Autoimmunity: Redoxing PTPN22 activity
The oxidative state of a critical cysteine residue determines the enzymatic activity of a phosphatase involved in T-cell immune responses.
- Molecular Physiology Division, Institute of Cardiovascular Physiology, University Medical Center, Georg-August University, Germany
A precisely tuned immune system is tremendously important for rapidly sensing and eliminating disease-causing pathogens and generating immunological memory. At the same time, immune cells need to be able to recognize the body’s own cells and distinguish them from foreign invaders. Even small dysregulations can result in the immune system attacking organs and tissues in the body by mistake, leading to conditions known as autoimmune diseases.
The incidence of autoimmune diseases worldwide has increased in recent years, leading scientists to investigate how genetic and environmental factors contribute to these pathologies (Ye et al., 2018). Amongst other findings, research has shown that an enzyme called PTPN22 (short for protein tyrosine phosphatase non-receptor type 22) is a risk factor in multiple autoimmune disorders, including rheumatoid arthritis, diabetes and systemic lupus erythematosus. PTPN22 prevents the overactivation of T-cells (cells of the adaptive immune system) by removing phosphate groups from phosphorylated proteins that are part of the T-cell receptor (TCR) signaling pathway (Figure 1; Bottini et al., 2006; Fousteri et al., 2013; Tizaoui et al., 2021).
Figure 1
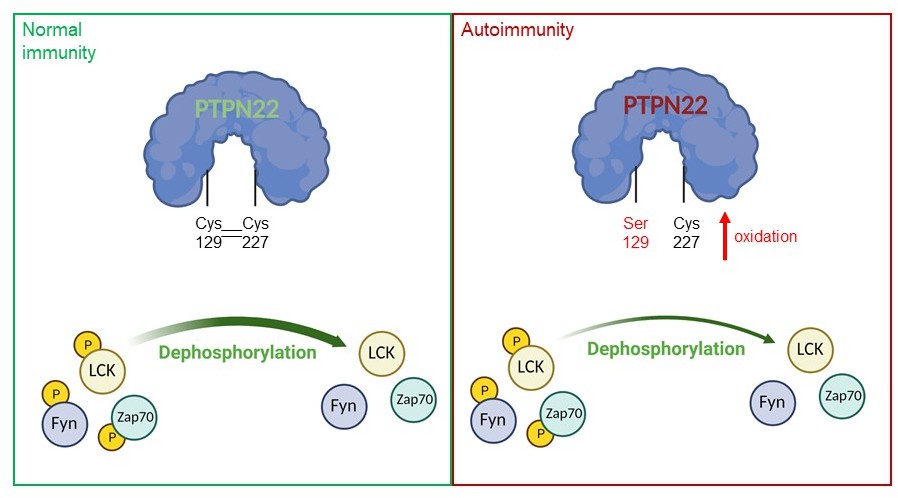
A model of PTPN22 redox regulation and its effect on T-cell activity.
In normal immunity, wildtype PTPN22 (left, blue protein with green lettering) is able to efficiently remove phosphate groups (yellow circles) from proteins downstream of the T-cell receptor (TCR), including LCK, Fyn and Zap70. Dephosphorylation inactivates these proteins, reducing T-cell activity. In this state, two PTPN22 cysteine residues (at positions 129 and 227) form a disulfide bond, which influences the redox state and the activity of the enzyme. If PTPN22 is mutated so that cysteine 129 becomes a serine (right, blue protein with red lettering, with the mutant serine residue shown in red), the disulfide bond cannot form, and the phosphatase becomes more sensitive to deactivation by oxidation. The mutant version of the phosphatase is also less efficient at dephosphorylating proteins, which increases TCR signaling and inflammation, leading to autoimmunity.
