Cis and trans RET signaling control the survival and central projection growth of rapidly adapting mechanoreceptors
- University of Pennsylvania, United States
- Max Planck Institute of Neurobiology, Germany
- Parkinson's Disease and Movement Disorder Center of Maryland, United States
Abstract
RET can be activated in cis or trans by its co-receptors and ligands in vitro, but the physiological roles of trans signaling are unclear. Rapidly adapting (RA) mechanoreceptors in dorsal root ganglia (DRGs) express Ret and the co-receptor Gfrα2 and depend on Ret for survival and central projection growth. Here, we show that Ret and Gfrα2 null mice display comparable early central projection deficits, but Gfrα2 null RA mechanoreceptors recover later. Loss of Gfrα1, the co-receptor implicated in activating RET in trans, causes no significant central projection or cell survival deficit, but Gfrα1;Gfrα2 double nulls phenocopy Ret nulls. Finally, we demonstrate that GFRα1 produced by neighboring DRG neurons activates RET in RA mechanoreceptors. Taken together, our results suggest that trans and cis RET signaling could function in the same developmental process and that the availability of both forms of activation likely enhances but not diversifies outcomes of RET signaling.
https://doi.org/10.7554/eLife.06828.001eLife digest
During development, cells send and receive numerous signaling molecules. In order to trigger a biological response, such signaling molecules must first bind to a specific receptor protein, often located on the cell surface. These receptor proteins can either work alone or with partner proteins called co-receptors. When the co-receptor is produced by the same cell as the receptor, it is called cis signaling. When the co-receptor is produced by other cells, it is called trans signaling.
RET is one such receptor that is important for the development of the nervous system and many other biological processes. It interacts with a particular family of signaling molecules, the glial cell line-derived neurotrophic factor (GDNF) family ligands, which first bind to a co-receptor, GFRα, before binding to RET. These co-receptors can come from the same cell as RET, or from a different cell.
Previous studies have indicated that RET can receive both cis and trans signals using cultured cells, but it was not clear whether both types of signal occur during normal development and contribute to the same biological processes. Fleming, Vysochan et al. investigated this question by analyzing the roles of RET signaling in a type of mouse neuron that is involved in sensing touch. RET is important for the survival and development of these neurons, which express both RET and its co-receptor GFRa2. Another RET co-receptor, GFRa1, is produced by other cells that are next to the cell bodies and projections of these touch-sensing neurons.
To investigate the roles of different GFRa co-receptors further, Fleming, Vysochan et al. generated a variety of mouse mutants, including mice with mutations in one or both types of co-receptor. The neurons in mice lacking both co-receptors shared the same defects as the neurons in the mice lacking RET. Loss of either co-receptor alone did not produce these abnormalities. This indicates that both co-receptors can mediate the normal development of these neurons, with GFRa2 signaling in cis and GFRa1 signaling in trans.
Fleming, Vysochan et al. propose that cis and trans RET signaling can lead to the same biological outcomes in these neurons. Future experiments should reveal if cis and trans RET signaling contribute towards common biological processes in other cell types inside the body as well. Such findings might also be important for understanding the role of RET signaling in cancer and other human diseases.
https://doi.org/10.7554/eLife.06828.002Introduction
The neurotrophic receptor tyrosine kinase RET plays critical roles in many biological processes, including kidney genesis, spermatogenesis, and development of enteric, sensory, autonomic, and motor neurons (Runeberg-Roos and Saarma, 2007; Ibáñez, 2013). Loss of RET signaling leads to Hirschprung's disease, while RET gain of function has been implicated in various human carcinomas (Runeberg-Roos and Saarma, 2007; Santoro and Carlomagno, 2013). In addition, activation of the RET signaling pathway has potential applications in the treatment of Parkinson's disease and promotion of spinal cord (SC) regeneration following injury (Bespalov and Saarma, 2007; Deng et al., 2013). Therefore, it is critical to thoroughly understand RET signaling mechanisms.
RET is the common signaling receptor for the glial cell line-derived neurotrophic factor (GDNF) family of ligands (GFLs), which includes GDNF, neurturin (NRTN), artemin, and persephin. For RET activation and signaling, GFLs first bind to a GPI-linked GDNF family receptor alpha (GFRa), which then associates with RET to form an active signaling complex (Airaksinen and Saarma, 2002). In vertebrates, the GFRas and their high-affinity ligand pairs are GFRa1 and GDNF (Jing et al., 1996; Treanor et al., 1996), GFRa2 and NRTN (Baloh et al., 1997; Buj-Bello et al., 1997; Klein et al., 1997), GFRa3 and artemin (Baloh et al., 1998), and GFRa4 and persephin (Yang et al., 2007).
RET can be activated by GFRas expressed in the same cell (cis signaling) or by GFRas (mainly GFRa1) produced from other sources (trans signaling) in vitro (Paratcha et al., 2001; Ledda et al., 2002). The existence of both cis and trans activation has been proposed to diversify RET signaling by either recruiting different downstream effectors or changing the kinetics or efficacy of kinase activation (Tansey et al., 2000; Paratcha et al., 2001). Consistent with the trans signaling model, Gfra1 is expressed in the target fields of many RET+ neurons during development and can promote axon growth upon GDNF treatment in culture (Trupp et al., 1997; Yu et al., 1998; Paratcha et al., 2001). However, the ‘cis-only’ mouse model, in which Gfra1 is expressed under the control of the Ret promoter in a Gfra1 null background, produced no overt phenotypes in many Ret-dependent developmental processes, suggesting that trans signaling may not play a major physiological role (Enomoto et al., 2004). Recently, trans RET signaling has been implicated in the development of inhibitory cortical interneurons, nigral dopaminergic neurons, and enteric lymphoids, and in perineural invasion by cancer cells (Canty et al., 2009; Kholodilov et al., 2011; Patel et al., 2012; He et al., 2014). Nevertheless, the physiological functions of trans RET signaling and whether cis and trans signaling lead to the same or different biological outcomes in vivo remain largely unresolved.
Aβ mechanoreceptors are large diameter somatosensory neurons mediating discriminative touch, which innervate layers III–V of the SC. They can be broadly divided into rapidly adapting (RA) and slowly adapting (SA) mechanoreceptors based on their adaptation properties to sustained mechanical stimuli (Fleming and Luo, 2013). Previously, we and other labs identified that a small population of mouse DRG (dorsal root ganglion) neurons, the early RET+ DRG neurons, develop into RA mechanoreceptors, and that Ret is required cell autonomously for the growth of their third order central projections innervating the dorsal SC (dSC) (Bourane et al., 2009; Luo et al., 2009; Honma et al., 2010).
RET in RA mechanoreceptors encounters environments in which both cis and trans activation are possible, providing a good model system to study the physiological functions of trans RET signaling. RA mechanoreceptors express Ret and Gfra2 (Bourane et al., 2009; Luo et al., 2009; Honma et al., 2010), whereas Gfra1 is highly expressed in their target field (Treanor et al., 1996; Yu et al., 1998) and by neighboring DRG neurons during development (Luo et al., 2009; Honma et al., 2010). Here, we found that the central projection deficit of RA mechanoreceptors is negligible in postnatal Gfra2 and Nrtn mutant mice, which is in great contrast to the severely affected Ret mutant mice. By genetically tracing RA mechanoreceptors in different mutant mouse lines during development, we showed that the initial growth of the third order central projections of RA mechanoreceptors depends on the cis activation of RET via GFRa2 and NRTN. However, central projections of Gfra2 null RA mechanoreceptors gradually recover during development. Gfra1 null mice show no obvious central projection deficit by itself, but Gfra1;Gfra2 double null mice have similar cell death and central projection deficits to those of Ret null mice. Moreover, we showed that Gfra1 is non-detectable in most RA mechanoreceptors, thus RET in RA mechanoreceptors is most likely activated by GFRa1 in trans. Finally, we determined that RET in Gfra2 null RA mechanoreceptors responds to GDNF in DRG explant culture, and this responsiveness is mediated by GFRa1 from neighboring DRG neurons (trans activation). Taken together, our results indicate that combinatorial cis and trans RET signaling promote survival and central projection growth of RA mechanoreceptors in vivo.
Results
Expression of Ret, Gfras, and GFLs in the developing mouse SC and DRGs
Since RET can be activated by GFLs/GFRas either in cis or in trans (mainly by GDNF/GFRa1) in vitro, we asked if the expression patterns of Gfra1, Gfra2, Gdnf, and Nrtn in the developing SC and DRGs would provide insight into RET signaling in RA mechanoreceptors in vivo. We performed in situ hybridization for Ret, Gfra1, Gfra2, Gdnf, and Nrtn on embryonic day 13.5 (E13.5) and E15.5 wild-type DRG and SC sections. Double in situ hybridizations that characterize the expression of Gfra1 and Gfra2 in different populations of DRG neurons have been previously conducted (summarized in Figure 1—figure supplement 1K [Luo et al., 2009]).
Similar to previous characterization (Molliver et al., 1997; Luo et al., 2007, 2009), Ret is expressed in motor neurons and a mix of small and large diameter DRG neurons at E13.5 and E15.5 (Figure 1—figure supplement 1A–B). Most large diameter RET+ DRG neurons at these stages are the early RET+ DRG neurons, which develop into RA mechanoreceptors (Bourane et al., 2009; Luo et al., 2009). Gfra1 is highly expressed in some DRG neurons and motor neurons as well, but these GFRa1+ DRG neurons come from NTRK1+ precursors and are not early RET+ RA mechanoreceptors (Yu et al., 1998; Luo et al., 2009; Honma et al., 2010). In addition, Gfra1 is highly expressed in the dorsal root entry zone and the dSC, which are the target fields of the central projections of RA mechanoreceptors (Figure 1—figure supplement 1C–D). Gfra2 is expressed in a small number of large diameter DRG neurons, which were previously shown to be RA mechanoreceptors (Bourane et al., 2009; Luo et al., 2009), and some SC cells and motor neurons at these stages (Figure 1—figure supplement 1E–F; Oppenheim et al., 2000).
Nrtn is diffusely expressed at a low level in the SC and DRGs at both E13.5 and E15.5; Gdnf transcript is barely detected at E13.5 but is clearly expressed in DRG and motor neurons at E15.5 (Figure 1—figure supplement 1G–J). Thus, based on the expression patterns of RET signaling components in the developing SC and DRGs, RET in the central projections and cell bodies of developing RA mechanoreceptors could potentially be activated in cis by NRTN/ GFRa2 or in trans by GDNF/ GFRa1, which may come from neighboring DRG neurons, dorsal root entry zone cells, or dSC cells.
Central projection deficit of RA mechanoreceptors is negligible in postnatal Gfra2 and Nrtn null mice
RA mechanoreceptors depend on RET for the growth of their third order central projections innervating layers III–V of SC. In postnatal Ret mutant mice, VGLUT1+ puncta, which label pre-synaptic terminals of mechanoreceptors and proprioceptors (Hughes et al., 2004; Paixão et al., 2013), are greatly reduced in layers III–V, indicating deficits in the third order central projections of RA mechanoreceptors (Luo et al., 2009). Since RA mechanoreceptors express a high level of Gfra2 but not any other Gfras (Luo et al., 2009), it is likely that RET in RA mechanoreceptors is activated by NRTN/GFRa2 in cis. Indeed, we previously found that Pacinian corpuscles, a subtype of RA mechanosensory end organs in the periphery, are not formed in Ret, Gfra2, or Nrtn mutant mice, supporting that NRTN/GFRa2-RET cis signaling occurs in RA mechanoreceptors (Luo et al., 2009). Here, we asked if NRTN-GFRa2/RET cis signaling is required for the growth of RA mechanosensory central projections as well. We performed immunostaining of VGLUT1 with postnatal day 7 (P7) Gfra2GFP/GFP null and Nrtn−/− null SC sections. No significant decrease of VGLUT1+ puncta in layers III–V of SC is observed in Gfra2 and Nrtn null mice (Figure 1A–C, Figure 1—source data 1 [p = 0.96], and data not shown). This result suggests that unlike RET signaling in the peripheral branches of RA mechanoreceptors, cis activation of RET by GFRa2 and NRTN may be dispensable for the normal development of central projections of RA mechanoreceptors.
Figure 1 with 3 supplements see all
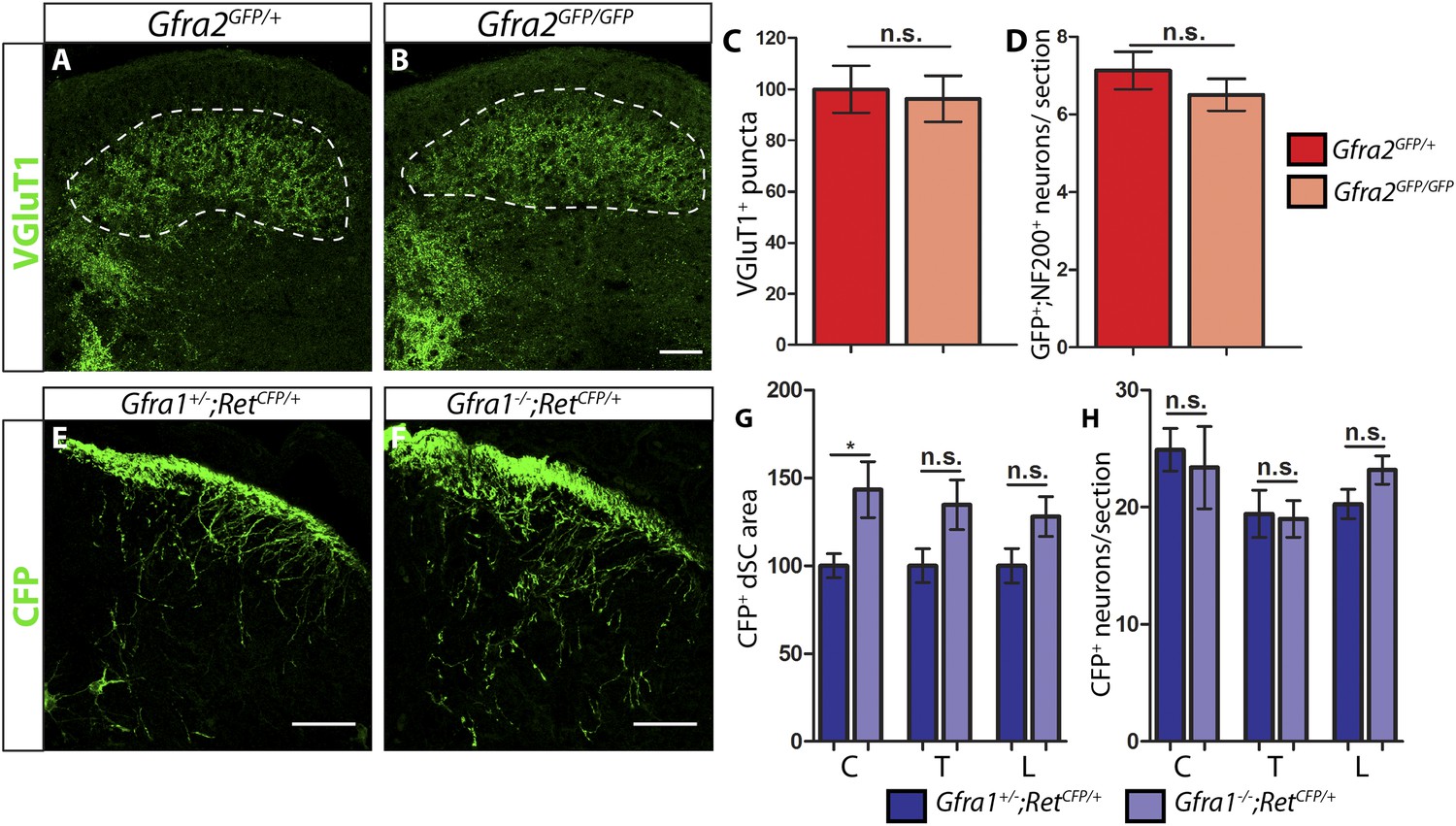
P7 Gfra2 mutant mice show normal dorsal spinal cord (dSC) VGLUT1 staining and Gfra1 null mice display normal rapidly adapting (RA) mechanoreceptor central projections at E13.5.
