dosage to levels below those commensurate with single-copy loss conferred a competitive advantage to hematopoietic progenitor cells in both primary and secondary transplantation assays and were associated with histopathologic changes typical of myeloid neoplasia. These findings implicate aberrantly low levels of MYBL2 expression as a central mechanism in the development of clonal dominance in MDS and other myeloid malignancies.
Results
MYBL2 identified as a potential tumor suppressor
We first studied the gene expression profiles of CD34+ hematopoietic progenitor cells from eight MDS cases with cytogenetically evident aberrations of chromosome 20q, as compared to CD34+ cells from normal individuals (Figure 1—figure supplement 1A). We found that MYBL2, which resides near the center of this region (Figure 1—figure supplement 2), was among the top-scoring genes downregulated in MDS cells compared to normal CD34+ controls. MYBL2 encodes a highly conserved transcription factor that acts as a major component of the dREAM complex, which controls the G2-to-M phase transition within the cell cycle (Korenjak et al., 2004; Lewis et al., 2004). The mean expression level of MYBL2 (39%) was less than that of normal CD34+ cells (Figure 1—figure supplement 1B), suggesting that mechanisms beyond the deletion of one allele might affect the remaining allele. Thus, on the assumption that some MDS cases with a normal karyotype might also have reduced expression of a gene or genes within the 20q CDR, we undertook an expression analysis of CD34+ cells from 18 MDS cases with a normal karyotype and the eight cases with a 20q aberration (Supplementary file 1A), compared to CD34+ cells from four normal bone marrow samples. MYBL2 was the top-scoring gene among 108 differentially expressed genes (Figure 1A). Further analysis showed that in 17 (65%) of 26 MDS patients (Figure 1—figure supplement 3A), MYBL2 expression levels in CD34+ cells were reduced to ≤45% of normal CD34+ cells levels (mean 32.8%). These microarray data were confirmed by QRT-PCR analysis of MYBL2 expression levels in 10 MDS cases and eight normal CD34+ bone marrow samples, including four additional normal control CD34+ samples that were not part of the original microarray analysis (Figure 1B). Importantly, MYBL2 levels measured by QRT-PCR in MDS samples were highly correlated (r = 0.966) with those determined by microarray analysis (Figure 1—figure supplement 3B). Hence, MYBL2 appears to be expressed at reduced levels in nearly two-thirds of MDS cases, including those with a normal karyotype, suggesting that it functions as a dose-dependent tumor suppressor gene.
Figure 1 with 3 supplements see all
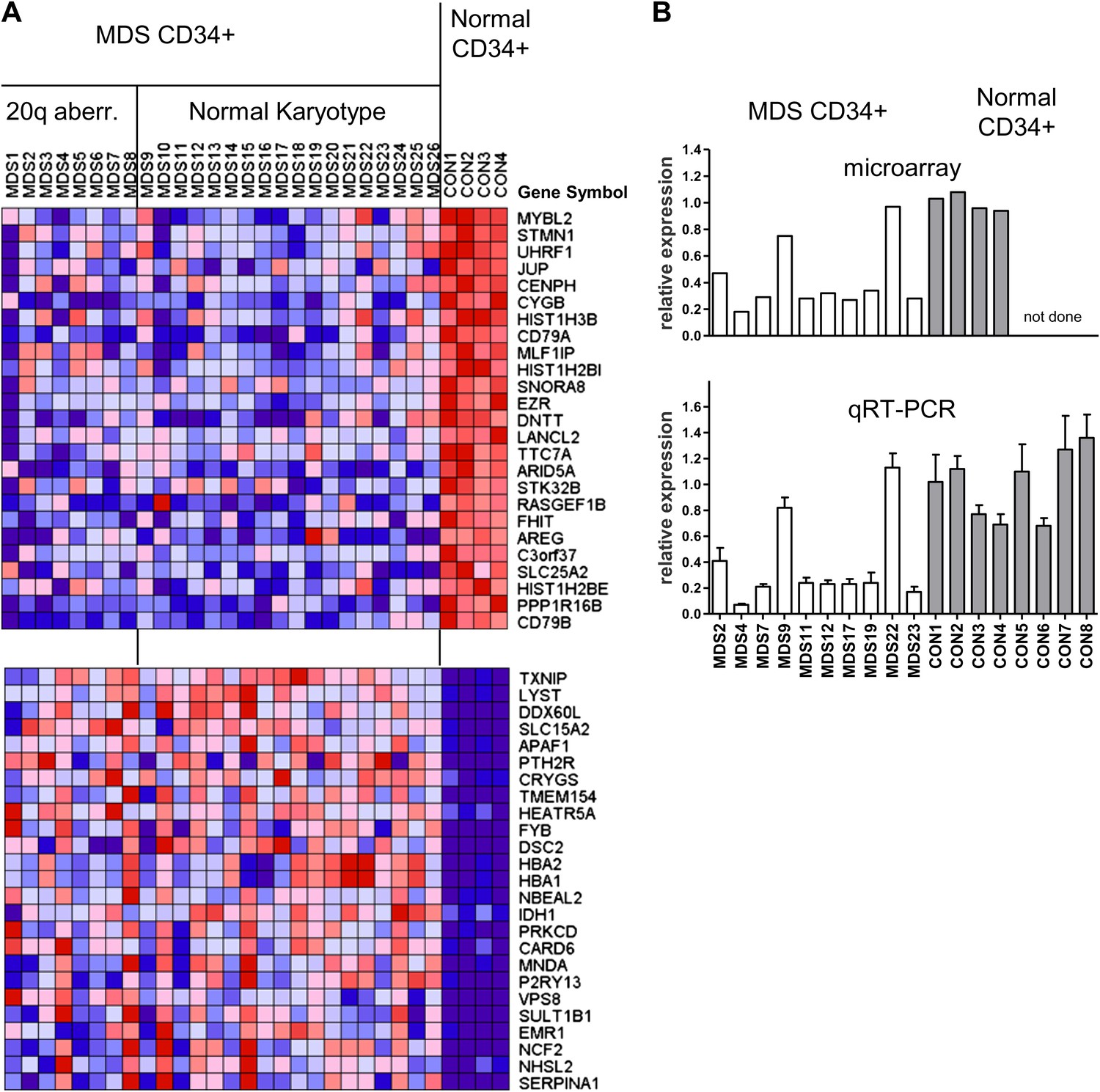
Gene expression analysis of CD34+ cells from MDS patients with a 20q aberration or a normal karyotype.
(A) Heat map showing the top-scoring 25 genes that were either up- or downregulated in CD34+ cells from 26 MDS patients vs normal donors (see Supplementary file 1A for patient characteristics). Genes were considered differentially expressed if they met two criteria: q ≤ 0.006 and fold-change ≥2.0 (108 genes). (B) Validation of microarray-based MYBL2 gene expression data for selected clinical samples (upper panel) by qRT-PCR analysis (lower panel), including four additional samples of normal CD34+ bone marrow cells. Relative MYBL2 expression results by qRT-PCR analysis were normalized to three control genes.
To address whether reduced levels of MYBL2 expression actively contribute to the MDS phenotype or merely constitute a compensatory response to other abnormalities, we asked if reduced MYBL2 transcriptional activity is reflected in the MDS gene expression signature. To model gene dose insufficiency by RNA interference (RNAi), we used a set of eight shRNA-expression vectors in the MDS/AML cell line SKM1, which reduced MYBL2 expression to 5–30% of the endogenous MYBL2 levels (Figure 2A), approximating the endogenous levels we had identified in CD34+ cells from patients (Figure 1B). To identify the genes most highly correlated with decreased MYBL2 expression, we introduced our shRNA-expression vectors into normal primary human CD34+ cells and measured gene expression at 48 hr post-transduction. Nearly all differentially expressed genes were downregulated after MYBL2 knockdown (81 of 89 genes; Figure 2B and Figure 2—figure supplement 1). Because these results are based on eight different MYBL2-specific shRNAs, each with different knockdown efficiency, this analysis ensures the robustness of the data and eliminates potentially confounding off-target effects by individual shRNAs. Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) identified mitosis as the key biological process reflected by the MYBL2 gene signature, specifically the G2-to-M phase transition (Figure 2—figure supplement 2) (Whitfield et al., 2002; Zhu et al., 2004; Shepard et al., 2005). Individual genes and their functions included the G2 cell cycle regulator cyclin B (CCNB1 and CCNB2), the early mitotic regulator FBXO5 and the coregulator of chromosome architecture CDCA2 (Supplementary file 1B).