Figure created using BioRender
Activation of the T-cell receptor is followed by the production of reactive oxygen species (ROS), highly reactive by-products of molecular oxygen, which can oxidize other molecules, including proteins. It is now clear that ROS have important roles in T-cell activation and that defects in ROS production may alter the immune system's responses (Simeoni and Bogeski, 2015; Kong and Chandel, 2018). However, high levels of ROS can also cause oxidative stress, leading to impaired cell activity and even death. Therefore, T-cells must optimally balance ROS production through antioxidative mechanisms and enzymes such as thioredoxin (Patwardhan et al., 2020).
Redox reactions (oxidation and its reverse reaction known as reduction) regulate many proteins, including phosphatases (Tonks, 2005), although how oxidation and reduction modulate PTPN22 activity remained unclear. Now, in eLife, Rikard Holmdahl and colleagues based in Sweden, China, Australia, Austria, France, Russia, Hungary and the United States – including Jaime James (Karolinska Institute) as first author – report that a non-catalytic cysteine may play an important role in the redox regulation of PTPN22 (James et al., 2022). Notably, this regulation was found to modulate inflammation in mouse models with severe autoimmunity.
The team genetically engineered mice that carried a mutated version of PTPN22, in which a non-catalytic cysteine at position 129 was replaced with a serine, preventing that residue from forming a disulfide bond with the catalytic cysteine at position 227 responsible for the enzymatic activity of PTPN22. Notably, this approach was based on a study in which the crystal structure of PTPN22 was examined and an atypical bond was observed between the non-catalytic cysteine at position 129 and the catalytic cysteine residue (C227; Orrú et al., 2009). In vitro experiments performed by James et al. revealed that the mutant enzyme was more sensitive to inhibition by oxidation than its wildtype counterpart. Interestingly, the results also showed that the mutant PTPN22 was less efficient at performing its catalytic role, and that it was less responsive to re-activation by antioxidant enzymes, such as thioredoxin.
To further test the role of cysteine 129 in PTPN22 redox regulation, James et al. used a mouse model that expressed the mutant protein and was susceptible to rheumatoid arthritis. These mice exhibited higher levels of inflammation in response to T-cell activation, which would be expected in animals that cannot downregulate TCR signaling. The mice also displayed more severe symptoms of arthritis, consistent with high immune activity. These effects were not observed when the experiment was repeated in mice that fail to produce high levels of ROS in response to TCR activation, confirming that the initial observations depend on the redox state of PTPN22.
Finally, James et al. performed in vitro experiments on T-cells isolated from mice carrying the mutant PTPN22. They found that when these cells became activated, the downstream targets of PTPN22 showed an increased phosphorylation status, consistent with lower PTPN22 activity.
Taken together, the elegant study of James et al. shows that cysteine 129 is critical for the redox regulation of PTPN22, and that its mutation impacts T-cell activity and exacerbates autoimmunity in mice (Figure 1). What still remains to be determined is why the mutant enzyme has lower catalytic activity, which may be due to the mutation affecting the structural conformation of PTPN22. Additionally, it will be important to assess other cysteines in PTPN22 to determine whether they are also partly involved in its redox regulation.
Understanding how the redox state of PTPN22 regulates the activity of T-cells may help researchers to develop new therapies for treating autoimmune diseases.
References
-
Role of PTPN22 in type 1 diabetes and other autoimmune diseasesSeminars in Immunology 18:207–213.https://doi.org/10.1016/j.smim.2006.03.008
-
Roles of the protein tyrosine phosphatase PTPN22 in immunity and autoimmunityClinical Immunology 149:556–565.https://doi.org/10.1016/j.clim.2013.10.006
-
Regulation of redox balance in cancer and T cellsThe Journal of Biological Chemistry 293:7499–7507.https://doi.org/10.1074/jbc.TM117.000257
-
A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosusHuman Molecular Genetics 18:569–579.https://doi.org/10.1093/hmg/ddn363
-
Redox regulation of regulatory T-cell differentiation and functionsFree Radical Research 54:947–960.https://doi.org/10.1080/10715762.2020.1745202
-
Redox regulation of T-cell receptor signalingBiological Chemistry 396:555–568.https://doi.org/10.1515/hsz-2014-0312
-
The role of PTPN22 in the pathogenesis of autoimmune diseases: a comprehensive reviewSeminars in Arthritis and Rheumatism 51:513–522.https://doi.org/10.1016/j.semarthrit.2021.03.004
Article and author information
Author details
Magdalena Shumanska

Publication history
- Version of Record published: May 19, 2022 (version 1)
Copyright
© 2022, Shumanska and Bogeski
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 572
- views
-
- 99
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Autoimmunity: Redoxing PTPN22 activity
eLife 11:e79125.
