G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases
- University of Cambridge, United Kingdom
Decision letter
-
W James NelsonReviewing Editor; Stanford University, United States
eLife posts the editorial decision letter and author response on a selection of the published articles (subject to the approval of the authors). An edited version of the letter sent to the authors after peer review is shown, indicating the substantive concerns or comments; minor concerns are not usually shown. Reviewers have the opportunity to discuss the decision before the letter is sent (see review process). Similarly, the author response typically shows only responses to the major concerns raised by the reviewers.
Editorial note dated 06 November 2014
Thank you for sending your work entitled “G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases” for consideration at eLife. Your article has been evaluated by Vivek Malhotra (Senior editor), a Reviewing editor, and three reviewers, one of whom, Richard Treisman, has agreed to share his identity.
The Reviewing Editor and the reviewers discussed their comments before we reached this decision, and the Reviewing editor has assembled the following comments to help you prepare a revised submission.
All three reviewers, with different degrees of enthusiasm, thought that the work in this manuscript was interesting and potentially important. One of the reviewers, Richard Treisman, had some minor comments, but stated: “This is a super paper. It is of interest as it presents a detailed molecular rationale for the biochemical function of actin in a non-cytoskeletal regulatory complex, adding to a number of recent structures in which G-actin plays a role as a regulator rather than regulate. Publish it!” The other two reviewers were less positive, and they have made a number of substantive criticisms that will need to be addressed directly before we can consider a revised manuscript. We have summarized their criticisms below:
Substantive comments:
Despite the extensive dataset and provocative finding that G-actin might regulate PP1/R15 substrate specificity, the potential impact of the work is undermined (1) by conceptual issues related to basic conclusion that PP1/R15 lacks substrate specificity, and (2) by technical limitations related to the phosphatase assays.
Building on a large body of biochemical studies, and substantiated by X-ray crystal structures of a growing number of PP1 complexes, the substrate specificity of PP1 is thought to occur through its interactions with its many 100s of PP1 binding proteins through both negative and positive selection. Like PP1-MYPT1, PP1-spinophilin and others, the interaction of the PP1 binding protein blocks dephosphorylation of substrates (typically phosphorylase is used), but allows either unaltered dephosphorylation of other substrates (e.g. PP1-spinophilin towards GluR1) or enhanced dephosphorylation of a specific target (e.g. PP1-MYPT toward myosin light chain). In the case of R15 early studies (e.g. Connor et al., 2001) showed that the activity of PP1 towards phosphorylase was blocked by GADD34, but activity towards eIF2α was maintained. Therefore the basic experimental set-up used does not address the selective blockade of non-preferred substrates; this is an important omission (our emphasis). The assessment of PP1 activity is also made more complicated by the known limitation that bacterial PP1 (generated in a Mn2+ form) possesses promiscuous activity that is not found for native enzyme. Thus the assay used here measures an activity which is not designed to address the question of what is driving preference for eIF2α (our emphasis). Indeed, an interpretation of the key data in Figure 4 is that addition of the cell lysate blocks the non-specific activity towards phospho-GST, not that it confers any change in eIF2α activity, though much more quantitative kinetic analysis is required to know exactly what is going on. On this point, the other reviewer also wondered why addition of lysate alone did not dephosphorylate the non-specific substrate, when there are many different phosphatases in cell lysates.
Both of the less positive reviewers agreed that there is a general lack of quantification (Vmax and Km data are needed to assess activities) and lack of analysis of replicated data for many of the figures. In short, all the phosphatase assays will need repeating to produce at least 3 replicates. The reviewers recognized that this is not trivial and will take time to set up and complete. Depending on the outcome, the conclusions of the paper and the significance of the structural interactions could change.
1) The phosphatase assays rely exclusively on a gel-shift assay that does not allow for assessment of linear rate kinetics, a requirement for a study of this type.
2) Figure 3B is supposed to show that the bacterial complex of PPP1R15A/PPPIc lacks specificity. But, the last two reactions are not relevant. There is apparently a huge amount of enzyme relative to substrate, and both substrates are completely dephosphorylated. What are the enzyme concentrations used for each reaction? More intermediate concentrations should be shown. Also, 3 time points are shown (5, 15 and 30 minutes) but there seems to be little difference in dephosphorylation between 15 and 30 minutes for either substrate suggesting the enzyme may be dying; the substrate seems to be at a high concentration relative to enzyme (2 micro molar substrate). Time points earlier than 5 minutes should be shown.
