Natural Killer Cells: Taking on SARS-CoV-2
A new study sheds light on how SARS-CoV-2 influences the way natural killer cells can recognize and kill infected cells.
- Center for Proteomics, Faculty of Medicine, University of Rijeka, Croatia
Responding to a viral infection is a complex, multistep process that involves a multitude of immune actors. Innate immunity acts first, deploying a battery of cellular and molecular entities which are not specific to the invading pathogen. Natural killer cells, for instance, are powerful antiviral agents which can recognize and kill cells infected with a broad range of viruses (Björkström et al., 2022). An adaptive immune response is then mounted, which specifically targets the virus causing the infection. For example, antibodies precisely selected to bind to a range of viral proteins are produced and released in large numbers. In the case of SARS-CoV-2, the virus that causes COVID-19, both innate and adaptive responses are considered to be essential for the control of infection (Merad et al., 2022).
For natural killer cells to eliminate their targets, a number of stress-induced molecules must first be displayed on the surface of infected cells; there, they can be recognized by receptors on natural killer cells, a process which activates the cells’ killing programme. However, some natural killer cells also recognize infected cells by harnessing virus-specific antibodies produced by the adaptive immune response. This mechanism, known as antibody-dependent cellular cytotoxicity (ADCC), involves natural killer cells expressing an activating receptor which interacts with the tail end of antibodies.
Despite the efficiency of natural killer cells, viruses often have a broad arsenal of strategies at their disposal to escape these cells. Whether SARS-CoV-2 actively evades early natural killer cell response, and whether antibodies engage these cells via ADCC to protect against COVID-19, remains unclear. Now, in eLife, Richard Stanton and colleagues at various institutions in the United Kingdom – including Ceri Fielding of Cardiff University as first author – report results showing how SARS-CoV-2 interferes with the recognition processes of natural killer cells during the early stages of infection (Fielding et al., 2022).
First, the team screened which proteins are expressed on the surface of infected cells. This showed that SARS-CoV-2 actively evades natural killer cells by preventing the synthesis of several ligands that bind to natural killer cell’s receptors (Figure 1A). Further experiments revealed the identity of the SARS-CoV-2 proteins which could be responsible for this effect: the viral proteins Nsp1 and Nsp14, which could cooperate to reduce the expression of a number of surface proteins recognized by natural killer cells. The viral proteins likely perform this role by degrading the mRNA coding for the ligands and inhibiting translation in the cell; according to previous reports, this strategy has also been used against other factors involved in the innate immune response (Hsu et al., 2021; Thoms et al., 2020). Interestingly, however, recent evidence suggests that the related viral protein Nsp13 can actually increase the activation of natural killer cells by interfering with a receptor which inhibits the cells’ killing response (Hammer et al., 2022). How these opposing effects of SARS-CoV-2 affect the way natural killer cells control infections in vivo remains to be determined.
Figure 1
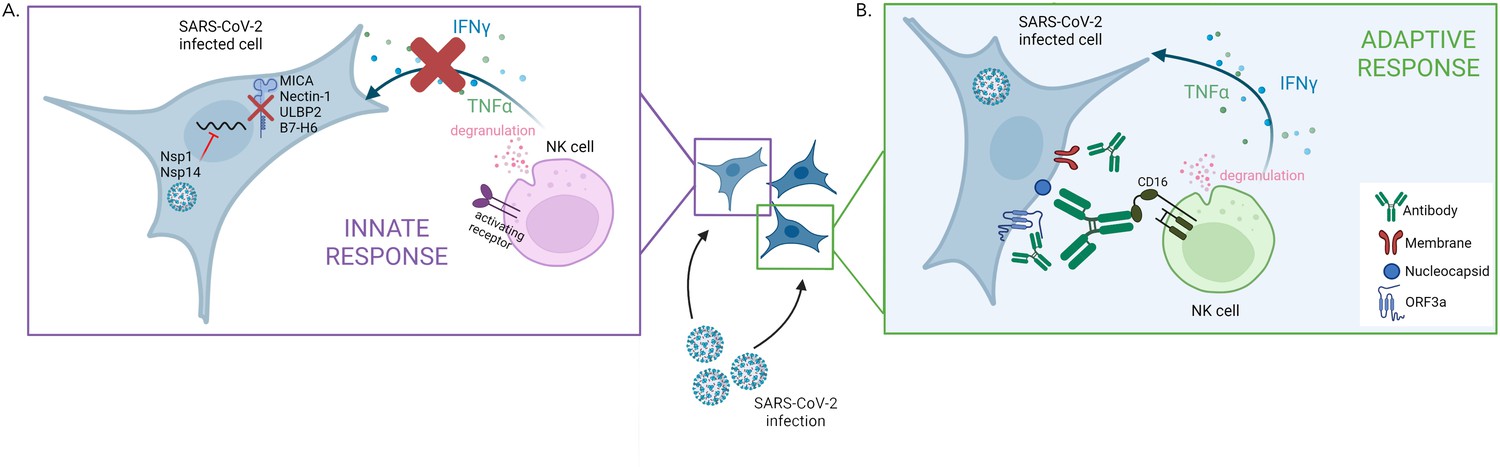
The two faces of the natural killer cell response against SARS-CoV-2.