(A–B) Anti-VGLUT1 immunostaining of P7 SC sections from Gfra2GFP/+ control (A) and Gfra2GFP/GFP null (B) mice. VGLUT1 staining labels presynaptic terminals of mechanosensory neurons, which are found in layers III–V of the dSC (outlined in white). Note that green fluorescent protein (GFP) driven from the Gfra2 locus cannot be visualized directly. Therefore, positive signal indicates presynaptic VGLUT1+ puncta and not GFRa2+ primary afferent axons. (C) Quantification of VGLUT1+ puncta in dSC, which is displayed as a percentage of VGLUT1+ pixels compared to the control pixel count. The similar density of VGLUT1+ puncta between mutant and control tissue suggests that cis RET signaling via GFRa2 is dispensable for the growth of RA mechanosensory central projections at P7. (D) Quantification of GFP+;NF200+ neurons, which indicate RA mechanoreceptors, per DRG section. The non-significant decrease in RA mechanoreceptor number per section in Gfra2 nulls suggests that most RA mechanoreceptors are not dependent on cis RET signaling for survival. (E–F) Anti-GFP immunostaining of RA mechanoreceptor central projections in E13.5 Gfra1+/−;RetCFP/+ control (E) and Gfra1−/−;RetCFP/+ mutant (F) SC sections. The increased CFP signal in Gfra1 null dSC is likely due to the precocious expression of Ret in some dSC neurons of Gfra1 mutants. (G) Quantification of CFP+ pixel number in dSC. The lack of a reduction in CFP+ axons in Gfra1 mutant dSC indicates that trans signaling via GFRa1 is not required for the initial growth of RA mechanosensory third order central projections. (H) Quantification of number of CFP+ neurons per DRG section indicates no loss of RA mechanoreceptors in Gfra1 mutants at E13.5. C: cervical level, T: thoracic level, L: lumbar level. Scale bars = 50 μm. Error bars represent SEM. n.s. = p > 0.05, * = p < 0.05. Source data are provided in Figure 1—source data 1, 2.
-
Figure 1—source data 1
VGLUT1 dSC staining and RA mechanoreceptor number in P7 Gfra2 mutants.
- https://doi.org/10.7554/eLife.06828.004
-
Figure 1—source data 2
RA mechanoreceptor central projections and cell number in E13.5 Ret, Gfra1, Gfra2, and Nrtn mutants.
- https://doi.org/10.7554/eLife.06828.005
To determine whether RA mechanoreceptors survive without Gfra2, we quantified the number of GFP+;NF200+ neurons per DRG section in P7 Gfra2GFP/+ controls and Gfra2GFP/GFP nulls. Green fluorescent protein (GFP) is expressed from the Gfra2 locus and most of GFP+;NF200+ neurons indicate RA mechanoreceptors in Gfra2GFP mice. In agreement with our previous findings at P0 (Luo et al., 2009), we found a slight but non-significant decrease in RA mechanoreceptor number between controls and mutants (Figure 1D, Figure 1—source data 1 [p = 0.34]). Therefore, cis RET signaling via GFRa2 does not seem to be critical for the early postnatal survival of RA mechanoreceptors.
Central projection deficit of Ret null mice at E13.5
To understand the mechanism of RET signaling that controls growth of RA mechanosensory central projections, we genetically traced RA mechanoreceptors in Ret, Gfra1, Gfra2, and Nrtn mutant mice at different developmental stages. We first used Ret mutant mice, which serve as a positive control for the central projection deficit, to determine a robust method for visualizing RA mechanosensory interstitial branches at E13.5. We compared two methods that have been previously used. One is immunostaining of neurofilament-200 (NF200), which is expressed by large diameter DRG neurons, including RA mechanoreceptors, SA mechanoreceptors, and proprioceptors (Bourane et al., 2009). The other is to use a knockin/null allele of Ret (Honma et al., 2010), RetCFP (Uesaka et al., 2008), in which cyan fluorescent protein (CFP, a variant of GFP) is expressed from the Ret locus. Although Ret is expressed in both RA mechanoreceptors and some other DRG neurons at E13.5 (Luo et al., 2009), central projections of non-RA mechanoreceptor RET+ neurons, most of which develop into nociceptors, do not innervate the dSC until E15.5 or later (Ozaki and Snider, 1997). In addition, the expression of Ret in dSC neurons is not obvious until E15.5 (Figure 1—figure supplement 1B). Thus, the RetCFP allele may allow us to specifically visualize central projections of RA mechanoreceptors at E13.5.
To compare these two methods, we performed anti-NF200 and anti-GFP immunostaining on SC sections of E13.5 RetCFP/+ control and RetCFP/CFP null embryos. We observed a decrease in the density of NF200+ fibers in the dorsal horn (Figure 1—figure supplement 2B,E). This decrease of NF200+ central projections, however, is not dramatic. This is because the NF200 antibody also recognizes central projections of SA mechanoreceptors and proprioceptors, which develop in a manner temporally comparable to RA mechanoreceptors. In contrast, CFP+ fibers innervating the dSC display a dramatic reduction in Ret null mice (Figure 1—figure supplement 2C,F). Ret null CFP+ fibers reach the dorsal surface of the SC but rarely grow interstitial branches innervating layers III–V. We quantified the number of CFP+ pixels in the dorsal horn (displayed as percentage of CFP+ pixels normalized to the control) as a proxy for the extent of axon growth and found a significant decrease in CFP+ fibers in Ret mutant dorsal horn (Figure 1—figure supplement 2H and Figure 1—source data 2, [p < 0.001]). This result suggests that the RetCFP allele is a valid tool for visualizing central projection deficits of RA mechanoreceptors at E13.5.
Since Ret signaling can positively regulate the expression of its own signaling components or control neuronal survival (Luo et al., 2007; Baudet et al., 2008; Golden et al., 2010), it is conceivable that the lack of dSC CFP+ fibers could be due to a downregulation of CFP expressed in Ret null RA mechanoreceptors or death of RA mechanoreceptors. To exclude these possibilities, we quantified the number of CFP+ neurons in DRGs. We found that the number of CFP+ neurons per DRG section was not statistically different between Ret heterozygotes and null mice (Figure 1—figure supplement 2I and Figure 1—source data 2). In addition, the intensity of GFP+ fibers at the dorsal surface of the SC is comparable between Ret mutant and control mice. Therefore, the loss of CFP+ fibers in the dorsal horn of E13.5 Ret mutants must mainly be due to a deficit in growth of interstitial central axons, but not due to the down-regulation of CFP expression or the death of RA mechanoreceptors.
Central projections of RA mechanoreceptors are normal in E13.5 Gfra1 null mice
The finding that dSC VGLUT1 staining is largely normal in postnatal Gfra2 and Nrtn null mice suggests that cis RET signaling may be dispensable for RA mechanosensory central projections. To determine if the development of RA mechanosensory central projections depends on the trans activation of RET via GFRa1 and GDNF, we generated Gfra1 null (Gfra1−) mice (Figure 1—figure supplement 3A–B and ‘Materials and methods’). In situ hybridization of Gfra1 control and null DRG sections showed that Gfra1 transcripts are not produced in mice homozygous for this mutant allele (Figure 1—figure supplement 3C–D). In addition, kidneys are not formed in these Gfra1 null mice (data not shown), a phenotype consistent with previously reported Gfra1 null mice (Cacalano et al., 1998; Enomoto et al., 1998). Thus, the Gfra1− allele we generated is a null allele.
If trans activation of RET via GFRa1 is required for the growth of interstitial central projections of RA mechanoreceptors, we expect to see a decrease of central projections of RA mechanoreceptors in the dSC of Gfra1 null mice. To test this idea, we generated E13.5 Gfra1+/−;RetCFP/+ control and Gfra1−/−;RetCFP/+ mutant embryos to examine RA mechanosensory central projections at this stage (Figure 1E–F). We found that innervation of dSC by CFP+ fibers was not reduced upon Gfra1 ablation (Figure 1G, Figure 1—source data 2). Additionally, the lack of Gfra1 function did not lead to a decrease of CFP+ DRG neurons (Figure 1H, Figure 1—source data 2). Together, our results suggest that trans activation of RET via GFRa1 is not required for the survival or central projection growth of RA mechanosensory neurons at E13.5.
Gfra2 and Nrtn mutant mice phenocopy central projection deficits of Ret mutant mice at E13.5
Since no deficit was observed in the central projections of RA mechanoreceptors in E13.5 Gfra1 mutants, we next asked whether cis RET signaling is required for the initial growth of RA mechanosensory third order central projections. We crossed the RetCFP allele into Gfra2 and Nrtn null mice and examined central projections of RA mechanoreceptors at E13.5 (Figure 2A–D). In contrast to what we observed at P7, at this early development stage CFP+ central projections of RA mechanoreceptors are greatly reduced in both Gfra2 and Nrtn null SC sections (Figure 2E–F, Figure 1—source data 2, Gfra2 mutant has 9.50 ± 1.44% of control staining at thoracic levels [p < 0.001]). In addition, similar to the E13.5 Ret mutant mice, the number of CFP+ DRG neurons in Gfra2 and Nrtn null mice is comparable to that of control mice (Figure 2G–H, Figure 1—source data 2), suggesting that the loss of CFP+ fibers in the dSC of these mutant mice is due to a deficit in the interstitial central projection growth of RA mechanoreceptors. Thus, at E13.5, Gfra2 and Nrtn null mice phenocopy the central projection deficit of Ret mutant mice, which suggests that RET is activated by NRTN/GFRa2 in cis for the initial growth of RA mechanosensory central projections.
Figure 2
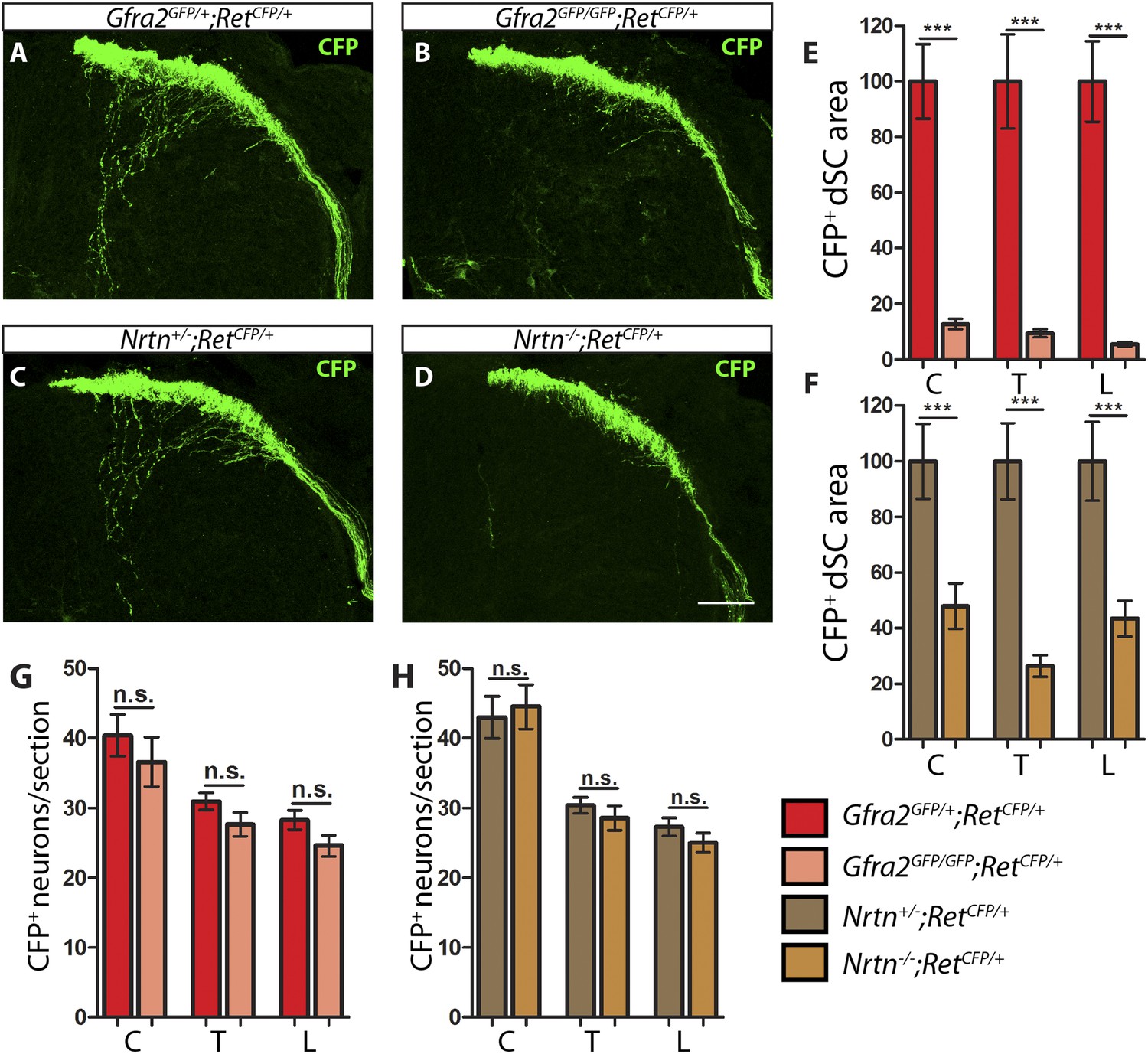
Gfra2 and Nrtn null mice show reduced RA mechanoreceptor central projections at E13.5.
