DNA Damage: Proteins pinpoint double strand breaks
- University of Wisconsin-Madison, United States
Every genome—whether it is bacterial, archaean or eukaryotic—is subjected to DNA damage on a regular basis. Thousands of DNA lesions can appear per cell per generation in an aerobic bacterial culture, and hundreds of thousands can appear in a single mammalian cell in a day (Hoeijmakers, 2009). Most cells have DNA repair systems to enforce genome stability and, in higher eukaryotes, to prevent cancer. However, these systems can break down (Putnam et al., 2012), and when they do, tumours form. The widespread mutations and rearrangements of chromosomes that are found in tumour cells prevail in what can only be described as genomic chaos (Carter et al., 2012).
Documenting these various genomic insults and their consequences represents a major challenge for medicine, and also for disciplines such as evolutionary biology and cell biology. Now, in eLife, Susan Rosenberg of Baylor College of Medicine and colleagues—including Chandan Shee as first author—have provided a promising new tool for the study of one such insult: the double strand break (Shee et al., 2013).
Double strand breaks are considered the most dangerous of all the DNA lesions. If left unrepaired, the resulting chromosome discontinuity often results in death. There are two main ways to repair a double strand break. Recombinational DNA repair is accurate but it relies on the presence of an unbroken homologous chromosome. Non-homologous DNA end-joining, on the other hand, repairs the break, but usually at the expense of adding or deleting genetic information (Chapman et al., 2012; Symington and Gautier, 2011).
Dangerous as they are, double strand breaks are sometimes deliberately introduced into a chromosome. During meiosis, for example, these ‘directed’ double strand breaks are introduced to initiate genetic crossovers between homologous chromosomes (Malkova and Haber, 2012). In yeast, directed double strand breaks are a prelude to an intrachromosomal exchange of genetic information that produces a mating type switch (Haber, 2012). Trypanosomes and other microbial pathogens can evade the immune system by periodically moving new genetic information from a “silent” gene to a highly transcribed locus that encodes a major cellular coat protein; this process is often initiated by a directed double strand break (Deitsch et al., 2009; Vink et al., 2012). Double strand breaks are also central to genetic elements called transposons, and in genomic rearrangements that are integral to the immune system.
Accurate real-time detection of double strand breaks in a cellular genome is thus of great interest in the continuing effort to understand genome maintenance and function. A variety of techniques have been developed to detect and quantify double strand breaks, but they all have one or more deficits in terms of utility, efficiency, sensitivity or specificity. Shee, Rosenberg and colleagues–including co-workers from the University of Texas, the MD Anderson Cancer Center and the University of Minnesota–now report a new approach, based on a protein called Gam, that offers some substantial advantages over existing approaches (Shee et al., 2013).
Gam is encoded by the bacteriophage Mu: this is basically a hybrid of a bacterial virus and a transposon, and it makes a living by moving efficiently within and between bacterial genomes (Baker, 1995; di Fagagna et al., 2003). When an integrated genomic copy of Mu replicates and transposes, the Gam protein protects the free ends of the Mu chromosome as they are transiently exposed. Gam is related to two eukaryotic proteins, Ku70 and Ku80, that are involved in non-homologous DNA end-joining. Whereas the Ku proteins bind to double strand ends, they also interact with an array of other eukaryotic proteins and DNA structures, rendering them less useful for development of a general reagent that binds to double strand breaks. Gam is a simpler system, a single protein with a high affinity for double stranded ends. Rosenberg and colleagues have fused Gam with green fluorescent protein (GFP) to generate GamGFP. When this protein is expressed in a cell, the double strand breaks light up when the cell is illuminated, and this allows the number of breaks to be counted.
In bacteria, GamGFP can detect double strand breaks arising from a variety of sources (Shee et al., 2013). For example, the double strand breaks that occur during DNA replication can be pinpointed (Figure 1), as can the sites where the restriction enzyme Scel cleaves a particular chromosome. Shee et al. also provide useful new estimates of the rate of spontaneous break generation. The overall detection efficiency (70–80% in the current study) bodes well for the application of this approach to the detection and quantification of breaks in research into the mechanisms responsible for genome maintenance in bacteria.