(A) SARS-CoV-2 infection interferes with the early activation of natural killer cells by having the viral proteins Nsp1 and Nsp14 prevent the synthesis of several surface proteins (MICA, Nectin1, ULBP2, B7–H6) that can activate natural killer cells. This leads to a dampening of that innate immune response, with impaired production of factors (IFNγ and TNFα) that promote the immune response and altered degranulation (a marker of the ability of natural killer cells to kill infected cells). (B) Antibodies produced against specific SARS-CoV-2 proteins – Nucleocapsid, Membrane and ORF3a – which are expressed on the membrane of infected cells, can efficiently trigger natural killer cell activation. This process takes place via CD16, an activating receptor on the surface of natural killer cells that interacts with the tail end portion of antibodies.
Figure created with Biorender.
Fielding et al. then showed that natural killer cells can be efficiently triggered by antibodies bound to SARS-CoV-2-infected cells (Figure 1B), demonstrating that the ADCC mechanism can activate these cells during COVID-19 infection. However, the antibodies triggering ADCC were not the ones targeting the spike protein, the viral component used in many current vaccines. In fact, further experiments revealed that vaccination-induced antibodies targeting the spike protein poorly engaged natural killer cells, a result in line with a study showing that vaccination-induced antibodies are not as good at mediating ADCC compared to infection-induced antibodies (Rieke et al., 2022). Fielding et al. then went on to reveal that the antibodies involved in ADCC were those produced in reaction to other viral proteins expressed at the surface of infected cells. In most COVID-19 patients, the infection-induced antibodies able to trigger ADCC persisted for at least six months.
Together, these results suggest that it could be possible to improve vaccine design by adding viral proteins which induce antibodies capable of triggering ADCC in natural killer cells to the current formulation. In addition, promoting natural killer cell activity by boosting ADCC response in patients with severe COVID-19 could become a therapeutic option, as these individuals show high levels of antibodies and impaired natural killer cell function (Merad et al., 2022; Witkowski et al., 2021).
References
-
Natural killer cells in antiviral immunityNature Reviews Immunology 22:112–123.https://doi.org/10.1038/s41577-021-00558-3
-
Natural killer cell-mediated antibody-dependent cellular cytotoxicity against SARS-CoV-2 after natural infection is more potent than after vaccinationThe Journal of Infectious Diseases 225:1688–1693.https://doi.org/10.1093/infdis/jiac060
-
Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2Science (New York, N.Y.) 369:1249–1255.https://doi.org/10.1126/science.abc8665
Article and author information
Author details
Publication history
- Version of Record published: June 28, 2022 (version 1)
Copyright
© 2022, Kučan Brlić and Brizić
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 896
- views
-
- 185
- downloads
-
- 2
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Natural Killer Cells: Taking on SARS-CoV-2
eLife 11:e80552.
https://doi.org/10.7554/eLife.80552
Further reading
-
- Cancer Biology
- Immunology and Inflammation
Solid tumors generally exhibit chromosome copy number variation, which is typically caused by chromosomal instability (CIN) in mitosis. The resulting aneuploidy can drive evolution and associates with poor prognosis in various cancer types as well as poor response to T-cell checkpoint blockade in melanoma. Macrophages and the SIRPα-CD47 checkpoint are understudied in such contexts. Here, CIN is induced in poorly immunogenic B16F10 mouse melanoma cells using spindle assembly checkpoint MPS1 inhibitors that generate persistent micronuclei and diverse aneuploidy while skewing macrophages toward a tumoricidal ‘M1-like’ phenotype based on markers and short-term anti-tumor studies. Mice bearing CIN-afflicted tumors with wild-type CD47 levels succumb similar to controls, but long-term survival is maximized by SIRPα blockade on adoptively transferred myeloid cells plus anti-tumor monoclonal IgG. Such cells are the initiating effector cells, and survivors make de novo anti-cancer IgG that not only promote phagocytosis of CD47-null cells but also suppress tumor growth. CIN does not affect the IgG response, but pairing CIN with maximal macrophage anti-cancer activity increases durable cures that possess a vaccination-like response against recurrence.
-
- Developmental Biology
- Immunology and Inflammation
Cardiac macrophages are heterogenous in phenotype and functions, which has been associated with differences in their ontogeny. Despite extensive research, our understanding of the precise role of different subsets of macrophages in ischemia/reperfusion (I/R) injury remains incomplete. We here investigated macrophage lineages and ablated tissue macrophages in homeostasis and after I/R injury in a CSF1R-dependent manner. Genomic deletion of a fms-intronic regulatory element (FIRE) in the Csf1r locus resulted in specific absence of resident homeostatic and antigen-presenting macrophages, without affecting the recruitment of monocyte-derived macrophages to the infarcted heart. Specific absence of homeostatic, monocyte-independent macrophages altered the immune cell crosstalk in response to injury and induced proinflammatory neutrophil polarization, resulting in impaired cardiac remodeling without influencing infarct size. In contrast, continuous CSF1R inhibition led to depletion of both resident and recruited macrophage populations. This augmented adverse remodeling after I/R and led to an increased infarct size and deterioration of cardiac function. In summary, resident macrophages orchestrate inflammatory responses improving cardiac remodeling, while recruited macrophages determine infarct size after I/R injury. These findings attribute distinct beneficial effects to different macrophage populations in the context of myocardial infarction.




{kind=link}