Figure 2 with 2 supplements see all
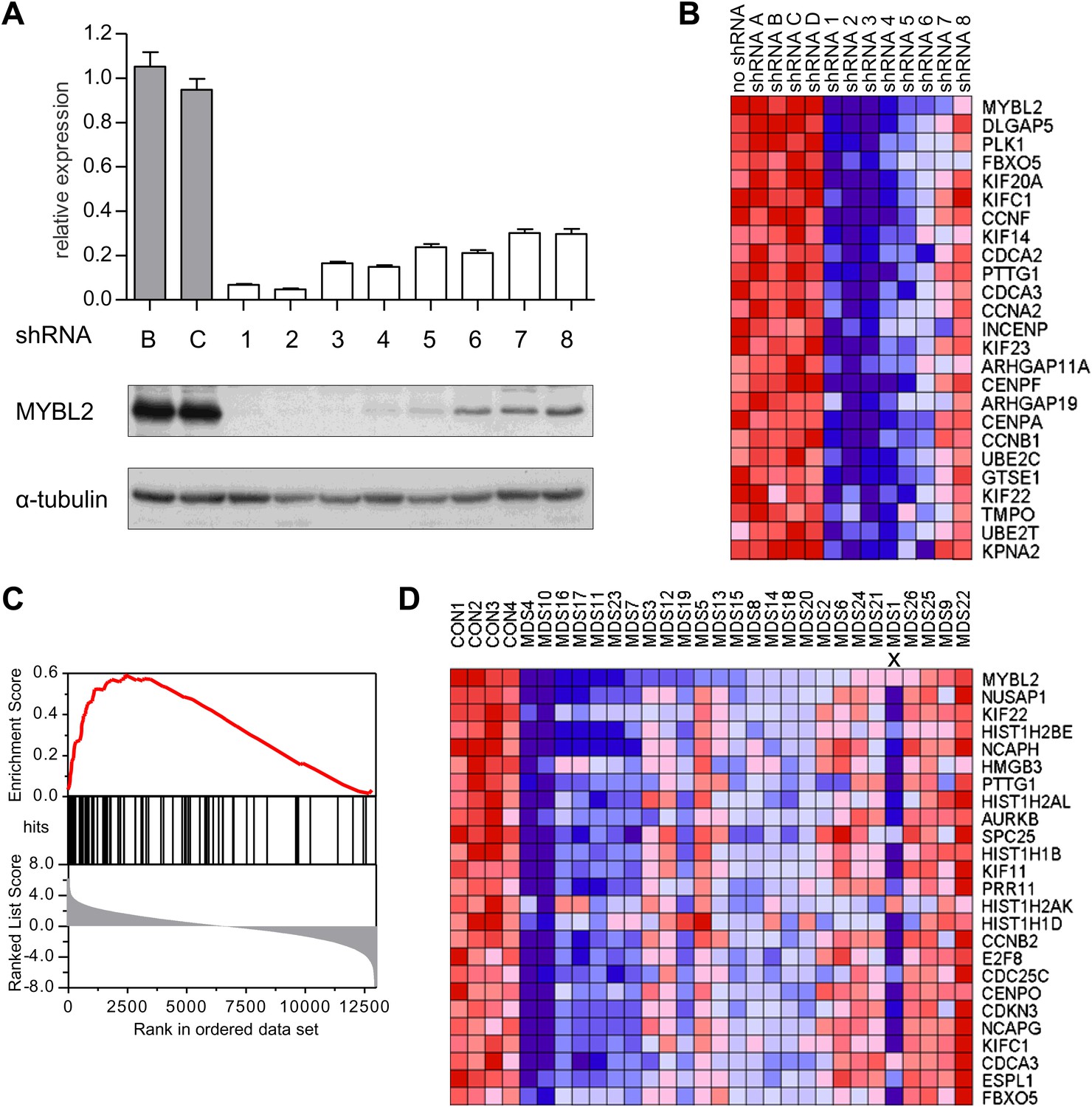
Identification of an MYBL2-regulated gene expression signature and its enrichment in CD34+ cells from MDS patients.
(A) MYBL2 knockdown in SKM1 cells by a series of shRNAs (control shRNAs are labeled B and C; MYBL2-specific shRNAs are labeled 1–8). MYBL2 expression levels were measured by qRT-PCR normalized to three control genes (upper panel) and by Western blotting with tubulin as a normalization control (lower panels). (B) Heat map of the top 25 genes whose expression levels positively correlated (score ≥ 0.8) with those of MYBL2. Gene expression in normal CD34+ cells was measured by microarray in one unperturbed sample (‘no shRNA’), four control shRNA-expressing samples (shRNAs A–D) and eight MYBL2-specific shRNA-expressing samples (shRNAs 1-8). (C) Gene set enrichment analysis (GSEA) of the MYBL2 gene signature (top 81 positively correlated genes) within the ranked gene expression results for CD34+ cells from MDS patients, compared to CD34+ cells from normal bone marrow. Enrichment of the MYBL2 signature in MDS bone marrow cells (see Figure 1A) is apparent. Genes are ranked by score and plotted on the x-axis; hits are indicated by vertical black bars and the enrichment score is plotted in red. (D) Heat map corresponding to the GSEA showing the 25 top-scoring genes. One sample, indicated by an ‘x’, shows enrichment of the MYBL2 signature, but relatively normal MYBL2 expression suggesting a measurement problem.
We finally performed GSEA to assess whether this MYBL2 gene expression signature (81 genes, Supplementary file 1B) was evident in MDS samples with intrinsically low expression of this gene. This analysis revealed robust enrichment of the MYBL2 signature in MDS CD34+ cells (Figure 2C). Furthermore, the expression levels of MYBL2-regulated genes corresponded closely with those of MYBL2 itself (Figure 2D). Together, these findings indicate that MYBL2 downregulation in CD34+ cells is reflected in the aberrant gene expression profile of a large fraction of MDS cases.
Low MYBL2 expression in cases with a normal karyotype
To determine which MDS patients with a normal karyotype have an ‘MYBL2-low’ expression signature, we first analyzed the expression levels of MYBL2-regulated genes in the CD34+ cells from our 18 normal-karyotype MDS cases, using a k-nearest neighbor (KNN) classifier and Euclidean distance (k = 3) to predict the class label by a majority vote. 10 of these cases (56%) were classified as MYBL2 low, while the remaining eight cases had an MYBL2 signature that was statistically similar to that of normal CD34+ controls (Figure 3—figure supplement 1). To validate our classification algorithm using a separate dataset, we then interrogated the normal karyotype cases (n = 94) reported by Pellagati et al. (Pellagatti et al., 2010), identifying a MYBL2-low signature in 36 cases (38%), most of which (30 cases) were identified at a confidence level of 1.0. 58 cases (62%) had a normal MYBL2 signature (Figure 3). Thus, we were able to confirm in an independent cohort that the ‘MYBL2 low’ signature is present in CD34+ cells from a substantial fraction of normal-karyotype MDS cases.
Figure 3 with 1 supplement see all
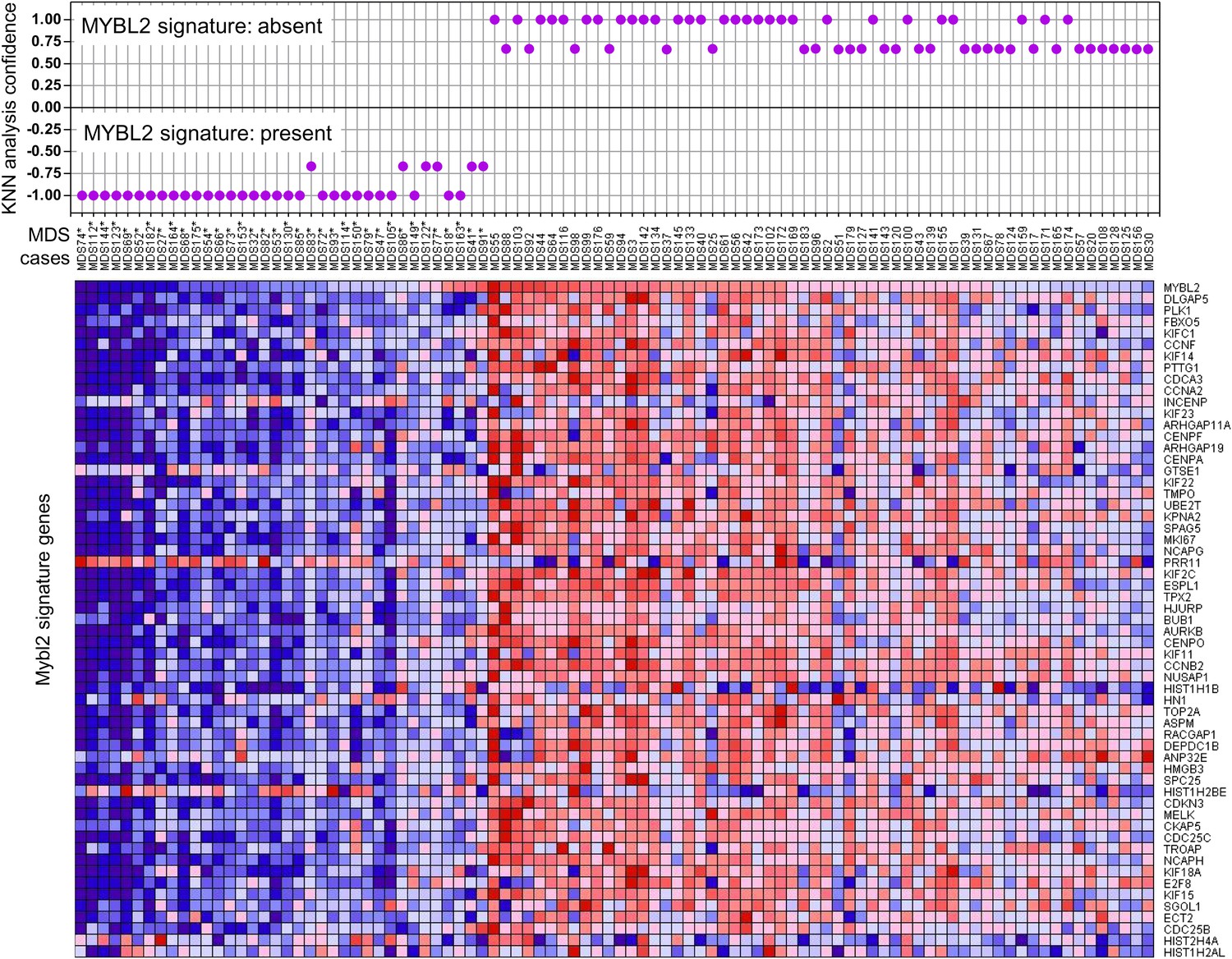
KNN classification for normal karyotype patient samples of an independent data set (Pellagatti et al., 2010).