Figure 1
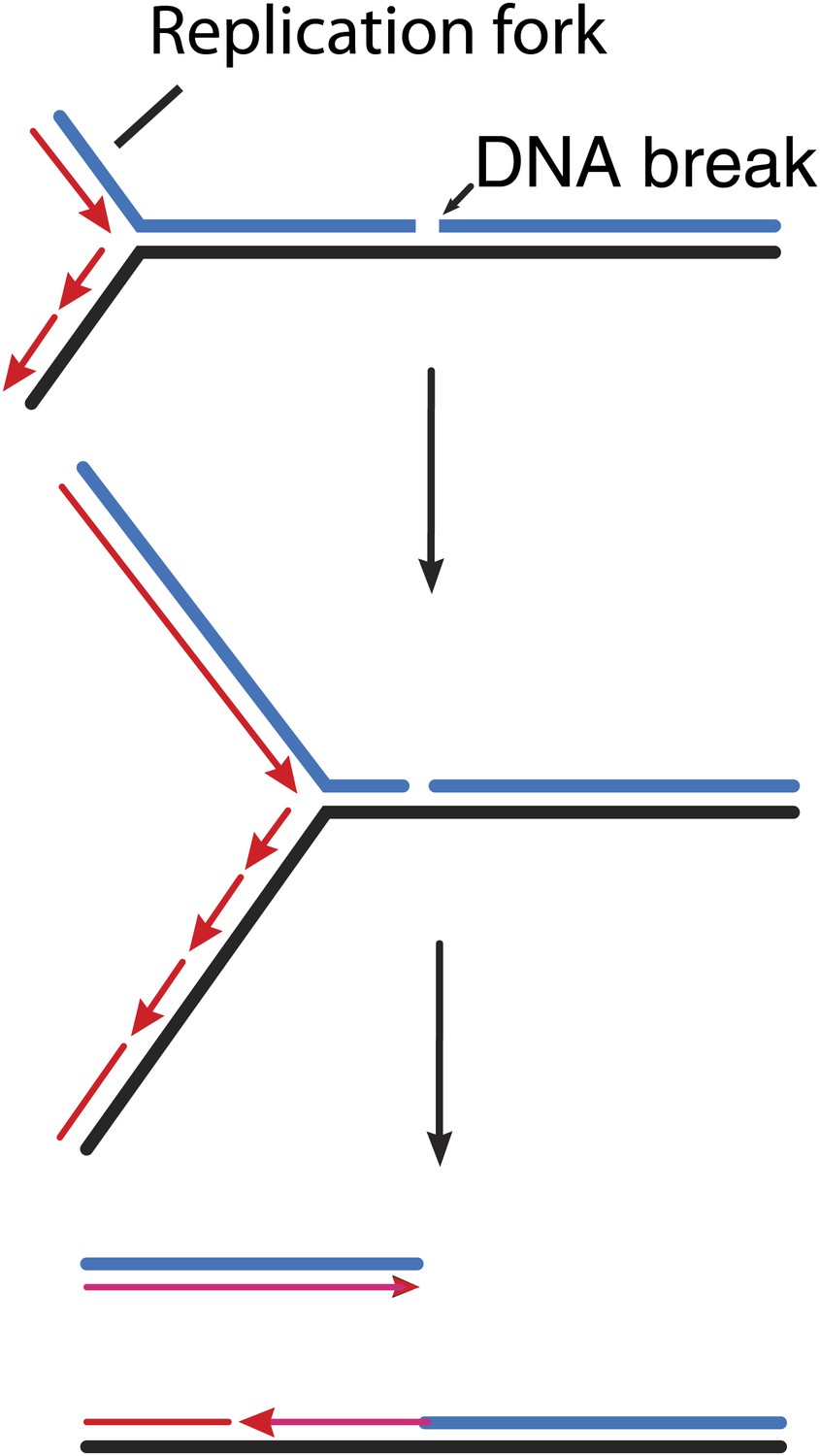
How double strand breaks occur during replication.
Many DNA repair processes generate transient single strand breaks in chromosomes. If a replication fork encounters such a break before it is repaired (top and middle), one arm of the replication fork separates to create a double strand break (bottom).
