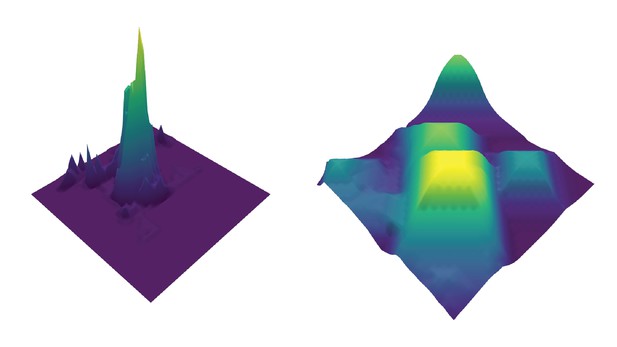
On the left, a depiction of a ‘fragile’ evolutionary landscape. The narrow, sharp peak represents a single mutation that provides viral blocking ability, which can easily be lost. On the right, a ‘permissive’ landscape. The many gently sloping peaks represent several mutations that lead to different levels of viral blocking ability, which is difficult to lose. Image credit: Jeanette Tenthorey (CC BY 4.0)
The evolutionary battle between viruses and the immune system is essentially a high-stakes arms race. The immune system makes antiviral proteins, called restriction factors, which can stop the virus from replicating. In response, viruses evolve to evade the effects of restriction factors. To counter this, restriction factors evolve too, and the cycle continues. The challenge for the immune system is that mammals do not evolve as fast as viruses. How then, in the face of this disadvantage, can the immune system hope to keep pace with viral evolution?
One human antiviral protein that seems to have struggled to keep up is TRIM5α. In rhesus macaques, it is very effective at stopping the replication of HIV-1 and related viruses. But in humans, it is not effective at all. But why? Protein evolution happens due to small genetic mutations, but not every mutation makes a protein better. If a protein is resilient, it can tolerate lots of neutral or negative mutations without breaking, until it mutates in a way that makes it better. But, if a protein is fragile, even small changes can render it completely unable to do its job. It is possible that restriction factors, like TRIM5α, are evolutionarily 'fragile', and therefore easy to break. But it is difficult to test whether this is the case, because existing mutations have already passed the test of natural selection. This means that either the mutation is somehow useful for the protein, or that it is not harmful enough to be removed.
Tenthorey et al. devised a way to introduce all possible changes to the part of TRIM5α that binds to viruses. This revealed that TRIM5α is not fragile; most random mutations increased, rather than decreased, the protein’s ability to prevent viral infection. In fact, it appears it would only take a single mutation to make TRIM5α better at blocking HIV-1 in humans, and there are many possible single mutations that would work. Thus, it would appear that human TRIM5α can easily gain the ability to block HIV-1. The next step was to find out whether these gains in antiviral activity are just as easily lost. To do this, Tenthorey et al. performed the same tests on TRIM5α from rhesus macaques and an HIV-blocking mutant version of human TRIM5α. This showed that the majority of random mutations do not break TRIM5α’s virus-blocking ability. Thus, TRIM5α can readily gain antiviral activity and, once gained, does not lose it easily during subsequent mutation.
Antiviral proteins like TRIM5α engage in uneven evolutionary battles with fast-evolving viruses. But, although they are resilient and able to evolve, they are not always able to find the right mutations on their own. Experiments like these suggest that it might be possible to give them a helping hand. Identifying mutations that help human TRIM5α to strongly block HIV-1 could pave the way for future gene therapy. This step would demand significant advances in gene therapy efficacy and safety, but it could offer a new way to block virus infection in the future.