A k-nearest-neighbor (KNN) classification model for patient samples was trained on our gene expression data set of CD34+ cells with or without MYBL2 knockdown by RNAi. Upper panel: KNN classification for the presence (negative values) or absence (positive values) for the MYBL2 signature using Euclidean distance (k = 3) to predict the class label by a majority vote. Lower panel: gene expression profile of MYBL2 signature genes. Note that some gene profiles (INCEP, GTSE1, PRR11, HIST1H1B, HIST1H2AL, HIST1H2BE, HIST2H4A, HN1) appear not to match the overall signatures most likely due to platform inconsistencies.
Mechanisms of MYBL2 dosage reduction
To begin to clarify the mechanism of reduced MYBL2 activity or aberrantly low MYBL2 expression in MDS, we resequenced DNA samples from 144 patients, identifying two cases with heterozygous somatic mutations within the coding sequence that were verified by analysis of patient-specific germline DNA collected from buccal swabs (Figure 4A). Both mutations, an amino acid substitution and a nonsense mutation, were located within the C-terminus. To test whether these mutations inactivate the MYBL2 protein, we replaced endogenous MYBL2 in SKM1 cells with MYBL2 carrying one of the mutations, using a knockdown/rescue approach. SKM1 cells died upon complete knockdown of MYBL2 with an shRNA targeting the 3′UTR (Figure 4B). This phenotype was rescued by re-expression of the wild-type MYBL2 cDNA and the cDNA from patient 28 (Figure 4A), but not by re-expression of the mutated, truncated MYBL2 cDNA of patient 27, which failed to restore the viability of the SKM1 cells. Thus, we have documented a somatically acquired gene-specific inactivating mutation that targets MYBL2 in one case of MDS. This specific case is highly informative because the inactivating mutation targets MYBL2 directly, and not an adjacent gene on 20q.
Figure 4 with 1 supplement see all
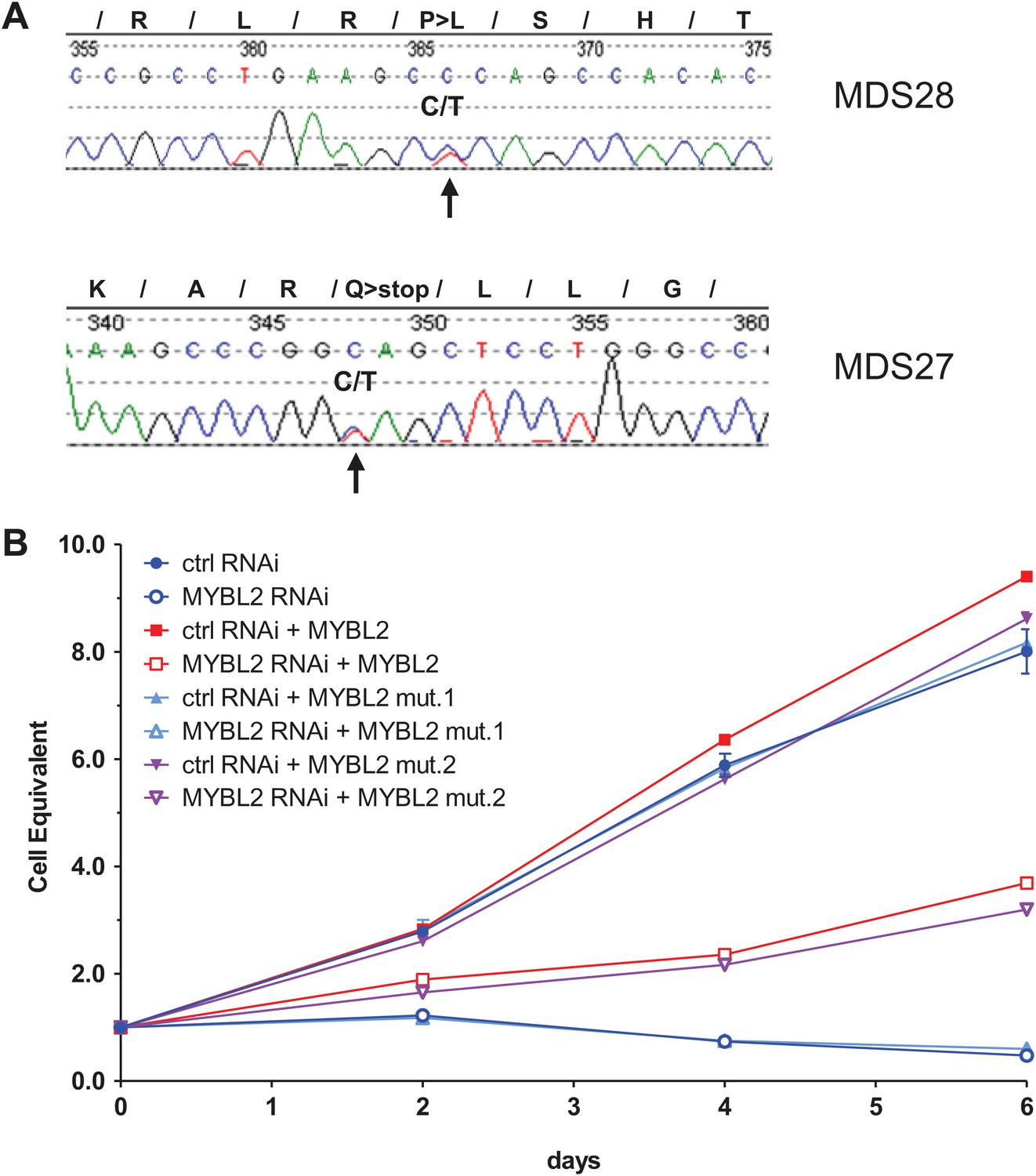
MYBL2 is mutated at a low frequency in MDS.
(A) MYBL2 exons were resequenced in the DNA of mononuclear bone marrow cells of 144 MDS patients. The results indicated a P-to-L aminoacid substitution (top, mutation 1, patient MDS28) and a nonsense mutation (bottom, mutation 2, patient MDS27). (B) Growth curves of SKM1 cells after transduction with a MYBL2 shRNA. SKM1 cells were transduced with an empty vector (circles), a vector expressing wildtype MYBL2 cDNA (squares), mutant MYBL2 cDNA (mut.1, triangles) or mutant MYBL2 cDNA (mut.2, diamonds). Upon transduction with a control (solid symbols) or a MYBL2-specific (open symbols) shRNA-expressing vector growth was monitored for 6 days.
Given that a substantial number of MYBL2-low cases lacked either a 20q deletion or gene-specific inactivating mutation, we hypothesized that additional mechanisms of MYBL2 downregulation exist in MDS. A potential candidate is aberrant DNA methylation, especially in view of the 921 base-pair CpG island that spans the proximal promoter and the first exon of the MYBL2 gene. However, analysis of this site using bone marrow cell DNA from 30 MDS patients failed to detect any methylated cytosines, indicating that DNA methylation is not a frequent mechanism for MYBL2 downregulation in MDS (JP Issa, personal communication, 2012). This finding is supported by a recently published study that did not identify methylation affecting the MYBL2 gene in cells from 83 MDS cases (Del Rey et al., 2012).
We also considered that the upregulation of one or more miRNAs targeting MYBL2 might explain our results. We pursued this notion in a subset of patient samples (seven with low MYBL2 expression and two with normal MYBL2 expression, together with three controls) by analyzing the expression levels of mir-29a and mir-30e. Both of these miRNAs target MYBL2 (Han et al., 2010; Martinez et al., 2011), with aberrant expression of mir-29a leading to clonal dominance and acute myeloid leukemia (Han et al., 2010). Our results show upregulation of either mir-29a or mir-30e in three cases with MYBL2 downregulation, but not in controls or cases with normal MYBL2 expression (Figure 4—figure supplement 1). Thus, overexpression of miRNAs targeting MYBL2 affords an attractive mechanism by which MYBL2 expression levels are lowered in a substantial fraction of MDS cases.