(A–D) Anti-GFP immunostaining to visualize RA mechanosensory central projections in E13.5 dSC sections of Gfra2GFP/+;RetCFP/+ control (A), Gfra2GFP/GFP;RetCFP/+ mutant (B), Nrtn+/−;RetCFP/+ control (C), and Nrtn−/−;RetCFP/+ mutant (D) mice. (E–F) Quantification of CFP+ pixel number in dSC of Gfra2 (E) and Nrtn (F) mice. The dramatic reduction in CFP+ axons in Gfra2 and Nrtn nulls at E13.5 suggests that cis activation of RET is required for the initial growth of RA mechanosensory third order central projections. (G–H) Quantification of number of CFP+ neurons per DRG section in Gfra2 (G) and Nrtn (H) mice. Similar number of CFP+ DRG neurons between control and mutant mice indicates that cell death of RA mechanoreceptors or downregulation of RetCFP allele do not occur at E13.5 when cis RET signaling is ablated. Scale bar = 50 μm. Error bars represent SEM. n.s. = p > 0.05, ** = p < 0.01 *** = p < 0.001. Source data are provided in Figure 1—source data 2.
Interstitial central projections of Gfra2 null RA mechanoreceptors begin to recover from E15.5
If Ret, Gfra2, and Nrtn null mice phenocopy each other at E13.5, why do their postnatal VGLUT1 staining patterns look so different (Figure 1 and [Luo et al., 2009])? One possibility is that since Ret has a much broader expression pattern than Gfra2 in the dSC and DRGs, the dramatic loss of VGLUT1 staining in layers III–V of SC may be caused by the loss of RET signaling both in RA mechanoreceptors and other RET+ cells. For Gfra2 and Nrtn mutant mice, though central projection deficits of RA mechanoreceptors may persist postnatally, VGLUT1+ puncta from SA mechanoreceptors could mask the phenotype. Alternatively, RA mechanosensory central projections in Gfra2 and Nrtn mutant mice could recover at later developmental stages due to the function of other RET signaling mechanisms.
To differentiate these possibilities, we examined central projections of RA mechanoreceptors in Gfra2 null mice through development. We focused on Gfra2 instead of Nrtn mutant mice because: (1) the cell autonomous requirement of a co-receptor is the key to differentiate cis vs trans RET signaling; and (2) Gfra2 and Nrtn null mice display very similar phenotypes of RA mechanoreceptors. Since Ret begins to be expressed in additional populations of DRG neurons (Molliver et al., 1997; Luo et al., 2007) and dSC cells (Figure 1—figure supplement 1B) from E15.5, we can't use the RetCFP allele to visualize the central projections of RA mechanoreceptors at late developmental stages. To overcome this problem, we used a tandem allele (see ‘Materials and methods’ and Figure 3—figure supplement 1) of an inducible Cre allele of Ret (RetCreERT) and Rosa26 conditional red fluorescent protein (RosaTdt). We combined these alleles with early (E11.5 and E12.5) 4-hydroxy tamoxifen (4-HT) treatment to specifically trace RA mechanoreceptors, as previously established (Luo et al., 2009).
We generated Gfra2GFP/+; RetCreERT; RosaTdt control and Gfra2GFP/GFP; RetCreERT; RosaTdt mutant mice and examined their SC and DRG sections at E15.5. Tdt+ fibers innervate layers III–V of the SC, which is consistent with specific genetic tracing of RA mechanoreceptors (Luo et al., 2009). In addition, the majority of Tdt+ DRG neurons are RET+, GFRa2+ (reported by the expression of GFP), but NTRK1− at E15.5 (Figure 3A–J), further supporting the specific labeling of RA mechanoreceptors. We found that central projections of Gfra2 null RA mechanoreceptors are also decreased at E15.5 (Figure 3K–N, Figure 3—source data 1, Gfra2 mutant has 55.13 ± 2.82% of control staining at the thoracic level [p < 0.001]). Since the number of labeled DRG neurons is not significantly reduced in the mutant mice (Gfra2 mutants have 79.52 ± 8.39% of control cell number [p = 0.06]), the central projection phenotype mostly reflects a growth deficit at this developmental stage. Noticeably, the relative reduction of innervation in Gfra2 null mice at E15.5 is less severe compared to that of E13.5 mutants (Figure 2), suggesting that central projections of Gfra2 null RA mechanoreceptors may start to recover at this stage.
Figure 3 with 1 supplement see all
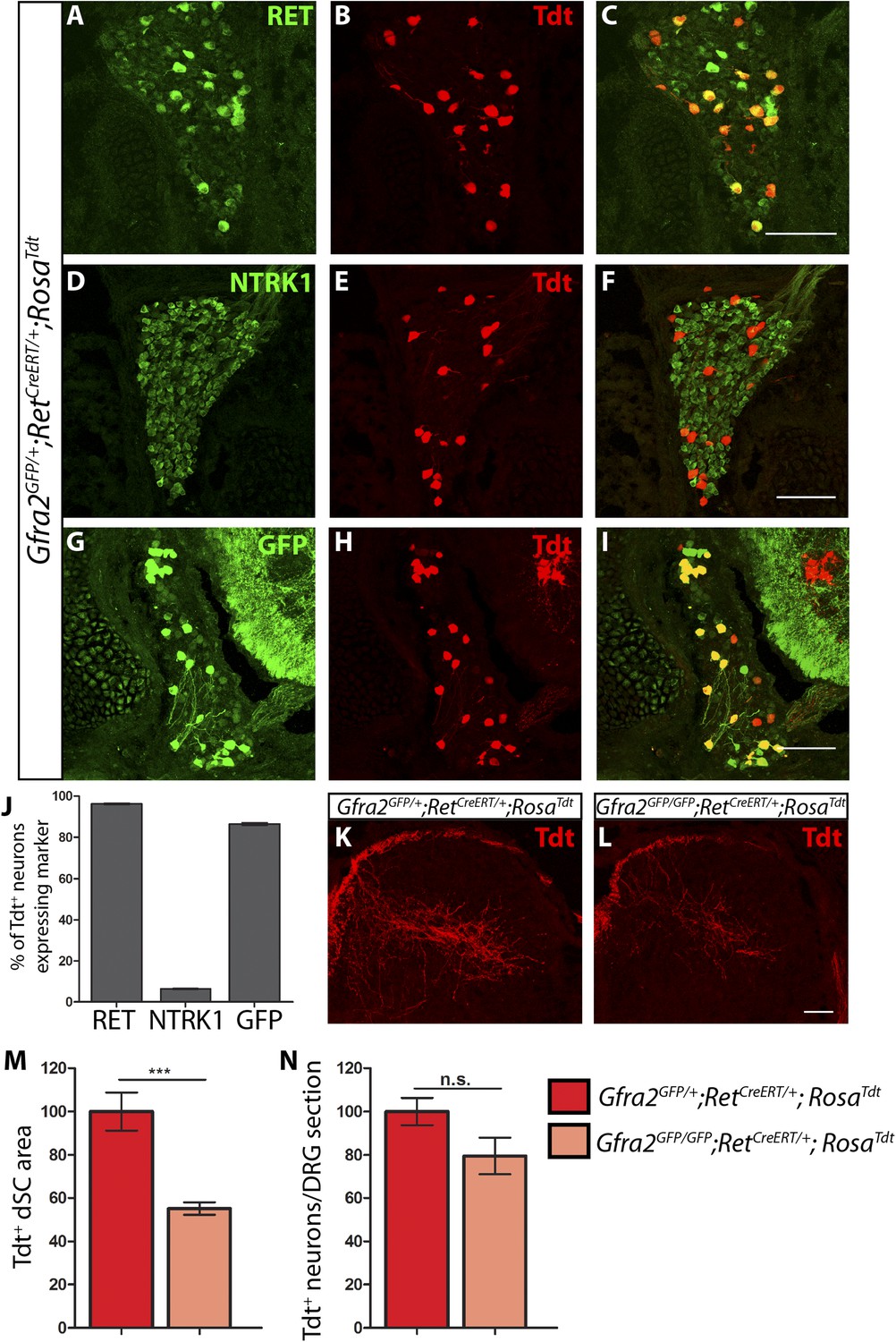
Central projection growth deficit of Gfra2 null RA mechanoreceptors at E15.5.
(A–I) E15.5 Gfra2GFP/+;RetCreERT/+;RosaTdt DRG sections stained with anti-RET (A–C), anti-NTRK1 (D–F), and anti-GFP (G–I). (J) Quantification of percentage of Tdt+ DRG neurons which co-express RET (96.16 ± 0.28%), NTRK1 (6.56 ± 0.18%), and GFP driven from the Gfra2 locus (86.48 ± 0.55%). The expression profile of Tdt+ neurons confirms that this genetic labeling strategy specifically labels RA mechanoreceptors. (K–L) Visualization of Tdt+ RA mechanosensory central projections in dSC of E15.5 Gfra2GFP/+; RetCreERT/+; RosaTdt control (K) and Gfra2GFP/GFP; RetCreERT/+; RosaTdt mutant (L) SC sections. (M) Quantification of Tdt+ pixels in dSC, which is displayed as a percentage normalized to dSC Tdt+ pixels of the within litter controls. Gfra2 mutant mice have 55.13 ± 2.82% of control staining (p < 0.001). Note that although Gfra2 null RA mechanoreceptors still have a central projection deficit at E15.5, the reduction at this stage is less severe than the deficit observed at E13.5. (N) Quantification of number of Tdt+ neurons per DRG section, which is displayed as a percentage normalized to Tdt+ neurons of the within litter controls. Gfra2 mutant mice have 79.52 ± 8.39% of control cell number (p = 0.06), which suggests that the survival of RA mechanoreceptors is not dependent on cis signaling at this stage. Scale bars = 100 μm (A–I), 50 μm (K–L). Error bars represent SEM. n.s. = p > 0.05, *** = p < 0.001. Source data are provided in Figure 3—source data 1.
-
Figure 3—source data 1
RA mechanoreceptor central projections and cell number in E15.5 Gfra2mutants.
- https://doi.org/10.7554/eLife.06828.011
Ret and Gfra2 null mice display different central projection and cell survival deficits at E18.5
To determine if RA mechanoreceptors require Ret but not Gfra2 for their central projection growth at later developmental stages, we generated E18.5 RetCreERT/+;RosaTdt control and RetCreERT/CreERT;RosaTdt mutant embryos (RetCreERT is a null allele of Ret). Consistent with previous results (Bourane et al., 2009; Luo et al., 2009; Honma et al., 2010), we found that RA mechanosensory central projections are greatly reduced in the Ret mutant mice (Figure 4A,C,I, Figure 4—source data 1, Ret mutant has 35.86 ± 4.97% of control staining at thoracic levels [p < 0.001]). In addition, we counted the number of Tdt+ neurons in L4/L5 DRGs and found that the number of Tdt+ RA mechanoreceptors is dramatically reduced as well (Figure 4B,D,J, Ret mutant has 52.52 ± 7.76% of control cell number [p < 0.001]). Taken together, these results suggest that Ret is absolutely required for both survival and central projection growth of RA mechanoreceptors at E18.5.
Figure 4 with 1 supplement see all
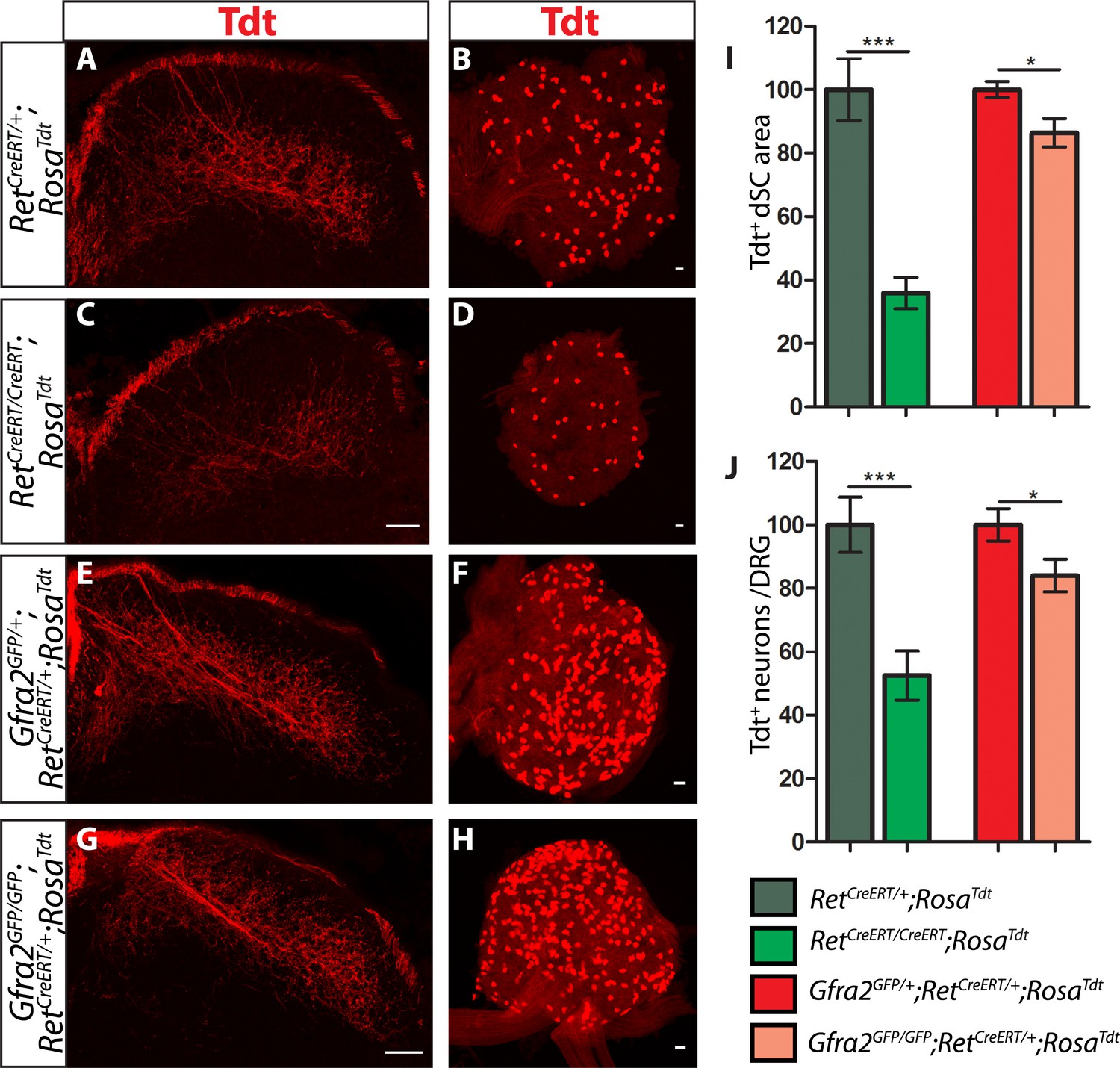
Ret and Gfra2 null mice display different central projection and cell survival deficits at E18.5.