3) A high concentration of actin is added to dephoshorylation assays (1 micro molar) and compared to assays with no actin (for example Figure 8). Is this really a specific effect? What happens if 1 micro molar concentration of a different purified protein is added?
4) It is unclear what the 'specificity factor“ calculated in Figure 8B really represents. The concentration of enzyme is different, the substrates are different and it is not clear how a velocity was calculated when some of the reactions have gone to completion at the first time point. For R66E the two reactions look identical on the gels shown. Only the R74E mutation provides useful information regarding the potential role of the interaction of R15 with G-actin. In contrast to the conclusions made by the authors (see also the Discussion), the other mutations do not seem to have much effect. Indeed there is not much difference between the minus and plus G-actin data in panels B, C and D, which is in contrast to data in earlier figures.
5) Information about the relative amounts of dimeric complexes that were used for the various mutants (in the subsection headed “G-actin, PPP1R15 and PP1 form a ternary holophosphatase that selectively dephosphorylates eIF2α” and Figure 6) should be given.
[Editors' note: further revisions were requested prior to acceptance, as described below.]
Author response
Authors response dated 19 December 2014 to editorial note dated 06 November 2014
Substantive comments:
[…] Both of the less positive reviewers agreed that there is a general lack of quantification (Vmax and Km data are needed to assess activities) and lack of analysis of replicated data for many of the figures. In short, all the phosphatase assays will need repeating to produce at least 3 replicates. The reviewers recognized that this is not trivial and will take time to set up and complete. Depending on the outcome, the conclusions of the paper and the significance of the structural interactions could change.
The key issues raised in review are related to the biochemical evidence for positive and negative selection of substrates by the binary complex of PP1-PPP1R15 versus the ternary complex of PP1-PPP1R15-Actin. A more formal quantitative analysis of enzyme kinetics of the different enzyme complexes with regard to eIF2α and non-specific substrates was also deemed important.
The revised version addresses these concerns with new experiments and a detailed quantitative analysis as noted below. The robustness of the measurements has been established through multiple replicates and is further attested to by the smooth fitting of multipoint data in the individual experiments. We have also showcased the reproducibility of our experimental setup by providing the reviewers several examples of duplicates of the experimental data submitted for publication (3Bdup, 3Cdup, 3Ddup, 4S1dup and 8Bdup). These are detailed below.
Revised Figure 3B presents a detailed comparison of the dephosphorylation reactions containing escalating concentrations of PP1-PPP1R15 binary complex and either the specific (eIF2αP) or the non-specific (GSTP) substrate. It reveals that over a range of enzyme concentrations that are limiting to the velocity of substrate dephosphorylation, the binary complex more rapidly dephosphorylates the non-specific substrate (GSTP) compared to the specific one (eIF2αP).
Revised Figure 3C presents a detailed comparison of the dephosphorylation reactions containing escalating concentrations of the specific (eIF2αP) or the non-specific (GSTP) substrate and fixed concentration of PP1-PPP1R15 binary complex. It reveals that over a range of substrate concentrations that are limiting to the velocity of dephosphorylation, the binary complex more rapidly dephosphorylates the non-specific substrate (GSTP) compared to the specific one (eIF2αP).
Together, revised Figures 3B and 3C prove that that the PP1-PPP1R15 binary complex has no selectivity towards eIF2αP compared to GSTP.
New Figure 4–figure supplement 1 A and B measures the rate of dephosphorylation of eIF2αP by the ternary complex of PP1 -PPP1R15-Actin across a rangeµM. of substrate concentrations from 0.5 to 15. The velocity of the dephosphorylation reaction is seen to increase linearly with substrate concentration. Thus, over the substrate concentration range accessible to testing, the PP1-PPP1R15-Actin enzyme cannot be saturated. This is not unusual in measurements of enzyme kinetics and whilst it precludes extraction of Km (for substrate) or a Vmax, it does enable us to compare the turnover number of the binary complex of PP1-PPP1R15 and the ternary complex of PP1- PPP1R15-Actin over a range of eIF2αP concentration, proving that the inclusion of G-actin in the enzyme complex selectively accelerates the de-phosphorylation of the specific substrate by a factor of at least 15X.