Clonal dominance of transplanted cells with low Mybl2 expression
If sub-haploinsufficient decreases in MYBL2 dosage contribute to MDS pathogenesis, a technique such as RNAi knockdown with a series of shRNA hairpins of graded potencies should provide an experimental model that can be used to demonstrate a clonal advantage within hematopoietic progenitor cells and implicate the level of Mybl2 knockdown that produces the maximum effect. Thus, using five independent, specific shRNA-expressing vectors to target Mybl2 in primitive hematopoietic cells, we established a competitive reconstitution assay in mice. These vectors downregulated Mybl2 expression to levels that ranged from 13% (M1) to 33% (M5) of those expressed by control shRNA-expressing vectors in 32D cells, a murine myeloid cell line (Figure 5—figure supplement 1). After transducing Lin− mononuclear bone marrow cells in separate vials with these five vectors, which coexpressed a green fluorescent protein (GFP), or with two different control shRNA vectors coexpressing a red fluorescent protein (RFP) (Strack et al., 2008), we washed the cells, pooled the five Mybl2-specific shRNA knockdown cell aliquots and the two control aliquots, and transplanted equal numbers of these two cell pools into lethally irradiated mice (Figure 5A). A second group of mice were transplanted with pooled GFP+ and RFP+ cells expressing control shRNAs only, generating control/GFP vs control/RFP recipients (Figure 5—figure supplement 2A). Analysis of GFP and RFP levels in peripheral blood at 12 weeks (Figure 5B) showed a strikingly higher median frequency of GFP+ vs RFP+ cells in the peripheral blood of the mice reconstituted with GFP+/Mybl2-shRNA vs RFP+/control shRNA (30.9% vs 2.6%, p<0.0001). By contrast, in mice injected with control/GFP vs control/RFP cells, the median frequencies of both RFP- and GFP-expressing cells were low (1.5% and 3.4%; Figure 5—figure supplement 2B).
Figure 5 with 3 supplements see all
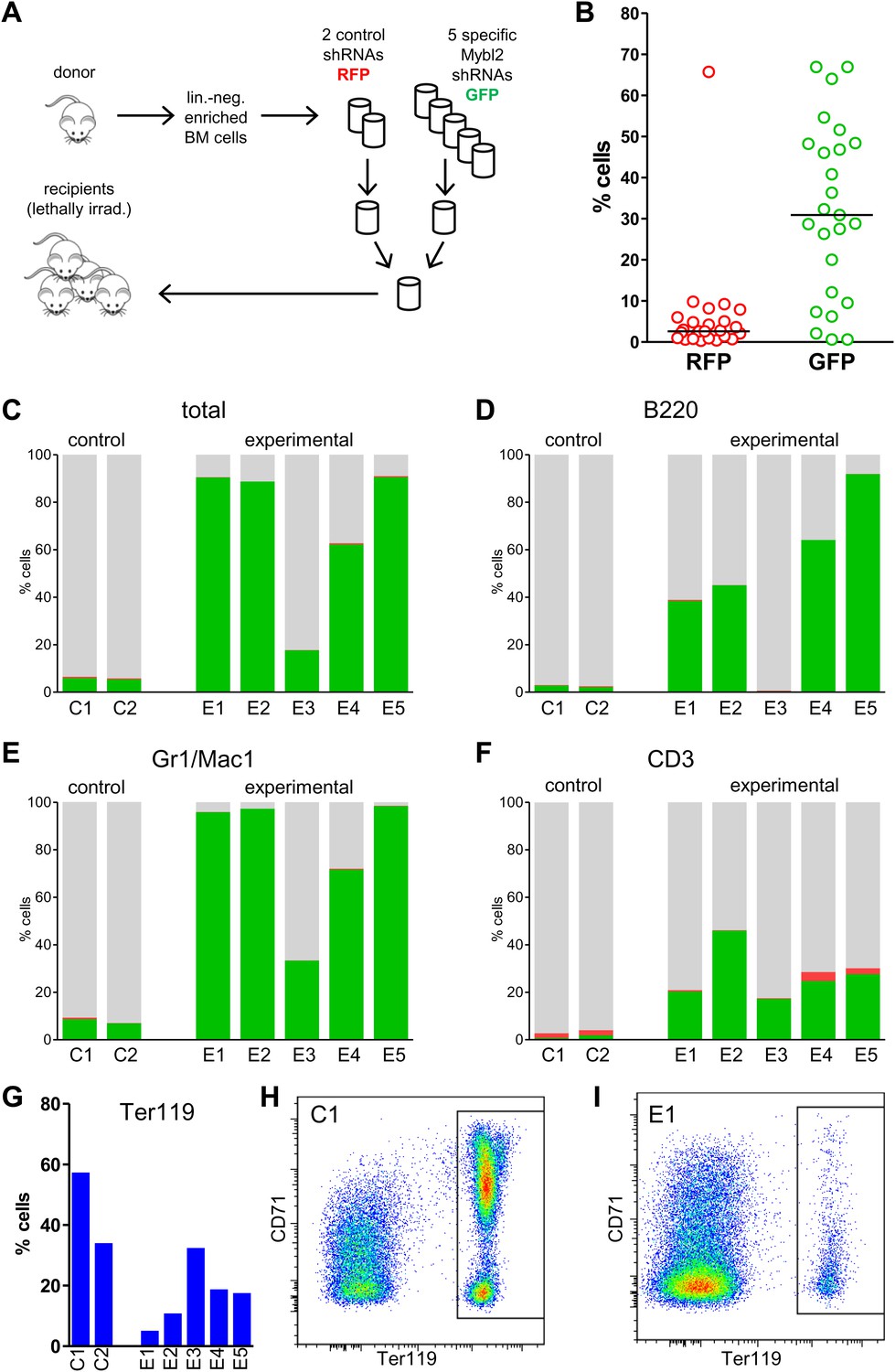
Competitive in vivo reconstitution assay testing expansion capacity of cells with low Mybl2 expression.
(A) Mononuclear bone marrow cells were collected from donor mice, enriched for lineage-negative cells and transduced in separate wells with specific Mybl2 shRNA/GFP vectors and control shRNA/RFP vectors. Equal numbers of cells were combined to obtain a pool with specific shRNA-expressing cells and a pool of control shRNA-expressing cells. Both pools were combined in equal parts and transplanted into lethally irradiated mice (n = 25) in two independent experiments with transduction efficiencies of 5–8%. (B) Analysis of peripheral blood mononuclear cells for GFP and RFP positivity at 12 weeks post-transplant as an indicator of long-term reconstitution (Morrison and Weissman, 1994) (GFP+ vs RFP+, p<0.0001 by paired t-test; horizontal bars denote median values). (C–F), Analysis of bone marrow samples from mice 6–7 months after competitive reconstitution. Flow cytometric analysis of mononuclear bone marrow cells from five experimental mice (E1–E5) transplanted with Mybl2-specific shRNA/GFP cells and control shRNA/RFP cells showed marked overrepresentation of GFP+ cells by comparison with results for control mice (C1, C2) transplanted with control shRNA/RFP- and shRNA/GFP-transduced cells. The fractions of GFP (green), RFP (red) and unlabeled cells (gray) are shown for the total cell populations (C), B220-positive population (D), Gr1/Mac1-positive population (E), and CD3-positive population (F). (G) Respective analysis of bone marrow erythropoiesis by CD71/Ter119 staining. Frequencies of Ter119-positive cells are shown for all animals, and representative plots presented for mouse C1 (H) and mouse E1 (I).
The possibility that these results were influenced by the use of pooled cells representing different shRNA sequences was excluded by transducing the Lin− cells with single hairpins specific for Mybl2. For this experiment (Figure 6A) we transduced Lin− bone marrow cells with either the M3 shRNA vector (n = 7 mice) or the M4 shRNA vector (n = 8 mice). These two shRNAs were chosen because QPCR analysis of the mice injected with pools of cells transduced with each of the five shRNAs (Figure 5A) showed that M3 and M4 were integrated into the DNA of the reconstituted cells that exhibited clonal dominance (Figure 6—figure supplement 1). The results of transplanting cells transduced with single Mybl2-specific hairpins clearly show clonal dominance by cells transduced with either of these vectors (Figure 6B). In a ‘color-swap’ experiment using the M3 shRNA, the bone marrow cells with Mybl2 knockdown became clonally dominant, whether the red or green fluorophore was used (Figure 6C). Together, these competitive reconstitution studies firmly establish the surprising finding that downregulation of Mybl2 levels to ∼30% of normal levels using two different Mybl2 shRNAs imparts a strong clonal advantage to hematopoietic progenitor cells, one that becomes reflected in the analysis of circulating white blood cells as early as 12 weeks after transplantation.