(A–H) SC sections and whole mount L4/L5 DRGs of Tdt labeled RA mechanoreceptor from E18.5 RetCreERT/+;RosaTdt control (A–B), RetCreERT/CreERT;RosaTdt mutant (C–D), Gfra2GFP/+;RetCreERT/+;RosaTdt control (E–F), and Gfra2GFP/GFP;RetCreERT/+;RosaTdt mutant (G–H) embryos. (I) Quantification of Tdt+ pixels in dSC, which is displayed as a percentage normalized to dSC Tdt+ pixels of the within litter controls. (J) Quantification of the number of Tdt+ DRG neurons per whole-mount L4/L5 DRG, which is displayed as a percentage normalized to Tdt+ neurons of the within litter controls. Ret mutants have significant decreases in RA mechanosensory axons innervating the dSC and in the number of Tdt+ RA mechanoreceptors, suggesting that Ret mutants have deficits in both the growth of third order central projections and the survival of RA mechanoreceptors at E18.5. In contrast, Gfra2 nulls have only minor deficits in RA mechanosensory central projection growth and the survival or RA mechanoreceptors, suggesting that an additional GFRa2 independent but RET-dependent mechanism functions in these processes. Scale bar = 50 μm. Error bars represent SEM. * = p < 0.05, *** = p < 0.001. Source data are provided in Figure 4—source data 1.
-
Figure 4—source data 1
RA mechanoreceptor central projections and cell number in E18.5 Ret, Gfra2, Gfra1, and Gfra1;Gfra2 mutants.
- https://doi.org/10.7554/eLife.06828.014
In contrast, central projections of Tdt+ Gfra2 null RA mechanoreceptors are only slightly reduced at E18.5 (Figure 4E,G,I, Figure 4—source data 1, Gfra2 mutant has 86.34 ± 4.48% of control staining at thoracic levels [p = 0.01]). At P7, almost no difference is observed (data not shown). Similarly, the number of Tdt+ RA mechanoreceptors is only slightly reduced in Gfra2 null mice (Figure 4F,H,J Gfra2 mutant has 84.01 ± 5.16% of control cell number [p = 0.04]), indicating that extensive cell death of RA mechanoreceptors resulting from an absence of RET signaling does not occur in Gfra2 null mice. The discrepancy between E18.5 Ret and Gfra2 mutant phenotypes suggests that RET signaling still occurs in neonatal Gfra2 null RA mechanoreceptors. To demonstrate this, we quantified the expression of phospho-S6 ribosomal protein, which is downstream of RET/PI3K/mTOR signaling (Plaza-Menacho et al., 2010), in RA mechanoreceptors. We found that the proportion of GFP+ RA mechanoreceptors which express phospho-S6 in P0 Gfra2GFP/+ control and Gfra2GFP/GFP mutant DRGs was similar (Figure 4—figure supplement 1 [p = 0.51]). This result is consistent with the idea that RET activation occurs in neonatal RA mechanoreceptors without Gfra2.
Collectively, our results suggest that Gfra2 null RA mechanoreceptors display a central projection deficit at E13.5 but recover during later development, which explains the almost normal VGLUT1 staining in layers III–V of SC at P7. In addition, our data indicate that from E15.5, an additional GFRa2 independent but RET-dependent mechanism begins to play a role in promoting the survival and central projection growth of RA mechanoreceptors.
RET in RA mechanoreceptors is activated via both GFRa1 and GFRa2
To determine if this GFRa2-independent but RET-dependent mechanism requires GFRa1, we examined genetically labeled Gfra1+/−;RetCreERT/+;RosaTdt control and Gfra1−/−;RetCreERT/+;RosaTdt mutant SC and DRGs at E18.5. Similar to E13.5, neither RA mechanosensory central projections nor their number is significantly decreased in Gfra1 null mice (Figure 5A–D,I–J, Figure 4—source data 1), suggesting that simply disrupting trans activation of RET via GFRa1 is not sufficient to block Ret signaling in RA mechanoreceptors.
Figure 5
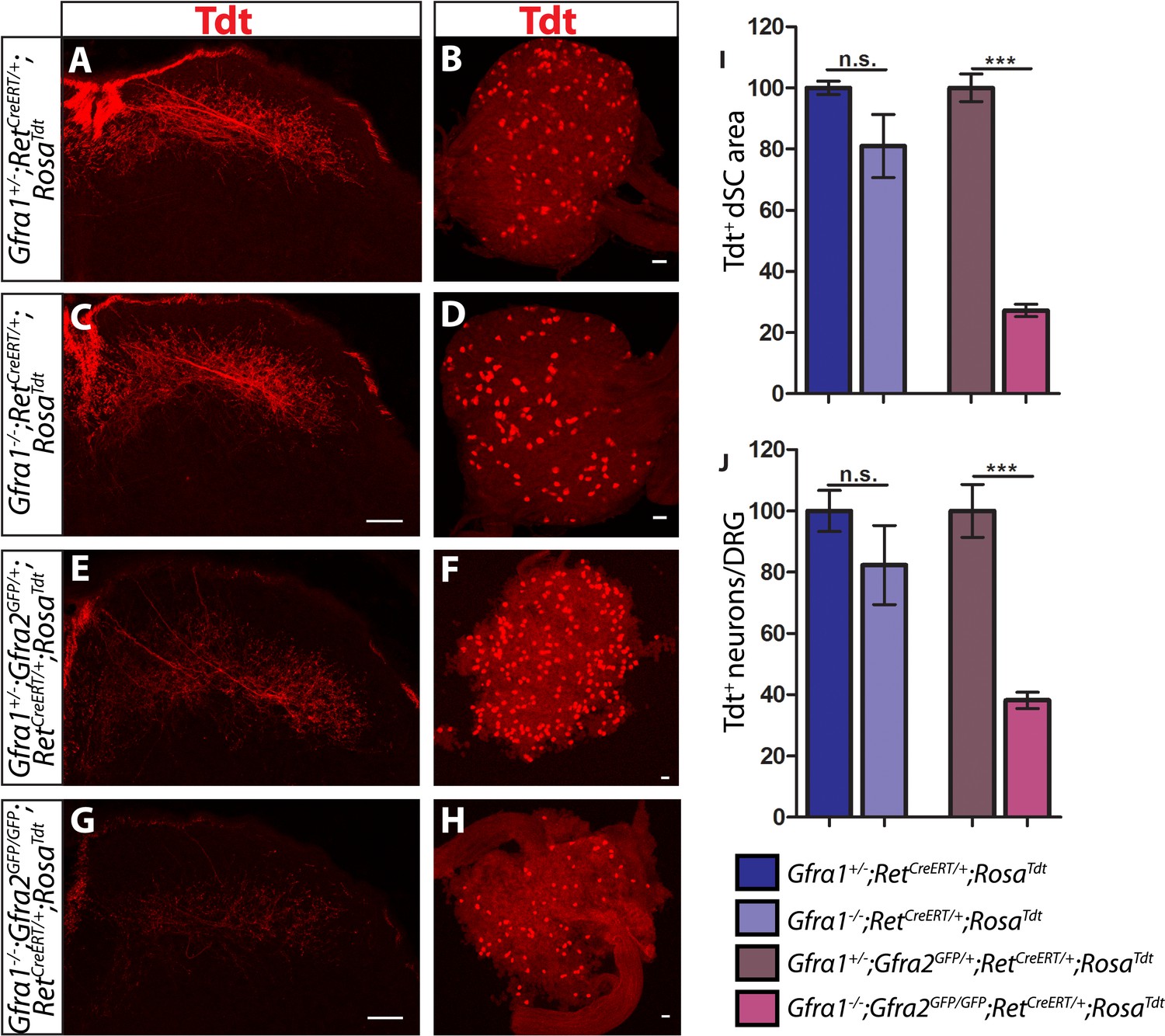
Gfra1;Gfra2 double null mice phenocopy Ret mutants at E18.5.
(A–H) SC sections and whole mount L4/L5 DRGs of Tdt labeled RA mechanoreceptors from E18.5 Gfra1+/−;RetCreERT/+;RosaTdt control (A–B), Gfra1−/−;RetCreERT/+;RosaTdt mutant (C–D), Gfra1+/−; Gfra2GFP/+;RetCreERT/+;RosaTdt control (E–F) and Gfra1−/−; Gfra2GFP/GFP;RetCreERT/+;RosaTdt double null (G–H) embryos. (I) Quantification of Tdt+ pixels in dSC, which is displayed as a percentage normalized to dSC Tdt+ pixels of the within litter controls. (J) Quantification of number of Tdt+ DRG neurons per DRG, which is displayed as a percentage normalized to Tdt+ neurons of the within litter controls. Gfra1 mutants have no significant deficits in RA mechanosensory third order projections or cell survival at E18.5, indicating that ablating trans signaling alone is not sufficient to disrupt the development of RA mechanoreceptors. However, loss of both cis and trans signaling in Gfra1;Gfra2 double nulls leads to a significant loss of RA mechanosensory third order projection growth and cell number, suggesting that both cis and trans RET signaling contribute to the development of RA mechanoreceptors. Scale bars = 50 μm. Error bars represent SEM. n.s. = p > 0.05, *** = p < 0.001. Source data are provided in Figure 4—source data 1.
The lack of Ret-mutant-like survival and central projection phenotypes of RA mechanoreceptors in both Gfra1 and Gfra2 single null mice made us wonder if cis and trans RET signaling function in the same developmental process and thus loss of one co-receptor can be compensated for by the other. To test this idea, we generated Gfra1;Gfra2 double knockout mice, in which RA mechanoreceptors were specifically labeled with Tdt using the RetCreERT;RosaTdt tandem allele. We examined control and double null SC sections and DRGs at E18.5. We found that Tdt+ RA mechanosensory central projections are greatly reduced in the dSC (Figure 5E,G,I, Figure 4—source data 1, Gfra1;Gfra2 double mutant has 27.25 ± 2.09% of control staining at thoracic levels [p < 0.001]). In addition, fewer Tdt+ RA mechanoreceptors remain in the double knockout DRGs (Figure 5F,H,J, Gfra1;Gfra2 double mutant has 38.17 ± 2.65% of control cell number [p < 0.001]), indicating that a significant number of RA mechanoreceptors die in the absence of Gfra1 and Gfra2. Strikingly, the extent of reduction in both cell number and central projections of RA mechanoreceptors is comparable between the Ret null and Gfra1:Gfra2 double null mice. Thus, our in vivo analyses strongly suggest that RET in RA mechanoreceptors is activated via both GFRa1 and GFRa2.
Gfra1 is not upregulated in Gfra2 null RA mechanoreceptors
Is RET in RA mechanoreceptors activated by GFRa1 in cis or trans? Although Gfra1 is not widely expressed in RA mechanoreceptors in wild-type mice, could it be upregulated to compensate for the loss of Gfra2 in the Gfra2 null mice? To address these questions, we conducted double fluorescent in situ hybridization of Gfra1 and GFP with E14.5 Gfra2GFP/+ control and Gfra2GFP/GFP null DRG sections. We found that a comparable low number of GFP+ neurons expressed Gfra1 transcript in both mutants and controls (Figure 6A–C [p = 0.52]), suggesting that Gfra1 is not upregulated in Gfra2 null RA mechanoreceptors. In addition, we performed in situ hybridization of Gfra1 with P0 Gfra2GFP/+;Ntrk1+/− control, Gfra2GFP/+;Ntrk1−/− null, and Gfra2GFP/GFP;Ntkr1−/− double null DRG sections. We previously showed (Luo et al., 2009) that Gfra1 is expressed in NTRK1+ DRG neurons and that the expression of Gfra1 is completely lost in Ntrk1 null mice. Here, we found that while Gfra1 expression was observed in Gfra2GFP/+;Ntrk1+/− control DRGs, no Gfra1 expression was observed in either Gfra2GFP/+;Ntkr1−/− null or Gfra2GFP/GFP;Ntkr1−/− double null DRGs (Figure 6D–F). This result indicates that the expression of Gfra1 in Gfra2 null DRG neurons still fully depends on NTRK1 signaling and thus it must be expressed in the non-RA mechanoreceptors. Moreover, we performed quantitative RT-PCR (QPCR) for Gfra1 transcripts in DRGs from E13.5, E15.5, and E18.5 Gfra2GFP/+ control and Gfra2GFP/GFP mutant embryos. We found no significant difference in the expression of Gfra1 between control and mutant DRGs at any stage (Figure 6—figure supplement 1, Figure 6—source data 1), suggesting that Gfra1 is not transcriptionally upregulated in DRG neurons upon Gfra2 ablation.