The reviewers also emphasized the importance of measurements aimed at detecting the reciprocal phenomenon, namely the ability of G-actin to selectively enfeeble the dephosphorylation of non-specific substrates. We have addressed this issue in Author response image 1, in which we measured the effect of addition of G-actin on the dephosphorylation of a structured substrate, phosphorylase A (PYGMP; phosphorylated on serine 15). In contrast to the marked acceleration of eIF2αP dephosphorylation by the addition of actin, the dephosphorylation of PYGMP was inhibited by G-actin. The extent of inhibition may have been limited by the fact that bacterially-derived PP1 may have intrinsically relaxed substrate specificity, a point emphasized by the knowledgeable reviewers. This has now been incorporated into the Discussion.
Author response image 1
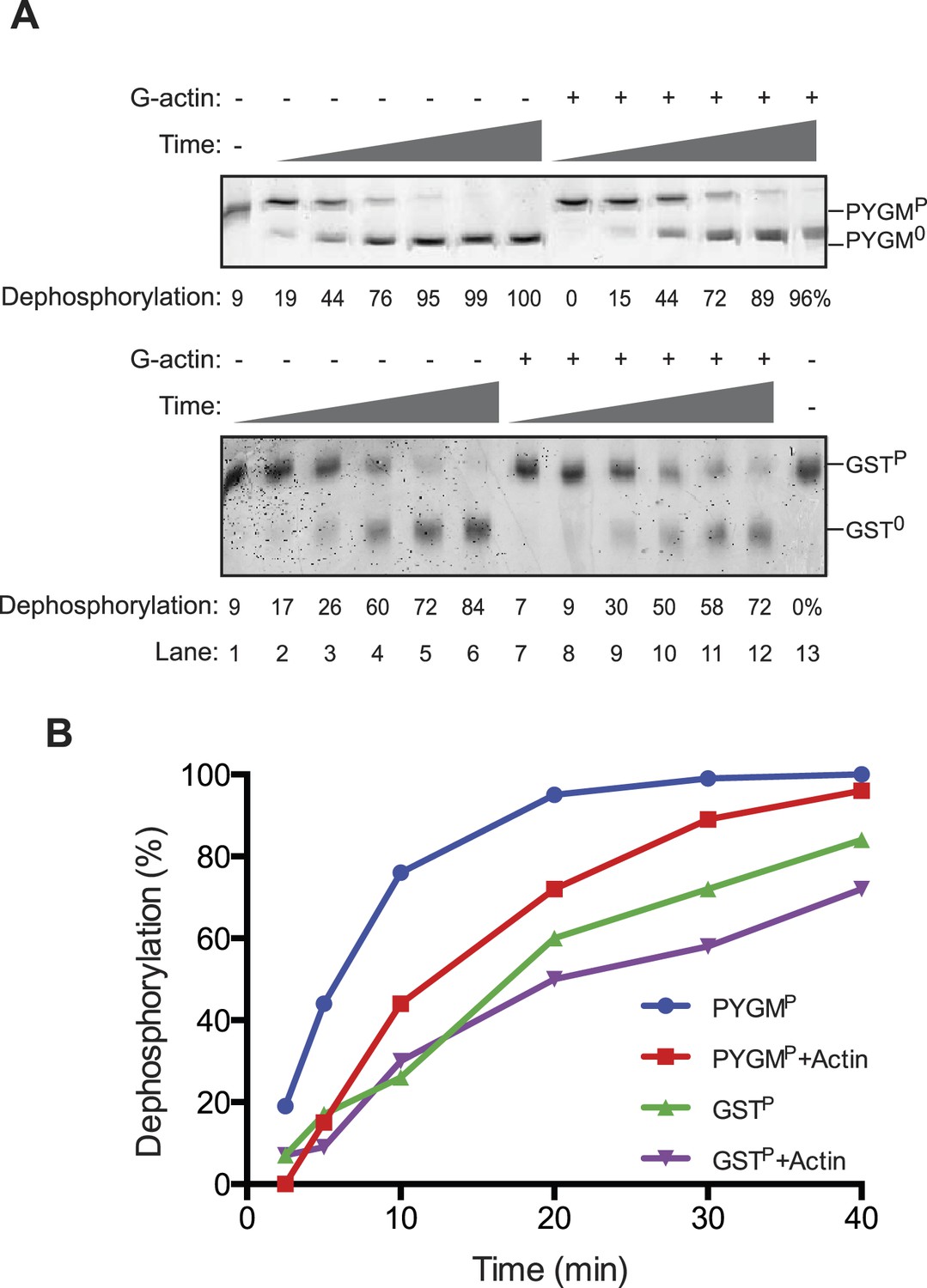
1) The phosphatase assays rely exclusively on a gel-shift assay that does not allow for assessment of linear rate kinetics, a requirement for a study of this type.