Figure 6 with 2 supplements see all
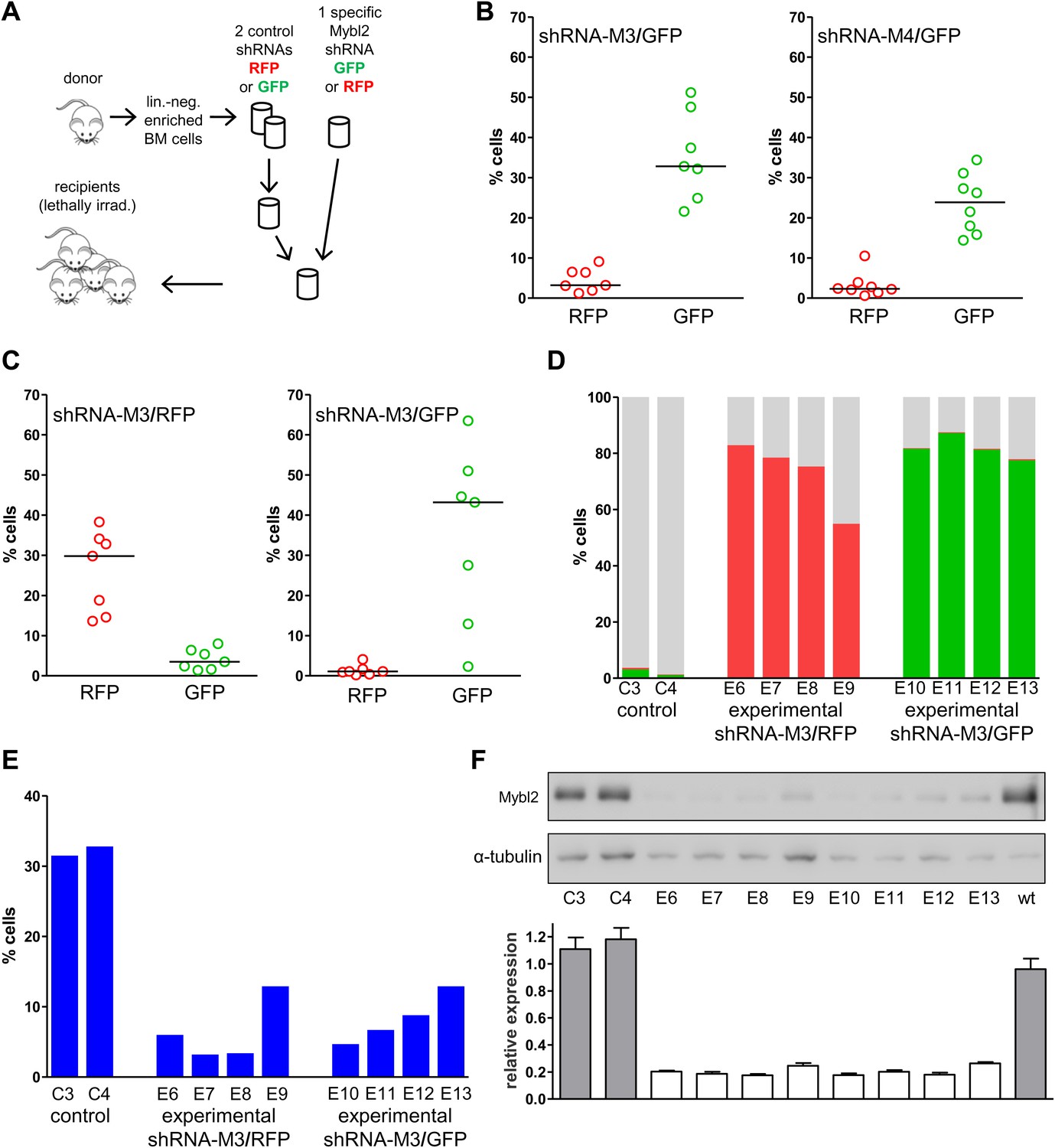
Analysis of peripheral blood and bone marrow samples from mice transplanted with single Mybl2-targeting shRNA vectors.
(A) Mononuclear bone marrow cells were collected from donor mice, enriched for lineage-negative cells and transduced in separate wells with a single, specific Mybl2 shRNA vector or control shRNA vector with an efficiency of <10%. The vectors allowed for co-expression of RFP or GFP. Equal numbers of control shRNA-expressing cells and Mybl2-specific shRNA-expressing cells were combined and transplanted into lethally irradiated mice in two independent experiments. (B) Experiment 1: Analysis of peripheral blood mononuclear cells for GFP and RFP positivity at 12 weeks post-transplant as an indicator of long-term reconstitution. Significant expansion of GFP-positive cells with use of either shRNA M3 (left, p=0.0004 by paired t-test) or M4 (right, p=0.0005). Horizontal bars denote median values. (C) Experiment 2, ‘color-swap’ experiment: Mybl2-specific shRNA M3 coupled with GFP (right) or RFP (left) expression associated with a significant expansion of Mybl2-downregulated cells (p=0.007 and p=0.001, respectively). (D) Flow cytometric analysis of mononuclear bone marrow cells from eight experimental mice (E6–E13) transplanted with cells expressing Mybl2-specific M3 shRNA/GFP and control shRNA/RFP cells (E6–E9) or Mybl2-specific M3 shRNA/RFP cells and control shRNA/GFP cells (E10–E13). Mybl2-RNAi cells were markedly overrepresented by comparison to results for control mice (C3, C4) transplanted with control shRNA/RFP- and shRNA/GFP-transduced cells. The fractions of GFP (green), RPF (red) and unlabeled cells (gray) are shown. (E) Analysis of bone marrow erythropoiesis by CD71/Ter119 staining with frequencies of Ter119-positive cell frequencies shown for all animals. (F) Analysis of Mybl2 levels of flow sorted cells by Western-blotting (upper panel) and qRT-PCR (lower panel).
Progression of Mybl2-low cells to myeloid malignancy
The marked competitive expansion of Mybl2 knockdown cells could represent either a benign process or a myeloid malignancy. Thus, we sacrificed 17 mice (nine transplanted with pooled cells [E1–E5, PD1–PD5] plus three controls and eight transplanted with cells transduced with a single vector [E6-E13] plus two controls) at 6 to 7 months post-transplantation, and studied their bone marrow cells by flow cytometry.
The results confirmed expansion of the Mybl2-knockdown cell population compared to control bone marrow cells (Figures 5C and 6D; Figure 8—figure supplement 1C), which surpassed even the percentage of GFP+ cells in the blood (Figure 5—figure supplement 3). Lineage analysis showed that this expansion included lymphocytes as well as myeloid cells (Figure 5D–F), indicating clonal dominance originating from transduced multilineage long-term repopulating cells. Cells of the myeloid lineage were most prominently affected, as the frequency of GFP+ cells in the Gr1+/Mac1+ population exceeded 90% in three of five experimental animals. Bone marrow erythropoiesis was reduced by Mybl2 knockdown, as indicated by the decreased percentages of Ter119+ cells (Figures 5G–I and 6E; Figure 8—figure supplement 1D). As expected, analysis by Western blotting and QRT-PCR of bone marrow cells expressing the M3 Mybl2-shRNA as a single hairpin showed that Mybl2 expression levels were reduced to approximately 20–30% of control levels after long-term reconstitution (Figure 6F). We did not observe any effect of this level of Mybl2 knockdown on the cell cycle-phase distribution of bone marrow cells from mice 6–7 months post-transplantation (Figure 6—figure supplement 2), indicating that this level of decreased Mybl2 expression does not induce a delay in the G2-to-M-phase transition.
We also evaluated the circulating blood counts in 13 of the 17 mice at the time of euthanasia 6–7 months after transplant. Reduced hemoglobin levels indicating anemia were observed in 12 of the 13 mice (Figure 7A), indicative of the ineffective erythropoiesis in the bone marrow observed by flow cytometry. Circulating white blood cell and platelet counts were in the normal range. Spleen weight was increased in 12 of the 13 mice, reflecting extramedullary hematopoiesis. A few of the mice showed hunched posture, weight loss and lethargy at the time of euthanasia (6–7 months post-transplant), but the majority of the recipient mice appeared well with no evidence of overt hematologic disease.