Figure 6 with 1 supplement see all
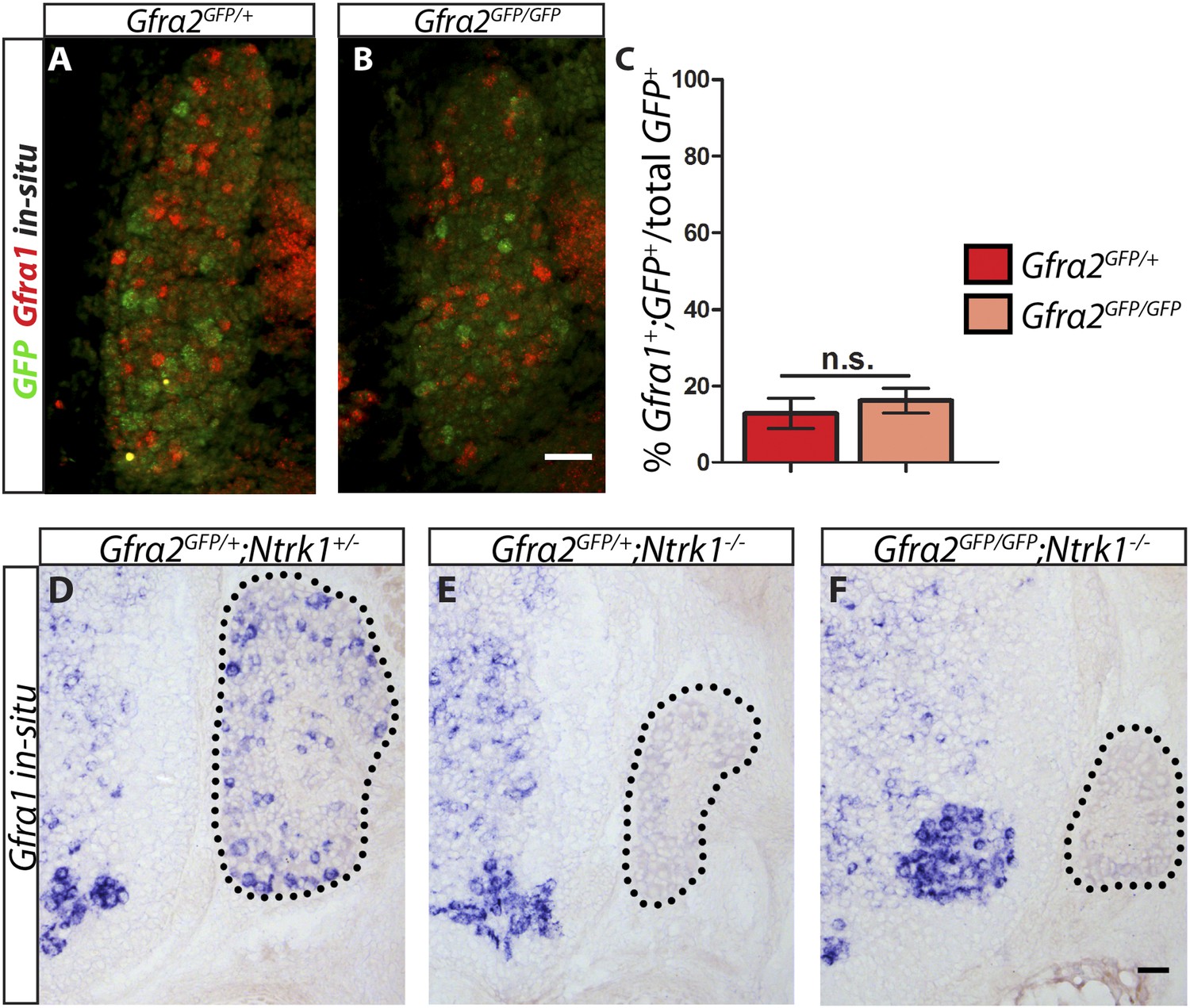
Gfra1 is not upregulated in Gfra2 null RA mechanoreceptors.
(A–B) Double fluorescent in situ hybridization against GFP and Gfra1 on E14.5 Gfra2GFP/+ control (A) and Gfra2GFP/GFP null (B) DRG sections. (C) Quantification of percentage of GFP+ neurons which co-express Gfra1. 12.81 ± 3.92% of control GFP+ neurons express Gfra1, and 16.17 ± 3.31% of Gfra2 null GFP+ neurons express Gfra1 (p = 0.52). The comparable low number of DRG neurons co-expressing GFP and Gfra1 in control and Gfra2 nulls suggests that Gfra1 normally is not expressed in most RA mechanoreceptors and that no upregulation of Gfra1 occurs in Gfra2 null RA mechanoreceptors. (D–F) In situ hybridization against Gfra1 in P0 Gfra2GFP/+;Ntrk1+/− control (D), Gfra2GFP/+;Ntrk1−/− null (E), and Gfra2GFP/GFP;Ntrk1−/− double null (F) DRG and SC sections. Black border outlines DRG. In control DRG sections, Gfra1 is expressed in some DRG neurons. In Gfra2GFP/+;Ntrk1−/− null DRG sections, Gfra1 transcript is not detected because the DRG neurons which normally express detectable levels of Gfra1 don't survive in the absence of Ntrk1. In Gfra2;Ntrk1 double null mice, no Gfra1 expression is detected in DRG neurons as well, which further supports that upregulation of Gfra1 doesn't occur in Gfra2 null RA mechanoreceptors. Scale bars = 50 μm. Error bars represent SEM. n.s. = p > 0.05.
-
Figure 6—source data 1
QPCR of Gfra1 in embryonic Gfra2 null DRGs.
- https://doi.org/10.7554/eLife.06828.018
GFRa1 produced by neighboring DRG neurons activates RET in RA mechanoreceptors in trans
Although Gfra1 transcript in most RA mechanoreceptors is below the detection level of in situ hybridization, it remains possible that an undetectable amount of GFRa1 could function in cis to promote RET signaling in RA mechanoreceptors. To exclude this possibility and to demonstrate that RET in RA mechanoreceptors is indeed activated by GFRa1 in trans, we used DRG explants from E14.5 embryos of different mutant backgrounds and treated these explants with NRTN, GDNF, GFRa1 plus GDNF, or GFRa1 alone.
In E14.5 explants harboring the RetCFP allele, the cell bodies and axons of RET+ neurons, some of which are RA mechanoreceptors, can be identified by anti-GFP immunostaining. We found that CFP+ neurons in RetCFP/+ control DRG explants grow long axons upon NRTN, GDNF, or GFRa1 plus GDNF, but not GFRa1 alone treatment (Figure 7—figure supplement 1A–D,I, and Figure 7—source data 1). In addition, the number of CFP+ DRG neurons is reduced in GFRa1 alone culture (Figure 7—figure supplement 1J, Figure 7—source data 2), suggesting that either cell death or down-regulation of Ret, and thus CFP expression, occur in the absence of RET signaling. Similarly, CFP+ neurons in RetCFP/CFP null DRG explants lost their responsiveness to GFLs completely (Figure 7—figure supplement 1E–H,I–J, Figure 7—source data 2), suggesting that this assay reflects RET-dependent signaling.
Next, we examined DRG explants harboring the Gfra2GFP allele, which drives a much lower level of GFP expression than RetCFP. Although some small diameter DRG neurons also express Gfra2 around P0 or later (Luo et al., 2007), in this Gfra2GFP mouse line GFP is mainly detected in the large diameter RA mechanoreceptors (Luo et al., 2009), which express a much higher level of Gfra2. Therefore, anti-GFP staining of E14.5 Gfra2GFP DRG explants should specifically show RA mechanoreceptors. Since GFP+ axons of these explants could not be reliably imaged and quantified due to the low level of GFP expression, we approximated the extent of RET signaling in Gfra2GFP DRG explants by quantifying the number of discernable GFP+ cell bodies. We found that Gfra2GFP/+ control DRG neurons show robust responses upon GFL application (Figure 7A–D,Q, Figure 7—source data 2). Interestingly, Gfra2GFP/GFP null DRG neurons lost their responsiveness to NRTN, but retain GFP expression in the presence of either GDNF or GFRa1 plus GDNF (Figure 7E–H,Q, Figure 7—source data 2). These results suggest that a GFRa2-independent but RET-dependent mechanism can mediate GDNF responsiveness of RA mechanoreceptors.
Figure 7 with 1 supplement see all
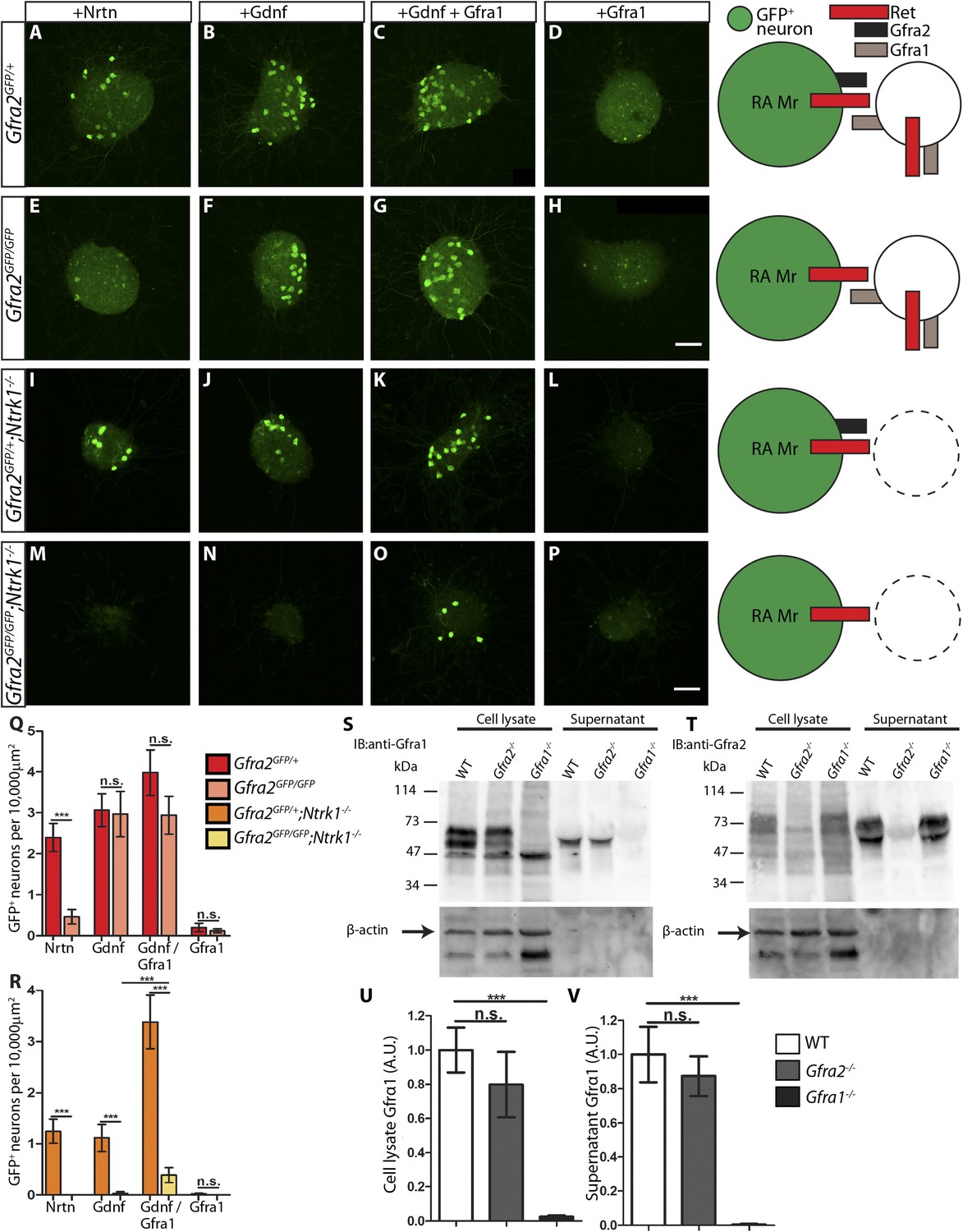
RA mechanoreceptors utilize GFRa1 produced by neighboring neurons to respond to GDNF.
(A–P) DRG explants from Gfra2GFP/+ control (A–D), Gfra2GFP/GFP null (E–H), Gfra2GFP/+;Ntrk1−/− null (I–L), and Gfra2GFP/GFP;Ntrk1−/− double null (M–P) embryos grown for 1 day in vitro and stained with anti-GFP antibody. Explants were treated with NRTN (50 ng/ml), GDNF (100 ng/ml), GDNF (100 ng/ml) plus GFRa1 (300 ng/ml), or GFRa1 (300 ng/ml), respectively. Schematic next to each genotype depicts the presence of RET and GFRas in each condition, and green color indicates cells detected by anti-GFP staining. (Q) Quantification of number of GFP+ neurons per 10,000 μm2 of explant in Gfra2GFP/+ control and Gfra2GFP/GFP null explants. GFP driven from the Gfra2 locus indicates RET signaling activity. Gfra2 control explants display many GFP+ neurons upon NRTN, GDNF, and GDNF plus GFRa1 treatment, but do not respond to GFRa1 alone. Gfra2 null explants lose their responsiveness to NRTN, but remain responsive to GDNF and GDNF plus GFRa1. (R) Quantification of number of GFP+ neurons per 10,000 μm2 of explant in Gfra2GFP/+;Ntrk1−/− null and Gfra2GFP/GFP;Ntrk1−/− double null explants. In a Ntrk1 null background, expression of Gfra1 is lost in non-RA-mechanoreceptor DRG neurons. Gfra2GFP/+;Ntrk1−/− null explants respond to NRTN, GDNF, and GDNF plus GFRa1. In this case, it is likely that GDNF activates RET signaling by interacting with GFRa2 (Jing et al., 1997; Sanicola et al., 1997; Rossi et al., 1999). In contrast, Gfra2;Ntrk1 double null DRG explants show GFP expression upon treatment with a combination of GDNF and GFRa1, but completely lose their responsiveness to GDNF. These results indicate that Gfra2 null RA mechanoreceptors do not express GFRa1 at a functional level and they depend on GFRa1 produced by neighboring NTRK1+ neurons to respond to GDNF. See Figure 7—source data 2 for quantification. (S–V) Western blot analysis of cell lysates and concentrated supernatants from cultured dissociated DRG neurons of E18.5-P1 wild-type, Gfra2 null, and Gfra1 null mice. (S) The specificity of the anti-GFRa1 antibody was confirmed by the loss of a doublet at the predicted size of GFRa1 in Gfra1 null cell lysates. GFRa1 was also detected in the supernatants of wild-type and Gfra2 null cultures, but not Gfra1 null cultures, indicating that GFRa1 is shed from the membrane of DRGs of both wild-type and Gfra2 mutants. Note that the size of cleaved GFRa1 is slightly smaller than that tethered to cells, which is consistent with previous publication (Paratcha et al., 2001). Following detection of GFRa1, membranes were stripped and probed for β-actin, which served as a loading control and confirmation that the supernatant fraction was not contaminated with cells or cellular debris (lower panel). (T) The specificity of the anti-GFRa2 antibody was confirmed by the loss of a band ∼75 kDa in Gfra2 null cell lysates. The larger than predicted size of GFRa2 may be due to post-translational modifications. Two GFRa2 specific bands were also detected in the supernatants of wild-type and Gfra1 null cultures, but not Gfra2 null cultures, indicating that GFRa2 is also shed from DRG cell membranes. The size of cleaved GFRa2 is also smaller than that tethered to cells. (U–V) Densimetric quantification of anti-GFRa1 blots shows no significant change in the level of GFRa1 produced by cells (U) or released into the media (V), which suggests that there is no compensation for the loss of GFRa2 through changes in the expression or release of GFRa1. See Figure 7—source data 3 for quantification. Error bars represent SEM. Scale bars = 50 μm. n.s. = p > 0.05, *** = p < 0.001. Source data are provided in Figure 7—source data 2, 3.