The Phos-tag gel-based method we use to distinguish the substrates and products of the dephosphorylation reaction is successfully applied as an endpoint assay and affords an accurate measurement of enzyme kinetics over the linear range. This is revealed by the linear regression coefficients of the correlation between rate of substrate to product conversion (reaction velocity) and substrate concentration (see Figure 3C and Figure 4–figure supplement 1, as examples).
2) Figure 3B is supposed to show that the bacterial complex of PPP1R15A/PPPIc lacks specificity. But, the last two reactions are not relevant. There is apparently a huge amount of enzyme relative to substrate, and both substrates are completely dephosphorylated. What are the enzyme concentrations used for each reaction? More intermediate concentrations should be shown. Also, 3 time points are shown (5, 15 and 30 minutes) but there seems to be little difference in dephosphorylation between 15 and 30 minutes for either substrate suggesting the enzyme may be dying; the substrate seems to be at a high concentration relative to enzyme (2 micro molar substrate). Time points earlier than 5 minutes should be shown.
The experiment has been redone in accordance with the reviewers’ suggestions. Lower concentrations of enzyme and earlier time points were included. The observations, presented in new Figure 3B reveal that over a range of enzyme concentrations that are limiting to the velocity of substrate dephosphorylation, the binary complex more rapidly dephosphorylates the non-specific substrate (GSTP) compared to the specific one (eIF2αP).
3) A high concentration of actin is added to dephoshorylation assays (1 micro molar) and compared to assays with no actin (for example Figure 8). Is this really a specific effect? What happens if 1 micro molar concentration of a different purified protein is added?
Figure 5 and Figure 6 present a detailed analysis of the effect of actin at concentrations ranging from low nanomolar to micromolar. As the figures and text make clear G-actin activates the phosphatase complex with an EC50 in the 10-7 M range. This does not strike us an un-physiologically high effective concentration for an abundant cellular constituent like actin.
New Figure 5–figure supplement 1 shows that bovine serum albumin in similar concentrations has no effect on eIf2αP dephosphorylation.
4) It is unclear what the 'specificity factor” calculated in Figure 8B really represents. The concentration of enzyme is different, the substrates are different and it is not clear how a velocity was calculated when some of the reactions have gone to completion at the first time point. For R66E the two reactions look identical on the gels shown. Only the R74E mutation provides useful information regarding the potential role of the interaction of R15 with G-actin. In contrast to the conclusions made by the authors (see also fifth paragraph of the Discussion), the other mutations do not seem to have much effect. Indeed there is not much difference between the minus and plus G-actin data in panels B, C and D, which is in contrast to data in earlier figures.
The experiments shown in Figure 8B have been re-done focusing on the most informative mutations and providing a time series over which the reactions initial velocity can be estimated. We have also revised the text to explain, in detail, the derivation of the ratiometric specificity factor used to quantify the impact of the mutations in eIF2α on its ability to serve as a specific substrate for the PP1-PPP1R15-Actin ternary complex.
As the revised figure makes clear, the concentration of enzyme in the upper (+Actin) and lower panel (-Actin) were set differently to ensure that both dephosphorylation reactions would proceed to a detectable level over the time period tested. However, as the same concentration of enzyme and substrate was used in comparing the different eIF2α mutants in each panel, the ratio of the velocity in the different reactions define the specificity factor as a valid measure of the ability of the mutant eIF2α to serve as a substrate of the specific ternary complex.
The specificity factor (SF) of a mutant substrate (mut) is based on the relative initial velocity (Vi) of its dephosphorylation by the PPP1R15B -MBP-PP1-actin ternary complex (TC) versus the PPP1R15B-MBP -PP1 binary complex (BC) and is normalized for the wildtype substrate (at identical enzyme and substrate concentrations):
SF = [ViTC(mut)÷ViBC(mut)] ÷ [ViTC(wt)÷ViBC(wt)]
5) Information about the relative amounts of dimeric complexes that were used for the various mutants (in the subsection headed “G-actin, PPP1R15 and PP1 form a ternary holophosphatase that selectively dephosphorylates eIF2α” and Figure 6) should be given.
This information is now provided in the figure legend and on the Results section.
[Editors' note: further revisions were requested prior to acceptance, as described below.]
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases
eLife 4:e04871.
https://doi.org/10.7554/eLife.04871




{kind=link}