Figure 7 with 2 supplements see all
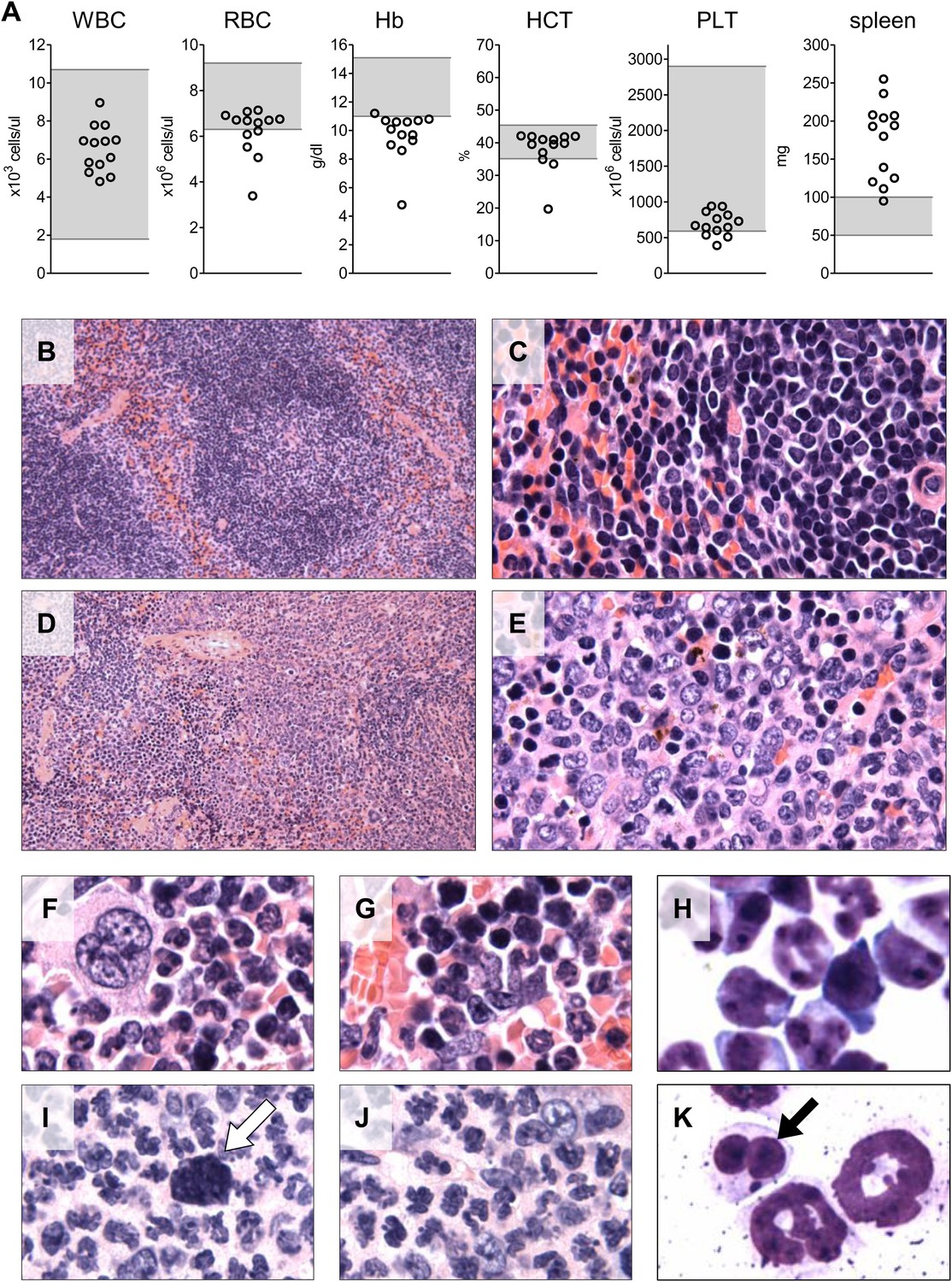
Analysis of blood counts and spleen weight (A), spleen (B–E) and bone marrow (F–K) samples from mice 6-7 months after competitive reconstitution.
(A) Analysis of peripheral blood parameters showing anemia of animals that received transplants of specific Mybl2-shRNA engineered cells: white blood cell count (WBC), red blood cell count (RBC), hemoglobin (HB), hematocrit (HCT), and platelet count (PLT). Spleen weights are shown as well. Grey areas indicate the normal range for each value. (B and C) Normal spleen histology at different magnification of control mouse C1 showing normal lymphoid-rich white pulp (50% spleen volume) and red pulp (50%). About 10% of the spleen consists of extramedullary hematopoiesis, which is in the red pulp and normal for mice. (D and E) Spleen histology of mouse E1 with specific Mybl2-knockdown at different magnification shows a marked depletion of white pulp. About 90% consists of red pulp, which is almost entirely extramedullary hematopoiesis. (F–K) Normal bone marrow histology (F and G) and cytology (H) of control mouse C1, compared to corresponding results for mouse E1 with specific Mybl2-knockdown (histology I, J; cytology K). Note marked shift to myeloid elements with loss of erythropoiesis and occasional dysplastic myeloid cells (white arrow: hypolobate megakaryocyte, black arrow: dysplastic neutrophil).
Histopathologic analysis of the spleen (Figure 7B–E) and the bone marrow (Figure 7F–K) at 6 to 7 months post transplantation showed marked changes in the overall tissue architecture and in specific cellular constituents. The bone marrow in experimental mice was hypercellular and contained more than 95% mature myeloid cells at the expense of erythroid cells (<5%), while 2% of the cells had a dysplastic morphology. The spleens were frequently enlarged (Figure 7A), the white pulp was virtually absent, and the whole organ showed evidence of extramedullary hematopoiesis. Notably, one mouse developed an erythroid leukemia characterized by a block of differentiation at the CD71-positive stage at 6 months post-transplantation (Figure 7—figure supplement 1). In summary, the histopathologic findings in these mice indicate a clonal myeloproliferative disorder with dysplastic features.
To identify the cells principally involved in the clonal dominance associated with this disorder, we analyzed the Lin−/Sca1+/Kit+ (LSK) bone marrow cells of the affected mice (n = 12) in comparison to controls. Within the LSK population of GFP+ or RFP+ Mybl2-knockdown bone marrow cells, HSCs (CD150+/CD48−) were diminished, MPPs (CD150−/CD48−) were maintained and LRPs (CD150−/CD48+) were increased compared to unlabeled, non-transduced cells. Thus, the clonal dominance due to low Mybl2 levels is first evident as an increased percentage of cells in the LRP fraction. (Figure 7—figure supplement 2).
We next performed secondary transplantation experiments to determine if self-renewing neoplastic cells were involved. We first analyzed four primary donors in detail and confirmed the presence of clonally dominant GFP+/Mybl2-RNAi cells in the peripheral blood and bone marrow of these mice (Figure 8—figure supplement 1A,C). Moreover, the mice were anemic based on low RBC and low hemoglobin levels (Figure 8—figure supplement 1B) and had markedly reduced erythropoiesis, as shown by low percentages of CD71neg/Ter119+ cells compared to that in a control transduced mouse (Figure 8—figure supplement 1D). For the secondary transplants, we injected 15 mice with bone marrow cells and eight with spleen cells that originated from these four individual donors (no pooling).
Analysis of blood samples collected from secondary recipients at 10 weeks revealed a marked clonal advantage for GFP+/Mybl2-RNAi cells transplanted from each of the four mice with primary disease, regardless of whether the cell source was spleen or bone marrow (Figure 8A). In addition, there was evidence of anemia (low RBC and low Hb levels) and thrombocytopenia in 9 of 13 animals (Figure 8B). The bone marrow of secondary recipients was also dominated by GFP+/Mybl2-RNAi cells (Figure 8C) and showed decreased levels of erythropoiesis (Figure 8D) as well as histologic evidence of a myeloproliferative disorder with dysplasia. Overall, the phenotypic consequences of Mybl2 knockdown were much more pronounced in secondary compared to primary recipients.
Figure 8 with 1 supplement see all
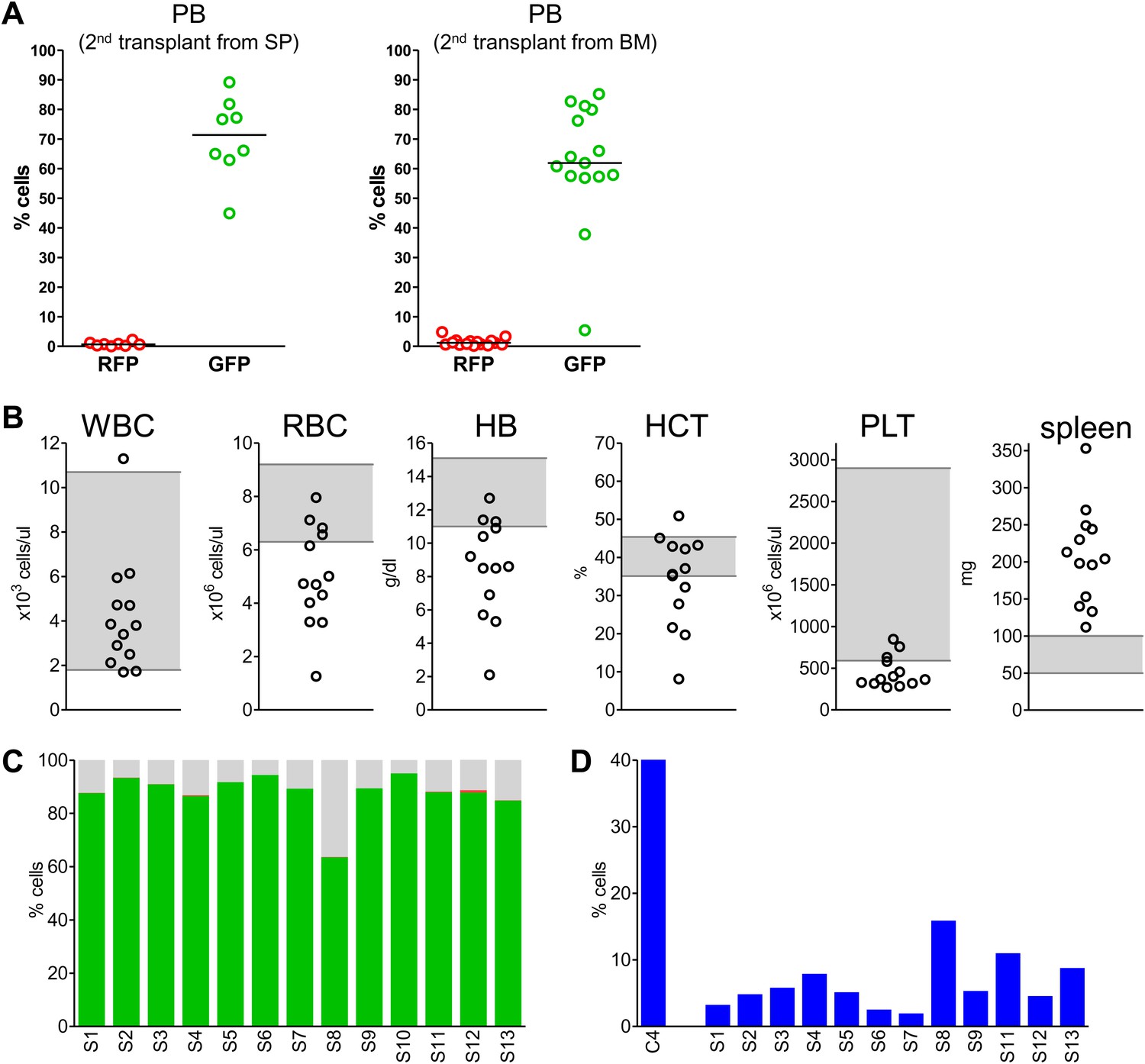
Secondary transplants: In vivo reconstitution assay testing capacity of cells with low Mybl2 expression to engraft in lethally irradiated mice.