-
Figure 7—source data 1
Quantification of axonal growth in Ret mutant DRG explants.
- https://doi.org/10.7554/eLife.06828.021
-
Figure 7—source data 2
GFP+ neuron number in Gfra2 null and Gfra2;Ntrk1 double null explants.
- https://doi.org/10.7554/eLife.06828.022
-
Figure 7—source data 3
Densimetric measurements of GFRa1 in DRG cell extracts and supernatants.
- https://doi.org/10.7554/eLife.06828.023
How can Gfra2 null RA mechanoreceptors retain their responsiveness to GDNF? It could be due to: (1) a very low level of GFRa1 is expressed in RA mechanoreceptors, which activates RET in cis in the presence of GDNF; or (2) GFRa1 expressed by neighboring DRG neurons binds GDNF and activates RET in RA mechanoreceptors in trans. To differentiate between these possibilities, we cultured E14.5 Gfra2GFP/GFP;Ntrk1−/− double mutant DRGs. Since the expression of Gfra1 in non-RA mechanoreceptor DRG neurons fully depends on NTRK1 signaling, as shown previously (Luo et al., 2009) and above (Figure 6D–F), GFRa1 should be depleted from non-RA mechanoreceptors in Gfra2GFP/GFP;Ntrk1−/− double mutant DRGs. Therefore, if GFRa1 is expressed at a low level in RA mechanoreceptors and activates RET in cis, the double null explants should retain their responsiveness to GDNF. On the other hand, if GFRa1 expressed by neighboring neurons activates RET in RA mechanoreceptors in trans, the GDNF responsiveness would be lost in the Gfra2;Ntrk1 double nulls. Here, we found that Gfra2GFP/+;Ntrk1−/− control explants were responsive to NRTN, GDNF, and GDNF plus GFRa1, but not GFRa1 alone (Figure 7I–L,R, Figure 7—source data 2). In contrast, Gfra2GFP/GFP;Ntrk1−/− double null explants only respond to GDNF plus GFRa1, but not to NRTN, GDNF, or GFRa1 (Figure 7M–P,R, Figure 7—source data 2). The loss of responsiveness of RA mechanoreceptors to GDNF in Gfra2;Ntrk1 double null DRG explants strongly suggests that Gfra1 is not expressed at a functional level in RA mechanoreceptors and that RET in Gfra2 null RA mechanoreceptors is activated by exogenous GFRa1 from the neighboring DRG neurons in trans.
GFRa1 and GFRa2 are normally shed by DRG neurons
Trans activation of RET could occur by direct contact between membranes of cells which express either RET or GFRa1, or by soluble GFRa1 which is shed from the cell surface. To determine whether GFRa1 is released by DRG neurons, we cultured dissociated DRGs from E18.5-P1 wild-type, Gfra2−/−, and Gfra1−/− mice. We collected cell lysates and concentrated media from days 3–6 in vitro and then performed Western blot analysis.
Immunoblotting with anti-GFRa1 revealed a doublet at ∼55–65 kDa in wild-type and Gfra2−/− cell lysates, which was absent in the Gfra1−/− samples (Figure 7S, lanes 1–3), confirming the specificity of the anti-GFRa1 antibody. A positive band of ∼55 kDa was present in concentrated supernatants of wild-type and Gfra2−/− but not Gfra1−/− cultures (Figure 7S, lanes 4–6), suggesting that soluble GFRa1 is shed from neonatal DRG cells. Together with reports of GFRa1 being shed by Sciatic nerve Schwann cells, immortalized neuronal progenitors (Paratcha et al., 2001), and adult DRG explants (He et al., 2014), our findings indicate that GFRa1 can be released by many cell types during both developmental and adult stages. Therefore, it is possible that RET in RA mechanoreceptors is activated in trans by both soluble GFRa1 and GFRa1 tethered to the membranes of neighboring cells.
In addition, although there is no significant increase of Gfra1 transcripts in Gfra2 null DRGs by in situ or QPCR (Figure 6 and Figure 6—figure supplement 1), it remains possible that post-transcriptional regulation may occur to alter the translation, perdurance, or release of GFRa1. To test this possibility, we quantified the amount of GFRa1 in cell lysates and supernatants of wild-type and Gfra2−/− cultures by densitometry. We found that the amount of GFRa1 expressed in the cell or shed into the media was not significantly different between wild-type and Gfra2 null cultures (Figure 7U–V, Figure 7—source data 3). Therefore, Gfra2 null DRGs do not produce or release more GFRa1 protein to compensate for the loss of Gfra2.
We also investigated whether GFRa2 is normally shed by DRGs. The specificity of the anti-GFRa2 antibody was confirmed by the absence of a ∼75 kDa band from Gfra2 null cell lysates, which was present in both wild type and Gfra1 null cultures (Figure 7T, lanes 1–3). Furthermore, secreted GFRa2 band was also present in the supernatants of wild-type and Gfra1 DRG cultures, but not in Gfra2 null cultures. Therefore, both GFRa1 and GFRa2 are normally released by DRGs during early postnatal development.
Dynamic expression of Gdnf during development
As described above, the central projections of Gfra2 null RA mechanoreceptors display a severe, Ret-like deficit at E13.5, but begin to recover from E15.5, which is due to compensation by trans signaling via GDNF/GFRa1. Why is trans RET signaling able to compensate for the loss of cis signaling during late embryonic development, but not at E13.5? One possible reason for the delay is the availability of trans signaling components. Our in situ hybridization data suggest that Gfra1 is expressed at high levels at both E13.5 and E15.5, but the expression of Gdnf is greatly increased in DRGs from E13.5 to E15.5 (Figure 1—figure supplement 1). To provide additional evidence for the dynamic expression of Gdnf during development, we examined DRG and SC sections of E13.5 and E15.5 GdnfLacZ/+ (Moore et al., 1996) embryos using X-Gal staining. We found that the expression of LacZ increased significantly in DRGs from E13.5 to E15.5 (Figure 8A–E [p < 0.001], Figure 8—figure supplement 1). In addition, X-Gal staining was found in the E15.5 dorsal root, the pathway through which DRG central projections travel to reach the dSC (Figure 8A–D, black arrows). Thus, the expression of Gdnf seems to significantly increase in both the DRG and dorsal root from E13.5 to E15.5, providing a possible explanation for why the trans compensation occurs from E15.5.
Figure 8 with 1 supplement see all
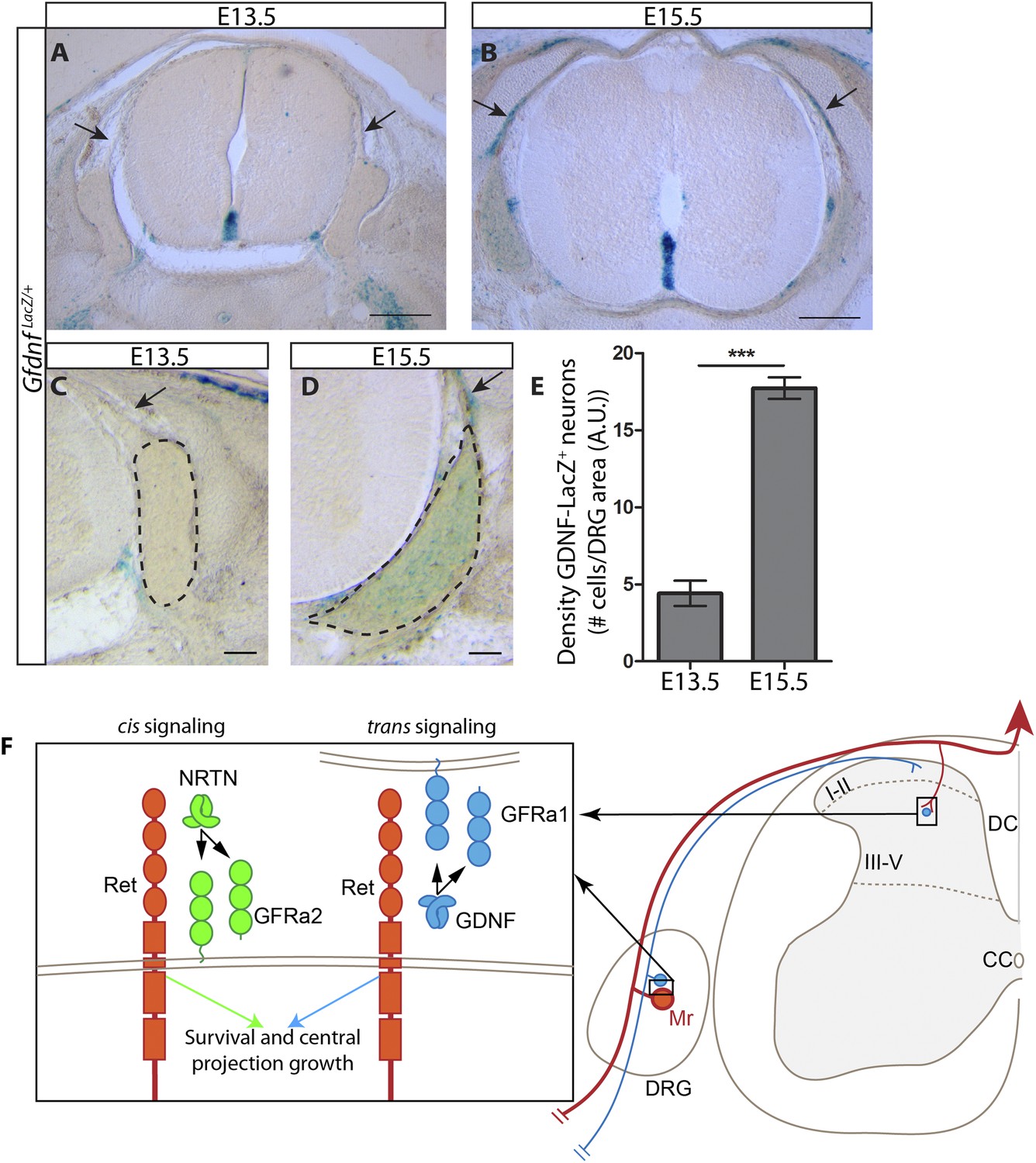
Dynamic expression of GDNF during development.
(A–D) X-Gal staining of E13.5 (A, C) and E15.5 (B, D) GdnfLacZ/+ DRG and SC sections (also see Figure 8—figure supplement 1). Arrows indicate dorsal roots, which express Gdnf at E15.5, but not E13.5. (E) Quantification of LacZ+ cells per DRG section, normalized to DRG area, reveals a significant increase in the number of cells expressing Gdnf from E13.5 to E15.5. E13.5 embryos have 4.41 ± 0.82 LacZ+ cells/unit area of DRG, E15.5 embryos have 17.73 ± 0.70 LacZ+ cells/unit area of DRG (p < 0.001). Error bars represent SEM. Scale bars = 200 μm (A–B), 100 μm (C–D). *** = p < 0.001 (F) Model of cis and trans signaling at cell bodies and central branches of RA mechanoreceptors. GFRa2 is co-expressed with RET in RA mechanoreceptors and can activate RET in cis. GFRa2 can also be shed from the membrane and may activate RET in its soluble form. GFRa1 is expressed in neighboring DRG neurons, dorsal root entry zone cells, and dSC cells. GFRa1 present at the membrane of these cells may directly contact the cell bodies or processes of RA mechanoreceptors to activate RET in trans. In addition, soluble GFRa1 released from these cells may also activate RET in RA mechanoreceptor in trans.
Discussion
In summary, we used RA mechanoreceptors as a model system to study the physiological functions of trans RET signaling and whether cis and trans activation of RET lead to the same or diversified biological outcomes in vivo. RA mechanoreceptors express Ret and Gfra2 and depend on Ret for their survival and the growth of central axonal projections into SC, whereas Gfra1 is highly expressed in the target field and neighboring DRG neurons. We found that the RA mechanosensory central projection deficit is negligible in postnatal Gfra2 and Nrtn mutant mice. We examined central projections of genetically traced RA mechanoreceptors in Ret, Gfra2, Nrtn, Gfra1, and Gfra1;Gfra2 double null mice and showed that only Gfra1;Gfra2 double null mice display similar cell death and central projection deficits to those of neonatal Ret mutant mice, indicating that RET in RA mechanoreceptors can be activated by both GFRa1 and GFRa2. Since Gfra1 is undetectable in control and Gfra2 null RA mechanoreceptors, it most likely activates RET in RA mechanoreceptors in trans. Finally, using DRG explant cultures, we determined that Gfra2 null RA mechanoreceptors respond to GDNF by utilizing GFRa1 produced by neighboring neurons, strongly suggesting that RET in RA mechanoreceptors is activated by GFRa1 in trans. Taken together, our results provide clear evidence that cis and trans RET signaling can function in the same development processes in vivo (Figure 8F) and that the existence of both cis and trans activation is likely to enhance but not diversify outcomes of RET signaling.
Trans activation of RET in vivo
Previous expression analyses revealed that Gfra1 is expressed more broadly than Ret, and cells which express Gfra1 usually lie adjacent to Ret-expressing cells (Trupp et al., 1997; Yu et al., 1998). This expression pattern suggests that GFRa1 may have RET-independent functions or that GFRa1 may interact with RET expressed on the surfaces of other cells in trans. Indeed, evidence for both ideas has been demonstrated. GFRa1 and GDNF interact with NCAM in neurons and Schwann cells to promote neurite outgrowth and Schwann cell migration (Paratcha et al., 2003; Nielsen et al., 2009). Additionally, GFRa1 and GDNF are required for the proper migration of cortical GABAergic interneurons and can act as ligand-dependent adhesion molecules for synapse formation, independent of RET and NCAM (Pozas and Ibáñez, 2005; Ledda et al., 2007). Recently, it was also shown that GFLs have additional roles in cortical development via interactions with Syndecan-3, likely independent of RET, GFRas, and NCAM (Bespalov et al., 2011). On the other hand, RET can be activated by GFRa1 and GDNF in trans using both heterologous cells and tissue explants (Paratcha et al., 2001; Ledda et al., 2002; Patel et al., 2012; He et al., 2014). Trans RET signaling may affect many cellular processes, including directional axonal outgrowth and promotion of axon regeneration (Paratcha et al., 2001; Airaksinen and Saarma, 2002; Ledda et al., 2002).