https://doi.org/10.7554/eLife.79125
Further reading
-
- Immunology and Inflammation
Environmental air irritants including nanosized carbon black (nCB) can drive systemic inflammation, promoting chronic obstructive pulmonary disease (COPD) and emphysema development. The let-7 microRNA (Mirlet7 miRNA) family is associated with IL-17-driven T cell inflammation, a canonical signature of lung inflammation. Recent evidence suggests the Mirlet7 family is downregulated in patients with COPD, however, whether this repression conveys a functional consequence on emphysema pathology has not been elucidated. Here, we show that overall expression of the Mirlet7 clusters, Mirlet7b/Mirlet7c2 and Mirlet7a1/Mirlet7f1/Mirlet7d, are reduced in the lungs and T cells of smokers with emphysema as well as in mice with cigarette smoke (CS)- or nCB-elicited emphysema. We demonstrate that loss of the Mirlet7b/Mirlet7c2 cluster in T cells predisposed mice to exaggerated CS- or nCB-elicited emphysema. Furthermore, ablation of the Mirlet7b/Mirlet7c2 cluster enhanced CD8+IL17a+ T cells (Tc17) formation in emphysema development in mice. Additionally, transgenic mice overexpressing Mirlet7g in T cells are resistant to Tc17 and CD4+IL17a+ T cells (Th17) development when exposed to nCB. Mechanistically, our findings reveal the master regulator of Tc17/Th17 differentiation, RAR-related orphan receptor gamma t (RORγt), as a direct target of Mirlet7 in T cells. Overall, our findings shed light on the Mirlet7/RORγt axis with Mirlet7 acting as a molecular brake in the generation of Tc17 cells and suggest a novel therapeutic approach for tempering the augmented IL-17-mediated response in emphysema.
-
- Immunology and Inflammation
SARS-CoV-2 vaccines have been used worldwide to combat COVID-19 pandemic. To elucidate the factors that determine the longevity of spike (S)-specific antibodies, we traced the characteristics of S-specific T cell clonotypes together with their epitopes and anti-S antibody titers before and after BNT162b2 vaccination over time. T cell receptor (TCR) αβ sequences and mRNA expression of the S-responded T cells were investigated using single-cell TCR- and RNA-sequencing. Highly expanded 199 TCR clonotypes upon stimulation with S peptide pools were reconstituted into a reporter T cell line for the determination of epitopes and restricting HLAs. Among them, we could determine 78 S epitopes, most of which were conserved in variants of concern (VOCs). After the 2nd vaccination, T cell clonotypes highly responsive to recall S stimulation were polarized to follicular helper T (Tfh)-like cells in donors exhibiting sustained anti-S antibody titers (designated as ‘sustainers’), but not in ‘decliners’. Even before vaccination, S-reactive CD4+ T cell clonotypes did exist, most of which cross-reacted with environmental or symbiotic microbes. However, these clonotypes contracted after vaccination. Conversely, S-reactive clonotypes dominated after vaccination were undetectable in pre-vaccinated T cell pool, suggesting that highly responding S-reactive T cells were established by vaccination from rare clonotypes. These results suggest that de novo acquisition of memory Tfh-like cells upon vaccination may contribute to the longevity of anti-S antibody titers.




{kind=link}