(A) Analysis of peripheral blood mononuclear cells for GFP and RFP positivity at 10 weeks post-transplant showing engraftment of cells originating from spleen (p<0.0001, left panel) or bone marrow cells (p<0.0001, right panel). Horizontal bars denote median values. (B) Analysis of peripheral blood parameters showing anemia of animals that received secondary transplants of specific Mybl2-shRNA engineered cells: white blood cell count (WBC), red blood cell count (RBC), hemoglobin (HB), hematocrit (HCT), and platelet count (PLT). Spleen weights are shown as well. Grey areas indicate the normal range for each value. (C) Flow cytometric analysis of mononuclear bone marrow cells 3 months after transplantation from 13 mice (S1–S13) demonstrating strong engraftment and expansion of cells with a specific Mybl2 knockdown (color code as in Figure 5). (D) Analysis of bone marrow erythropoiesis by CD71/Ter119 staining showing the frequencies of CD71−/Ter119+ cells and revealing a strong reduction of Ter119+ cells in all experimental animals in comparison to a wild-type control animal.
Thus, our secondary transplantation assays show that either spleen or bone marrow cells from mice with myeloid neoplasia due to shRNA-mediated reduced Mybl2 expression can efficiently transmit the blood disorder to secondary recipients. This indicates that the disorder is cell autonomous and that hematopoietic progenitor cells with reduced Mybl2 expression can self-renew after transplantation and undergo sustained expansion leading to clonal dominance.
Discussion
Recent molecular studies of MDS have uncovered a number of previously unrecognized genes whose aberrant inactivation through mutation or other means can disrupt hematopoietic cell development, leading to ineffective hematopoiesis and a myelodysplastic phenotype (Shih et al., 2012). The picture beginning to emerge from these investigations is that a broad array of molecular changes have the potential to drive the biology and clinical phenotypes of MDS in unique ways. Yet, the disease remains very heterogeneous and none of the molecular lesions that have been convincingly linked to MDS pathogenesis affect more than a fourth of cases overall, with a prevalence of 1–15% being much more common (Bejar et al., 2011; Shih et al., 2012).
In this study, we identified MYBL2 as a dosage-dependent tumor suppressor gene that was reduced in expression to sub-haploinsufficient levels in 17 (65%) of 26 human MDS cases. This result is somewhat surprising given that MYBL2 has been implicated as a gene that is typically overexpressed (rather than downregulated) in several types of cancer and whose elevated expression correlates with a poor prognosis in human breast cancer and other malignancies (Amatschek et al., 2004; Paik et al., 2004). However, no direct evidence demonstrating oncogenic activity by overexpressed MYBL2 has been reported to date. High levels of expression of this transcription factor in other tumor types may reflect the greater proliferative rate of the tumor cell population compared to resting cells, so that the apparently levels of high MYBL2 activity may be a consequence rather than a cause of malignant transformation. By contrast, the proliferative fraction of cells in myeloid neoplasia is generally quite low.
Our findings demonstrate the surprising finding that abnormally low levels of MYBL2 expression have the unique property of imparting a clonal advantage to multipotent hematopoietic progenitors, leading rapidly to the clonal dominance of such cells within the myeloid, and B- and T-lymphocyte cell lineages. The persistence of clonal dominance in recipient mice for more than 6 months after bone marrow cell transplantation, and in recipients of secondary transplants, indicates that the affected multipotent hematopoietic cells have both self-renewal and long-term repopulating ability (Morrison and Weissman, 1994). Hence, our finding establishes MYBL2 as a key tumor suppressor gene of the 20q CDR affected in human MDS and MPD. However, we cannot exclude the possibility that haploinsufficiency for other genes within the 20q CDR cooperates with aberrantly low expression of MYBL2 to move the cells toward malignant transformation, as has been shown for the CDR associated with the 5q-syndrome, where haploinsufficiency of the gene RPS14 as well as loss of expression of an miRNA (Ebert et al., 2008; Barlow et al., 2010; Starczynowski et al., 2010) are critical steps in MDS pathogenesis.
Most intriguing is our finding of decreased MYBL2 expression in MDS cases with a normal karyotype, which suggests that interstitial deletion of chromosome 20q is not the only mechanism by which MYBL2 activity can be reduced in MDS. Indeed, low levels of MYBL2 were apparent in 10 (56%) of 18 of our normal-karyotype MDS cases, and 38% of those reported by Pellagatti et al. (Pellagatti et al., 2010). We were unable to implicate aberrant methylation of the MYBL2 genomic locus as an additional mechanism of gene-dosage reduction; instead, we found elevated expression levels of mir-29a and mir-30e, which are known to target MYBL2 (Martinez et al., 2011), in CD34+ cells from patients with MDS and low levels of MYBL2 expression. In addition, others have shown that overexpression of mir-29a in murine bone marrow cells leads to acute myeloid leukemia (Han et al., 2010). Combined with our results, this observation suggests that mir-29a may be oncogenic, at least in part, because it targets Mybl2 transcripts. Finally, we identified a case with heterozygous missense mutation in MYBL2 that led to the loss of gene function, analogous to the effect of monoallelic gene deletion. Thus, our results indicate that the MYBL2 transcription factor gene is targeted by diverse molecular events in MDS, including not only deletion within the 20q chromosomal region, but also heterozygous loss-of-function mutations and the repression of gene expression by dysregulated miRNAs.
Clarke et al. (Clarke et al., 2012) have also described an association between MYBL2 expression levels and the occurrence of hematologic disorders, including MDS, in aged Mybl2 haploinsufficient mice. These authors were able to show that haploinsufficiency for the Mybl2 gene increases susceptibility to age-related myeloid neoplasia. Our data indicate that more pronounced gene dosage reduction of Mybl2 might be even more advantageous for clonal dominance. Although our findings do not rule out a causal role for straight-forward haploinsufficiency in myeloid malignancies, they strongly suggest that the oncogenic effects of reduced MYBL2 dosage are more pronounced as the dosage level falls to near 30% of normal, in keeping with the continuum model of tumor suppression (Berger et al., 2011). We would also stress that in the range of MYBL2 dosage reductions in this study were modeled based on those found in CD34+ cells from MDS clinical samples, in which the reductions of MYBL2 gene dosage were predominately in the range of 20–30% of normal levels, and thus were considerably lower than the 50% predicted by classical haploinsufficiency. Importantly, expression levels lower than 20% appear incompatible with cell growth (Figure 4B), consistent with the known function of MYBL2 as a key regulator of G2-to-M transition and the fact that biallelic inactivation of this gene causes early embryonic lethality in the mouse due to a block in inner cell mass formation during embryonic development (Tanaka et al., 1999).
Our results implicating a transforming role for sub-haploinsufficient levels of MYBL2 build upon earlier studies implicating progressive inactivation of CTNNA1 and the graded reduction of PU.1 expression in the molecular pathogenesis of myeloid malignancies (Rosenbauer et al., 2004; Liu et al., 2007). Our approach, which tested the effects of graded levels of Mybl2 knockdown of hematopoietic progenitors in vivo, provides compelling evidence for the importance of reductions of tumor suppressor gene levels below the 50% level associated with classical haploinsufficiency in the molecular pathogenesis of myeloid malignancies.
Materials and methods
Clinical samples
Request a detailed protocolBone marrow samples represented 28 MDS patients (Supplementary file 1A) seen at the MD Anderson Cancer Center (MDACC). The diagnosis and classification of MDS followed criteria of the World Health Organization (WHO), the French-American–British Cooperative Group (FAB) and the International Prognostic Scoring System (IPSS). Mononuclear cells were isolated from bone marrow aspirates by Ficoll centrifugation, and further purified by magnetic beads (Miltenyi Biotec, Auburn, CA) to obtain the CD34+ fraction. Normal CD34+ cells were processed in exactly the same manner.
Competitive reconstitution assays
Request a detailed protocolMononuclear cells were isolated from the femurs and tibias of syngeneic mice (C57BL/6), and were depleted of lineage-marked cells using sheep anti-rat IgG-coupled Dynabeads (Life Technologies, Carlsbad, CA) together with affinity-purified rat anti-mouse antibodies (eBioscience, San Diego, CA) against CD3, CD5, B220, Gr1, Mac1, Ter119. Cells were cultured for 24 hr in StemSpan SFEM (Stem Cell Technologies, Vancouver, BC, Canada) supplemented with 100 U/ml penicillin/streptomycin, 2 mM glutamine, 80 ng/ml Scf, 40 ng/ml TPO and 40 ng/ml Flt3l. Cells were aliquoted and transduced in separate wells with single shRNA- or control shRNA-expressing vectors, extensively washed and cultured separately for an additional 24 hr before transplantation. Single vector transduced cells were pooled according to the scheme in Figure 5 just before transplantation. Recipient C57BL/6 mice were lethally irradiated (9.5 Gy) at a dose rate of approximately 1 Gy/min, delivered as a single dose. The next day, mice were reconstituted by retro-orbital venous sinus injection of 2 × 105 lentivirus-transduced cells mixed with a 1 × 105 freshly isolated whole bone marrow cells for radioprotection.