Evidence for physiologically relevant in vivo function of trans RET signaling, however, has remained less conclusive. Enomoto et al. (2004) generated a ‘cis-only’ mouse model in which Gfra1 is expressed in all RET-expressing cells, but not in cells that do not express RET. Using this model, they found that the major RET-dependent developmental processes were completely normal, suggesting that trans signaling is likely to be irrelevant for most RET-dependent processes. Results from this model, however, may not necessarily preclude a physiological role for trans signaling. Not only does this model present a loss of trans signaling, but it also presents a gain of function: Gfra1 is expressed at a high level in RET+ cells which may not normally express this co-receptor. If cis and trans RET activation lead to similar physiological outcomes, any deficits due to the loss of trans signaling may be masked by a gain of cis signaling. Indeed, the gain of function of GFRa1 was recently demonstrated in enteric hematopoietic cells derived from cis-only mice (Patel et al., 2012). Thus, whether trans RET signaling has any physiological function in development has remained an open question.
We aimed to address this question by analyzing the survival and growth of RA mechanosensory central projections using loss-of-function mouse lines. Here, we found that loss of cis signaling, via ablation of either Gfra2 or Nrtn, produces a central projection deficit during early embryonic development (Figure 2). Our findings are consistent with previous observations using a different Ret knock-in line and NRTN ectopic expression (Honma et al., 2010), but differ from the findings using anti-GFRa2 staining to visualize RA mechanosensory central projections at E13.5 (Bourane et al., 2009). This is likely due to different subcellular localization of CFP and GFRa2, as well as the expression of GFRa2 by some dSC cells (Figure 1—figure supplement 1E,F), which may mask the RA mechanoreceptor phenotype. Interestingly, this phenotype recovers during late embryonic development and the central projections of Gfra2 null mice seem nearly normal postnatally (Figures 1, 4). Thus, our results suggest that cis RET signaling is required for the initial growth of RA mechanosensory central projections, but an additional cis signaling independent process takes place during later development. Indeed, the loss of both cis and trans signaling in Gfra1;Gfra2 double mutants recapitulates the Ret phenotype (Figures 5, 6). Furthermore, using DRG explant and dissociated culture, we demonstrated that soluble GFRa1 is normally released by DRGs and that GFRa1 produced by NTRK1+ DRG neurons present a potential source to activate RET in RA mechanoreceptors in trans to promote their survival (Figure 7). Taken together, our results suggest that trans RET signaling contributes to the development of RA mechanoreceptors in vivo.
Nevertheless, the exact subcellular locus of trans RET activation in RA mechanoreceptors remains speculative. The expression pattern of Gfra1 suggests that trans RET activation is possible in the axons of RA mechanoreceptors along their path to dSC, and/or at the cell body within the DRG. Although individual DRG cell bodies are surrounded by satellite glial cells, large macromolecules and proteins are able to invade the space between the neuron and satellite cell (Hanani, 2005), suggesting trans RET activation by soluble GFRa1 could occur within DRGs.
RET signaling and the survival of RA mechanoreceptors
Signaling of neurotrophic RTKs, such as NTRK1, NTRK2, and NTRK3, is critical for the specification and survival of numerous classes of neurons (Ernsberger, 2009). RET signaling also plays important roles in survival, differentiation, and specification of distinct neuronal classes (Enomoto, 2005). For example, RET signaling components are absolutely required for the survival of enteric neurons (Taraviras et al., 1999), but their roles in DRG neuron survival are more complicated to dissect. Previously, it was reported that the number of total DRG neurons is not significantly reduced in neonatal and early postnatal Ret mutants (Luo et al., 2007). At that time, specific molecular markers or genetic approaches for labeling RA mechanoreceptors had not been identified, so it was impossible to specifically assay the role of RET signaling in the survival of this neuronal population. Given that RA mechanoreceptors represent a very small proportion of the total DRG neurons (Molliver et al., 1997; Luo et al., 2007), a partial loss of this population may not lead to a significant change in cell counts of total DRG neurons.
In another paper (Luo et al., 2009), it was proposed that RA mechanoreceptors depend on NRTN-GFRa2/Ret signaling for their development. This was based on the findings that GFRa2 is the only co-receptor expressed in RA mechanoreceptors and that Ret, Gfra2, and Nrtn null mice display the same no-Pacinian-corpuscle phenotype. Since RET can't be used as a molecular marker to quantify the number of RA mechanoreceptors in Ret null mice, the number of P0 RET+/NTRK1− and Gfra2GFP DRG neurons, most of which indicate the RA mechanoreceptors, was quantified in Nrtn and Gfra2 nulls. No significant change in cell number between mutants and controls was found, suggesting that Gfra2 and Nrtn are not required for survival of neonatal RA mechanoreceptors. These results are interesting in light of the current findings. Here, we show that when cis signaling via NRTN/GFRa2 is perturbed (as was tested in [Luo et al., 2009]), trans signaling via GDNF/GFRa1 can activate RET in RA mechanoreceptors to support their survival and central projection growth. When the number of genetically labeled RA mechanoreceptors was quantified in different mutant backgrounds, we found only marginal changes in Gfra2 nulls but drastic decreases in Ret mutants at E18.5 (Figure 4). The slight difference between the current and previous findings regarding the loss of RA mechanoreceptors in Gfra2 nulls at P0 (Luo et al., 2009) is likely due to different methods in identifying and quantifying RA mechanoreceptors. In short, the current study clarifies that RET signaling is required for RA mechanoreceptor survival but simple disruption of cis RET signaling components may not reveal this deficit.
Co-existence of cis and trans RET signaling
What is the purpose for RET to be activated both in cis and in trans? Do cis and trans signaling activate different cellular responses and influence distinct developmental processes, or do cis and trans signaling exert similar physiological effect? Here, we found that, in RA mechanoreceptors, cis and trans signaling seem to produce similar biological outputs in vivo. Our results demonstrate that cis and trans signaling can compensate for the loss of each other to promote both the central projection growth and survival of RA mechanoreceptors (Figure 8F). This compensatory ability suggests that the existence of both cis and trans activation is likely to enhance but not diversify outcomes of RET signaling. Consistent with this notion, a recent study found that peripheral nerves secrete both GDNF and GFRa1, which attracts perineural invasion of heterogeneous cancer cells, some of which expresses Ret and Gfras, while some express only Ret (He et al., 2014).
In an attempt to show that GFRa1 is normally released from wild-type DRG cells for trans RET signaling, we found the same for GFRa2 (Figure 7). Given that soluble GFRa2 could also activate RET in trans with NRTN (Worley et al., 2000), our finding raises many interesting questions, such as whether all GFRas are secreted and whether ‘cis’ and ‘trans’ RET signaling normally co-exist even when RET and GFRas are expressed in the same cells. It seems plausible that even for GFRas co-expressed with RET (usually defined as ‘cis’ signaling), such as GFRa2 in RA mechanoreceptors, it could be secreted and then upon NRTN binding activates RET in the cell from which it was released (‘trans’ activation).
Although cis and trans activation of RET lead to a similar biological outcome in the growth and survival of RA mechanoreceptors, it is worth noting that substantial differences likely exist between the signaling processes of cis and trans RET activation. Cis RET activation might be more efficient, given that GFRas and RET are located in the same membrane. In addition, cis and trans signaling could differ in the kinetics of recruitment of RET to lipid rafts upon GFLs stimulation, interactions with downstream-associated proteins, and the longevity of activated RET and downstream effectors (Tansey et al., 2000; Paratcha et al., 2001). Additionally, it remains possible that although the gross structure of the RA mechanosensory central projections seems mostly normal in mice lacking either cis or trans signaling, more precise aspects of circuit formation, such as specific synapse formation, which are beyond the resolution of current analysis, may differentially depend on cis or trans signaling. Finally, it will be interesting to see whether cis and trans signaling can produce similar biological outcomes in other systems, as we have shown here for RA mechanoreceptors. Future experiments to carefully dissect cis and trans RET signaling in other types of cells and tissues will address these issues.
Materials and methods
Mouse strains
Request a detailed protocolMice except GDNFlacZ line were raised in a barrier facility in Hill Pavilion, the University of Pennsylvania. All procedures were conducted according to animal protocols approved by Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and National Institutes of Health guidelines. GDNFlacZ mice were raised in accordance with the European Community Council Directive of November 24, 1986 (86/609/EEC), and approved by the ethics. Most mice used in this paper were described previously: RetCreERT, RetCFP, Nrtn+/− (purchased from the Jackson lab), Gfra2GFP (re-derived using sperm provided by Dr Jeffery Milbrandt at the Washington University), Rosa26Tdt, and GdnfLacZ mice (Moore et al., 1996; Heuckeroth et al., 1999; McDonagh et al., 2007; Uesaka et al., 2008; Luo et al., 2009; Madisen et al., 2010). The Ntrk1− allele was generated by crossing the floxed TrkAF592A allele (Chen et al., 2005) to germline Cre mice. The generation of Gfra1 conditional and null mice and the RetCreERT;RosaTdt tandem allele are described below. All mice except GdnfLacZ were maintained on a mixed C57BL/6J and CD1 background. GdnfLacZ mice were maintained on a C57BL/6N background. Except for Gfra1;Gfra2 double null animals (n = 2), at least three animals per genotype were examined. N-values for explants are listed in Figure 7—source data 1, 2.
Generation of Gfra1 conditional and null alleles
Request a detailed protocolWe generated Gfra1 conditional knockout mice, in which loxP sites flank exon 6 of Gfra1, by homologous recombination. Mice harboring the floxed allele were crossed to germ line Cre mice, resulting in a Gfra1 allele lacking exon 6. The loss of Gfra1 transcript in Gfra1−/− mice was confirmed by in situ hybridization of mutant and control DRGs (see Figure 1—figure supplement 3).
Generation of RetCreERT;RosaTdt tandem allele
Request a detailed protocolSince Ret and Rosa26 loci are located only ∼5 megabases apart on mouse chromosome 6, we generated a tandem configuration of RetCreERT and RosaTdt alleles, which are linked during meiosis and became a great genetic advantage for our experiments (Figure 3—figure supplement 1). We used this tandem RetCreERT;RosaTdt allele to specifically label RA mechanoreceptors in different mutant mouse lines described in the text.
Genetic labeling of RA mechanoreceptors
Request a detailed protocolWe set up timed pregnancy mating for mice in the evening and checked mice for vaginal plugs the following morning. The time when a female mouse was found to have a plug was counted as embryonic day 0.5 (E0.5). We treated embryos harboring the RetCreERT;RosaTdt reporter allele with 4-hydroxy-tamoxifen (4-HT, 2 mg and 1 mg at E11.5, and E12.5, respectively) by oral gavage to pregnant female mice to specifically label RA mechanoreceptor population.
Tissue preparation and histology
Request a detailed protocolSpinal columns of embryos and neonatal mice at the desired developmental stages were dissected out and directly immersed in PBS/4% paraformaldehyde (PFA) for 2 to 4 hr at 4°C. Postnatal mice were sacrificed with CO2, transcardially perfused with 4% PFA, and spinal columns were dissected out and post-fixed with 4% PFA for 2 hr at 4°C. They were then cryo-protected in 1× PBS, 30% sucrose overnight. 20-µm frozen sections of SC and DRGs were cut using a CM1950 cryostat (Leica, Buffalo Grove, IL). Immunostaining of SCs and DRG sections were performed as described previously (Niu et al., 2013). Antibodies used are as follows: rabbit anti-GFP (A-11122, 1:2000, Invitrogen, Carlsbad, CA), chicken anti-GFP (GFP-1020, 1:1000, Aves, Tigard, OR), chicken anti-NF200 (NF-H, 1:500, Aves), rabbit anti-NF200 (N4142, 1:1000, Sigma, St. Louis, MO), rabbit anti-cRet (18121, 1:50, IBL, Minneapolis, MN), rabbit anti-NTRK1 (06-574, 1:1000, Millipore, Temecula, CA), guinea pig anti-VGLUT1 (AB5905, 1:1000, Millipore), rabbit anti-phospho-S6 (2215s, 1:200, Cell Signaling, Beverly, MA), and Alexa Fluorescent conjugated Goat or Donkey secondary antibodies (1:500, Invitrogen or Jackson ImmunoResearch, West Grove, PA).
LacZ color reaction
Request a detailed protocolEmbryos of the desired age were eviscerated and fixed in 1% PFA, 2 mM MgCl2, 5 mM EGTA, 0.02% NP40, and 0.2% glutaraldehyde in phosphate buffer (pH 7.4) for 1.5 to 2 hr at 4°C. Vibratome sections were incubated for 30 min in washing solution (2 mM MgCl2, 0.02% NP-40 in phosphate buffer pH 7.4). LacZ reaction was developed with X-gal (1 mg/ml) at 37°C.
In situ hybridization
Request a detailed protocolDIG- or FITC-labeled riboprobes were synthesized using a DIG or FITC RNA labeling kit (11175025910, Roche, Indianapolis, IN). Template for GFP was amplified by PCR and subcloned into vector pGEM-T Easy (A1360, Promega, Madison, WI). Antisense RNA probes for Ret, Gfra1, Gfra2, Gdnf, and Nrtn were generated as previously described (Luo et al., 2009). The detailed procedures of in situ hybridization and double fluorescent in situ hybridization were performed as described previously (Fleming et al., 2012).
QPCR
Request a detailed protocolDRGs from E13.5, E15.5, and E18.5 Gfra2GFP/+ and Gfra2GFP/GFP embryos were dissected and rapidly frozen on dry ice. RNA was extracted with the GeneJet RNA Purification Kit (K0731, Fermentas, Vilnius, Lithuania) and cDNAs were generated using Super-Script III First-Strand Synthesis System (18080-51, Invitrogen). 500 ng of total RNA was used for each RT reaction in a total volume of 25 μl. QPCR reactions were performed in triplicate for three samples of each age and genotype. QPCR reactions contained SYBR Green PCR master mix (4309155, Life Technologies, Carlsbad, CA), 0.5 μM of each primer, and 3 μl (for Gfra1) or 1 μl (for Gapdh) of cDNA template per 15 μl reaction. Reactions were run and analyzed on a StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Primers used were Gapdh (5′-CCACCAACTGCTTAGCCCCC-3′ and 5′-GCAGTGATGGCATGGACTGTGG-3′) and Gfra1 (5′-TGTCTTTCTGATAATGATTACGGA-3′ and 5′-CTACGATGTTTCTGCCAATGATA-3′). p-values between samples were calculated from ΔCT values with the Student's t-test, and relative concentrations were calculated by the 2−ΔΔCT method (Livak and Schmittgen, 2001).