The detection of double strand breaks in the milieu of a eukaryotic chromosome is a much bigger task. Breaks may be buried in chromatin and/or blocked by proteins such as Ku binding to them. In the first round of meiotic cell division, for example, directed double strand breaks are introduced by the protein Spo11, which remains covalently linked to the DNA at the breakage site. Will GamGFP recognize such sites? And in yeast, will GamGFP be able to detect the breaks that are induced to initiate a mating type switch?
Shee et al. demonstrate that GamGFP binds to laser-generated double strand breaks in human HeLa cells, but they find that the recruitment of GamGFP to these sites is inhibited by competition with Ku. Overall, the efficiency of double strand break detection by GamGFP in eukaryotic cells is difficult to assess, and may depend on the source of the double strand breaks that one may want to detect. Nevertheless, this new technology is destined for creative application to important problems in eukaryotic cell biology. The list of potential experiments seems endless.
References
-
Absolute quantification of somatic DNA alterations in human cancerNat Biotechnol 30:413–421.https://doi.org/10.1038/nbt.2203
-
Molecular origins of cancer: DNA damage, aging, and cancerN Engl J Med 361:1475–1485.https://doi.org/10.1056/NEJMra0804615
-
Bioinformatic identification of genes suppressing genome instabilityProc Natl Acad Sci USA 109:E3251–E3219.https://doi.org/10.1073/pnas.1216733109
-
Microbial antigenic variation mediated by homologous DNA recombinationFEMS Microbiol Rev 36:917–948.https://doi.org/10.1111/j.1574-6976.2011.00321.x
Article and author information
Author details
Publication history
- Version of Record published: October 29, 2013 (version 1)
Copyright
© 2013, Cox
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,482
- views
-
- 119
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
DNA Damage: Proteins pinpoint double strand breaks
eLife 2:e01561.
https://doi.org/10.7554/eLife.01561
Further reading
-
- Cancer Biology
- Chromosomes and Gene Expression
The chromatin-associated protein WD Repeat Domain 5 (WDR5) is a promising target for cancer drug discovery, with most efforts blocking an arginine-binding cavity on the protein called the ‘WIN’ site that tethers WDR5 to chromatin. WIN site inhibitors (WINi) are active against multiple cancer cell types in vitro, the most notable of which are those derived from MLL-rearranged (MLLr) leukemias. Peptidomimetic WINi were originally proposed to inhibit MLLr cells via dysregulation of genes connected to hematopoietic stem cell expansion. Our discovery and interrogation of small-molecule WINi, however, revealed that they act in MLLr cell lines to suppress ribosome protein gene (RPG) transcription, induce nucleolar stress, and activate p53. Because there is no precedent for an anticancer strategy that specifically targets RPG expression, we took an integrated multi-omics approach to further interrogate the mechanism of action of WINi in human MLLr cancer cells. We show that WINi induce depletion of the stock of ribosomes, accompanied by a broad yet modest translational choke and changes in alternative mRNA splicing that inactivate the p53 antagonist MDM4. We also show that WINi are synergistic with agents including venetoclax and BET-bromodomain inhibitors. Together, these studies reinforce the concept that WINi are a novel type of ribosome-directed anticancer therapy and provide a resource to support their clinical implementation in MLLr leukemias and other malignancies.
-
- Chromosomes and Gene Expression
- Immunology and Inflammation
Ikaros is a transcriptional factor required for conventional T cell development, differentiation, and anergy. While the related factors Helios and Eos have defined roles in regulatory T cells (Treg), a role for Ikaros has not been established. To determine the function of Ikaros in the Treg lineage, we generated mice with Treg-specific deletion of the Ikaros gene (Ikzf1). We find that Ikaros cooperates with Foxp3 to establish a major portion of the Treg epigenome and transcriptome. Ikaros-deficient Treg exhibit Th1-like gene expression with abnormal production of IL-2, IFNg, TNFa, and factors involved in Wnt and Notch signaling. While Ikzf1-Treg-cko mice do not develop spontaneous autoimmunity, Ikaros-deficient Treg are unable to control conventional T cell-mediated immune pathology in response to TCR and inflammatory stimuli in models of IBD and organ transplantation. These studies establish Ikaros as a core factor required in Treg for tolerance and the control of inflammatory immune responses.




{kind=link}