RNA preparation and qRT-PCR
Request a detailed protocolRNA was extracted from Trizol (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions, and 500 ng were used for reverse transcription (Quantitect kit, Qiagen, Hilden, Germany). Real-time PCRs were performed on an ABI PRISM 7700 system using SYBR green-based assays with AmpliTaq Gold (Life Technologies). All reactions were performed in triplicate. Quantitative data were calculated from the Ct-values for each reaction using the mean reaction efficiency for each primer pair. Data were normalized to expression levels of PAPOLA, UBQLN1 and VPS39 (housekeeping genes) and scaled to the mean of the controls. Sequences of primer sets are available in Supplementary file 1C.
Microarray analysis
Request a detailed protocolTotal RNA was isolated from cells lysed in Trizol (Life Technologies). 100–500 ng of each sample was converted into fragmented, biotinylated cDNA hybridized to a microarray chip (Gene 1.0 ST) and fluorescently labeled according to the standard protocol (Affymetrix, Santa Clara, CA) at the Dana-Farber microarray core facility. Raw data were processed in Expression Console (Affymetrix) using RMA normalization. Expression values for each gene were annotated by mapping all probe sets to the human genome version hg19. Data complexity was reduced to one canonical transcript per gene, resulting in a single identifier per gene (17,982 total). The expression data were processed in Gene Pattern (Reich et al., 2006) (Broad Institute, Cambridge, MA). Non-expressed genes were filtered out, and the resulting expression matrix was analyzed with the comparative marker module (two class comparison with 10,000 permutations) or the gene neighbor module in Gene Pattern. GSEA (Broad Institute) was performed as described previously (Subramanian et al., 2005). Data are available at Gene Expression Omnibus, accession number GSE43401.
A k-nearest neighbor (KNN) algorithm was used to classify normal-karyotype MDS cases by using Euclidean distance and k = 3 to predict the class label by a majority vote. To generate a training set, the 89 genes most correlated with MYBL2 (Pearson correlation coefficient >0.80; 81 positively correlated and 8 negatively correlated) based on the short hairpin knock-down experimental data were reduced to a 59 gene signature positively correlated with MYBL2 by removing 14 genes which hierarchically clustered with the 8 negatively correlated genes and 9 genes having low expression (≥3 out of 4 controls) in the CD34+ control samples in the 30 patient samples arrays. This signature was also examined in an independent cohort of 94 normal karyotype patients using KNN.
Western blotting
Request a detailed protocolProtein extracts were prepared by SDS lysis and normalized by total protein content (BCA assay). Total protein (5–25 µg) was separated by SDS polyacrylamide gel electrophoresis and blotted on PVDF membranes. Specific proteins were visualized by chemiluminescence using antibodies for MYBL2 (ab12296, Abcam, Cambridge, United Kingdom) or Tubulin (B-5-1-2, Sigma, St. Louis, MO).
Cell culture
Request a detailed protocolCryopreserved human bone marrow CD34+ cells were obtained from Lonza and cultured in StemSpan SFEM (Stem Cell Technologies) supplemented with 100 U/ml penicillin/streptomycin, 2 mM glutamine, 50 µg/ml lipoproteins (EMD Millipore, Billerica, MA), 80 ng/ml SCF, 40 ng/ml TPO, 40 ng/ml FLT3L, 10 ng/ml IL3 and 10 ng/ml IL6. SKM1 cells were obtained from the DSMZ (Braunschweig, Germany) and maintained in RPMI medium supplemented with 10% fetal bovine serum.
Lentiviral vectors and transduction
Request a detailed protocolOligonucleotides encoding shRNAs were cloned into pLKO1. pLKO1 derivatives were obtained by replacing the puromycin resistance marker with EGFP or DsRedExpress2 (Strack et al., 2008). Lentiviral RNAi vectors are available on request. The MYBL2 cDNA including a 3xFLAG tag was cloned into derivate of the pLenti6.3 (Life Technologies) vector under control of the SFFV promoter. Lentiviral particles were produced in 293T cells by cotransfection of pMD2G, pCMV-dR8.9 and the lentiviral vector. Cells were spin-transduced in the presence of polybrene (4 µg/ml) and selected at 24 hr post-transduction with puromycin (1.5 µg/ml) or blasticidin (3 µg/ml). Growth curves were obtained with the CellTiter-Glo luminescent assay (Promega, Madison, WI).
Flow cytometry
Request a detailed protocolA FACSCanto II machine (BD Biosciences, San Jose, CA) equipped with a 407 nm, 488 nm and 633 nm laser was used. Staining was performed with the following antibodies (clone id in parentheses), all purchased from eBioscience: Mac1 (M1/70), Gr1 (RB6-8C5), B220 (RA3-6B2), CD3 (145-2C11), CD71 (R17217) and Ter119 (Ter119).
Competitive reconstitution assays
Request a detailed protocolC57BL/6 mice, purchased from Jackson Laboratories, were housed in individually ventilated cages in the Dana-Farber Cancer Institute animal facility in accord with guidelines of the Institutional Animal Care Use Committee. Transplanted mice received Baytril-containing water (Bayer, Robinson Township, PA) to reduce the probability of infection from opportunistic pathogens in first 30 days after transplantation. For peripheral blood analysis, tailvein blood was collected and RBCs were lysed with ammonium chloride buffer prior to staining for flow cytometry. For bone marrow analysis, cells were isolated from femurs and tibias with RBCs lysis buffer used for all subsequent procedures except analysis of erythropoiesis by Ter119/CD71 flow cytometry.
Cytomorphologic and histologic evaluation
Request a detailed protocolCytospin slides were prepared from isolated mononuclear bone marrow cells and stained with Wright-Giemsa stain. Bone and spleen samples were fixed in 10% formalin, washed with PBS and stored to 80% ethanol. Sections were stained with hematoxylin and eosin.
Statistical analysis
Request a detailed protocolAll data were analyzed with GraphPad Prism, version 5.04 for Windows (http://www.graphpad.com). p-values (two-tailed) for competitive reconstitution were calculated by a paired t-test.
Data availability
-
MYBL2 Is a Sub-haploinsufficient Tumor Suppressor Gene in Myeloid MalignancyID GSE43401. Publicly available at GEO (http://www.ncbi.nlm.nih.gov/geo/).
-
Expression data from bone marrow CD34+ cells of MDS patients and healthy controlsID GSE19429. Publicly available at GEO (http://www.ncbi.nlm.nih.gov/geo/).
References
-
Mutation and cancer: statistical study of retinoblastomaProc Natl Acad Sci USA 68:820–823.https://doi.org/10.1073/pnas.68.4.820
-
miR-29 and miR-30 regulate B-Myb expression during cellular senescenceProc Natl Acad Sci USA 108:522–527.https://doi.org/10.1073/pnas.1017346108
-
A zebrafish bmyb mutation causes genome instability and increased cancer susceptibilityProc Natl Acad Sci USA 102:13194–13199.https://doi.org/10.1073/pnas.0506583102
-
The role of mutations in epigenetic regulators in myeloid malignanciesNat Rev Cancer 12:599–612.https://doi.org/10.1038/nrc3343
-
A noncytotoxic DsRed variant for whole-cell labelingNat Methods 5:955–957.https://doi.org/10.1038/nmeth.1264
-
Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profilesProc Natl Acad Sci USA 102:15545–15550.https://doi.org/10.1073/pnas.0506580102
Article and author information
Author details
Funding
National Institutes of Health (NIH P01 CA-108631)
- Stefan Heinrichs
- Lillian F Conover
- Carlos E Bueso-Ramos
- Outi Kilpivaara
- Kristen Stevenson
- Donna Neuberg
- Mignon L Loh
- Guillermo Garcia-Manero
- Hagop M Kantarjian
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank John R Gilbert for editorial review.
Ethics
Human subjects: The specific ethical approval was obtained for work with human samples from the institutional ethics committee at the M.D. Anderson Cancer Center in accordance with the Declaration of Helsinki by the WMA. Guidelines were strictly followed. Informed consent was obtained for all human samples.
Animal experimentation: Animal care was in strict compliance with institutional guidelines established by the Dana-Farber Cancer Institute, the National Academy of Sciences Guide for the Care and Use of laboratory Animals, and the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Dana-Farber Cancer Institute (protocol #05-048)
Copyright
© 2013, Heinrichs et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,506
- views
-
- 171
- downloads
-
- 34
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 34
- citations for umbrella DOI https://doi.org/10.7554/eLife.00825
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
MYBL2 is a sub-haploinsufficient tumor suppressor gene in myeloid malignancy
eLife 2:e00825.
https://doi.org/10.7554/eLife.00825




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}