DRG explant culture and immunostaining
Request a detailed protocolE14.5 embryos were removed from the dam and placed in F-12 media (11765-047, Invitrogen) on ice. SCs with attached DRGs were dissected from the spinal column, and individual DRGs were removed and placed in fresh F-12 on ice. Using a dissecting needle, DRGs were cleaned and bisected, and then placed in fresh F-12. Explants were grown on Superfrost Plus slides (22-034-979, Fisher, Waltham, MA) coated with poly-L-lysine (P1274, Sigma, 0.1 mg/ml in ddH2O overnight at 4°C) and laminin (354232, BD, Franklin Lakes, NJ), 20 μg/ml in HBSS [14170122, Invitrogen] at 37°C for one to 3 hr). Immediately before placing explants on the slide, slides were washed with HBSS and DRG culture medium (Neurobasal medium [21103-049, Invitrogen], 1× B27 [17504-044, Invitrogen], 100 U/ml penicillin/streptomycin [15140-122, Invitrogen], 2 mM L-glutamine [25030-081, Invitrogen], and 35 mM glucose). DRG culture media supplemented with the appropriate recombinant proteins (50 ng/ml Nrtn [477-MN-025, R&D, Minneapolis, MN], 100 ng/ml GDNF [512-GF-010, R&D], 300 ng/ml GFRa1 [560-GF-100, R&D], or 100 ng/ml GDNF and 300 ng/ml GFRa1) were added to the culture dish. Four to six DRG explants were placed on each slide and the culture dishes were carefully moved to a 37°C incubator and left undisturbed overnight. Following 16–24 hr of incubation, cultures were rinsed with PBS and fixed in 4% PFA in PBS for 30 min at room temperature. Immunofluorescence was then performed directly in the culture dish using antibody dilutions described above. Following secondary antibody, explant slides were mounted on microscope slides using Superglue, and coverslipped with Fluoromount-G (0100-01, Southern Biotech, Birmingham, AL) and 22 × 22 mm coverglass.
Dissociated DRG cultures and biochemistry
Request a detailed protocolDRGs from E18.5-P1 mice were collected into HBSS on ice. DRGs were first digested in 0.5 mg/ml collagenase (LS4186, Worthington, Lakewood, NJ) plus 100 U/ml penicillin/streptomycin, 10 mM HEPES, and 1× MEM vitamins (M6895, Sigma) in MEM (11095072, Invitrogen) at 37°C for 30 min, followed by a second digestion with 0.05% trypsin (25200056, Invitrogen) plus 100 U/ml penicillin/streptomycin, 10 mM HEPES, and 1× MEM vitamins in MEM at 37°C for 30 min. Digestion was stopped by adding 5% FBS and 10 mM HEPES in HBSS. Cells were then triturated with a fire polished Pasteur pipette to a homogenous solution. The cells were then pelleted at 500×g for 5 min and resuspended in DRG culture media, as described above, supplemented with 50 ng/ml NRTN, 100 ng/ml GDNF, and 50 ng/ml NGF (556-NG-100, R&D). Cells were plated in six-well collagen-coated plates (Millipore, PICL06P05) and cultured at 37°C and 5% CO2. After 2 days, media were removed and cells were rinsed with warmed Neurobasal media. 2 ml of fresh DRG culture media supplemented with NRTN, GDNF, and NGF (but without B27) was added to each well. After 2 days, media were removed and saved at 4°C with added protease inhibitors (P8340, Sigma). Fresh media supplemented with growth factors but without B27 were then added to each well. After an additional 2 days, media were removed and pooled with previously collected media, and additional protease inhibitor was added. The cells were rinsed twice with PBS, and then lysed directly in the well by the addition of 70 μl 2× sample buffer (0.125 M Tris pH 6.8, 20% glycerol, 4% SDS, 0.16% bromophenol blue, 10% 2-mercaptoethanol) and scraping, followed by heating at 95°C for 5 min. All cell lysates were then brought to a total volume of 140 μl with 1× PBS. Supernatants were centrifuged at 14,000×g for 15 min to clear cellular debris, and then were concentrated to ∼30 μl with Amicon 30 kDa filters (UFC503024, Millipore), then mixed with an equal volume of 2× sample buffer and heated at 95°C for 5 min.
Duplicate 4–15% gradient mini-Protean TGX gels (456-1084, Biorad, Hercules, CA) were used to run samples. 40 μl of cell lysate of each genotype or one third of the total volume of concentrated supernatant of each genotype was used. Both gels were then transferred to nitrocellulose membrane and blocked in 3% BSA in TBS plus 0.1% Tween-20 (TBST) for 1 hr at room temperature. Membranes were then incubated overnight with either goat anti-GFRa1 (0.2 μg/ml, AF560, R&D) or goat anti-GFRa2 (0.2 μg/ml, AF613, R&D) in blocking solution overnight at 4°C. Following washes with TBST, membranes were incubated with donkey anti-goat-AP (SC-2022, 1:5000, Santa Cruz Biotechnology, Santa Cruz, CA) in blocking solution for 1 hr at room temperature. After washes, AP was detected with CDP-Star (T2218, Applied Biosystems) and membranes were imaged with a Chemi-Doc system (BioRad).
Following imaging, membranes were stripped with 2× 10 min stripping buffer (0.2 M glycine, 0.1% SDS, 1% Tween-20, pH 2.2), followed by 2× 10 min wash with PBS and 2× 5 min wash with TBST. Membrane was then probed with rabbit anti-β-actin (sc-130656, 1:400, Santa Cruz Biotechnology) and goat anti-rabbit-AP (T2191, 1:5000, Applied Biosystems) following the above procedure, except that all blocking and antibody incubations were performed in 5% milk in TBST.
Western blot densitometry was performed with ImageJ. Three cultures per genotype were analyzed. Densitometry measurements for each antibody were performed on three blots running independent culture samples. Relative quantifications were performed using β-actin in the cell lysates as a measure of total protein per lane, and optical density values for GFRa1 were scaled accordingly. Because an equal proportion of total lysates was run in each lane, total β-actin per cell lysate lane was used as a proxy for cell number, and was therefore used to normalize protein levels in the supernatant lanes (equal proportion of total supernatant volume were run in each lane). Cell lysate and supernatant samples were scaled to wild-type quantifications of respective sample type and reported in arbitrary units. Student's t-test was used to measure significance of differences between genotypes.
Image acquisition
Request a detailed protocolFluorescent images of SC/DRG sections were acquired on a Leica SP5II confocal microscope. DRG explant cultures were imaged on Leica DM5000B microscope. Bright field images were taken using Leica DM5000B microscope.
Quantification and Statistics
Request a detailed protocolFor histological analysis, at least six sections per specified spinal/DRG level per animal were analyzed. For quantification of genetically labeled neuron number in E18.5 embryos, whole-mount L4/L5 DRGs were imaged and total Tdt+ cell number was counted in each DRG. Except for Gfra1;Gfra2 double null animals (n = 2), at least three animals per genotype were examined. N-values for explants are listed in Figure 7—source data 1 and 2. Pixel counts for central projections were generated by counting the number of pixels at each intensity level (0–256) in an outlined immunoreactive area in ImageJ. Background staining was subtracted by counting pixel number of each intensity level in a non-immunoreactive region of the tissue. The minimal intensity level at which two consecutive levels displayed a pixel count of zero was taken as the threshold cut of background fluorescence. Pixel counts of real staining were then calculated by summing the pixel counts for all intensity levels above the defined background level. Column graphs were generated in GraphPad Prism 5. All error bars are ± standard error of the mean (SEM), unless otherwise specified. All statistical analyses were performed using SAS version 9.3 (SAS Inc., Cary, NC). Due to differences in labeling efficiency across litters in 4-HT treated mice, quantification for SC section staining and whole mount DRGs were performed by normalizing to controls within the same litter. For all explant quantifications, GFP+ neuron number per 10,000 μm2 was calculated for each explant. For RetCFP explants, a circle with a radius 200 μm larger than the explant was drawn around the explant in ImageJ, and the number of CFP+ axons which crossed the circle was counted.
References
-
The GDNF family: signalling, biological functions and therapeutic valueNature Reviews Neuroscience 3:383–394.https://doi.org/10.1038/nrn812
-
Retrograde signaling onto Ret during motor nerve terminal maturationThe Journal of Neuroscience 28:963–975.https://doi.org/10.1523/JNEUROSCI.4489-07.2008
-
GDNF family receptor complexes are emerging drug targetsTrends in Pharmacological Sciences 28:68–74.https://doi.org/10.1016/j.tips.2006.12.005
-
Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and arteminThe Journal of Cell Biology 192:153–169.https://doi.org/10.1083/jcb.201009136
-
Regionalized loss of parvalbumin interneurons in the cerebral cortex of mice with deficits in GFRalpha1 signalingThe Journal of Neuroscience 29:10695–10705.https://doi.org/10.1523/JNEUROSCI.2658-09.2009
-
Regulation of neural development by glial cell line-derived neurotrophic factor family ligandsAnatomical Science International 80:42–52.https://doi.org/10.1111/j.1447-073x.2005.00099.x
-
RET signaling is required for survival and normal function of nonpeptidergic nociceptorsThe Journal of Neuroscience 30:3983–3994.https://doi.org/10.1523/JNEUROSCI.5930-09.2010
-
Satellite glial cells in sensory ganglia: from form to functionBrain Research Brain Research Reviews 48:457–476.https://doi.org/10.1016/j.brainresrev.2004.09.001
-
GFRalpha1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signalingProceedings of the National Academy of Sciences of USA 111:E2008–E2017.https://doi.org/10.1073/pnas.1402944111
-
Structure and physiology of the RET receptor tyrosine kinaseCold Spring Harbor Perspectives in Biology 5:.https://doi.org/10.1101/cshperspect.a009134
-
GFRalpha-2 and GFRalpha-3 are two new receptors for ligands of the GDNF familyThe Journal of Biological Chemistry 272:33111–33117.https://doi.org/10.1074/jbc.272.52.33111
-
GDNF and GFRalpha1 promote formation of neuronal synapses by ligand-induced cell adhesionNature Neuroscience 10:293–300.https://doi.org/10.1038/nn1855
-
Role of glial cell-line derived neurotropic factor family receptor alpha2 in the actions of the glucagon-like peptides on the murine intestineAmerican Journal of Physiology Gastrointestinal and Liver Physiology 293:G461–G468.https://doi.org/10.1152/ajpgi.00424.2006
-
Role of glial cell line-derived neurotrophic factor (GDNF)-neural cell adhesion molecule (NCAM) interactions in induction of neurite outgrowth and identification of a binding site for NCAM in the heel region of GDNFThe Journal of Neuroscience 29:11360–11376.https://doi.org/10.1523/JNEUROSCI.3239-09.2009
-
Modality-based organization of ascending somatosensory axons in the direct dorsal column pathwayThe Journal of Neuroscience 33:17691–17709.https://doi.org/10.1523/JNEUROSCI.3429-13.2013
-
Glial cell line-derived neurotrophic factor and developing mammalian motoneurons: regulation of programmed cell death among motoneuron subtypesThe Journal of Neuroscience 20:5001–5011.
-
Initial trajectories of sensory axons toward laminar targets in the developing mouse spinal cordThe Journal of Comparative Neurology 380:215–229.https://doi.org/10.1002/(SICI)1096-9861(19970407)380:23.0.CO;2-6
-
Glial cell line-derived neurotrophic factor-dependent RET activation can be mediated by two different cell-surface accessory proteinsProceedings of the National Academy of Sciences of USA 94:6238–6243.https://doi.org/10.1073/pnas.94.12.6238
-
Central role of RET in thyroid cancerCold Spring Harbor Perspectives in Biology 5:a009233.https://doi.org/10.1101/cshperspect.a009233
-
Signalling by the RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous systemDevelopment 126:2785–2797.
-
Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-alpha indicates multiple mechanisms of trophic actions in the adult rat CNSThe Journal of Neuroscience 17:3554–3567.
-
Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in miceThe Journal of Clinical Investigation 118:1890–1898.https://doi.org/10.1172/JCI34425
-
Developmental regulation of GDNF response and receptor expression in the enteric nervous systemDevelopment 127:4383–4393.
-
Expression of GDNF family receptor components during development: implications in the mechanisms of interactionThe Journal of Neuroscience 18:4684–4696.
Article and author information
Author details
Funding
National Institutes of Health (NIH) (R00NS069799)
- Wenqin Luo
March of Dimes Foundation (Basil O'Connor Starter Scholar Research Award, 5-FY12-109)
- Wenqin Luo
Klingenstein Third Generation Foundation (KTGF) (Klingenstein-Simons Fellowship Awards in the Neurosciences)
- Wenqin Luo
Max-Planck-Gesellschaft (Research Grant)
- Sόnia Paixão
- Rüdiger Klein
National Institutes of Health (NIH) (F31NS086168)
- Michael S Fleming
Hearst Foundations (Student Fellowship)
- Michael S Fleming
National Institutes of Health (NIH) (1R01NS083702)
- Wenqin Luo
National Institutes of Health (NIH) (T32HD007516)
- Michael S Fleming
National Institutes of Health (NIH) (T32GM07517)
- Michael S Fleming
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Drs G Bashaw, DD Ginty, M Ma, B Pierchala, and other Luo lab members for helpful comments on the manuscript. We thank Ming Lu for statistical consultation. WL is supported by National Institutes of Health (NIH, R00NS069799 and 1R01NS083702), the Basil O'Connor Starter Scholar Research Award (Grant No. 5-FY12-109 from the March of Dimes Foundation), and the Klingenstein-Simons Fellowship Awards in the Neurosciences. MF is supported by NIH (F31NS086168, T32HD007516, and T32GM07517) and the Hearst Foundation. Work in the Klein lab is supported by the Max-Planck Society.
Ethics
Animal experimentation: Mice except GDNFlacZ line were raised in a barrier facility in Hill Pavilion, the 618 University of Pennsylvania. All procedures were conducted according to animal protocols 619 approved by Institutional Animal Care and Use Committee (IACUC) of the University of 620 Pennsylvania and National Institutes of Health guidelines. GDNFlacZ mice were raised in 621 accordance with the European Community Council Directive of November 24, 1986622 (86/609/EEC), and approved by the ethics.
Copyright
© 2015, Fleming et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,670
- views
-
- 481
- downloads
-
- 33
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 33
- citations for umbrella DOI https://doi.org/10.7554/eLife.06828
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cis and trans RET signaling control the survival and central projection growth of rapidly adapting mechanoreceptors
eLife 4:e06828.
https://doi.org/10.7554/eLife.06828




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}