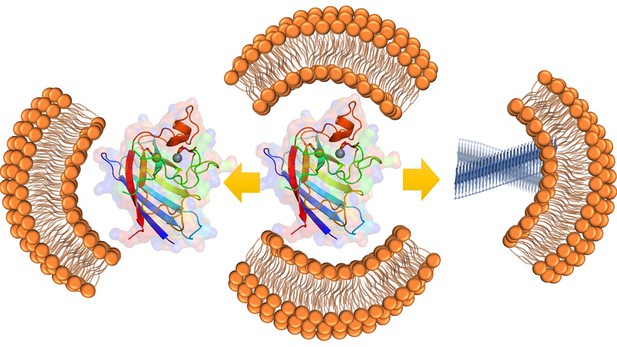
When the SOD1 protein (molecule in center) contains a zinc and copper metal ion (gold and silver spheres), it behaves normally and does not bind to the mitochondrial membrane (left). However, if the zinc ion is removed (right), this causes the SOD1 protein to attach to the membrane and form toxic aggregates (shown in blue). Image credit: Achinta Sannigrahi (CC BY 4.0)
Amyotrophic lateral sclerosis, or ALS, is an incurable neurodegenerative disease in which a person slowly loses specialized nerve cells that control voluntary movement. It is not fully understood what causes this fatal disease. However, it is suspected that clumps, or aggregates, of a protein called SOD1 in nerve cells may play a crucial role.
More than 140 mutations in the gene for SOD1 have been linked to ALS, with varying degrees of severity. But it is still unclear how these mutations cause SOD1 aggregation or how different mutations influence the survival rate of the disease. The protein SOD1 contains a copper ion and a zinc ion, and it is possible that mutations that affect how these two ions bind to SOD1 influences the severity of the disease.
To investigate this, Sannigrahi, Chowdhury, Das et al. genetically engineered mutants of the SOD1 protein which each contain only one metal ion. Experiments on these mutated proteins showed that the copper ion is responsible for the protein’s role in neutralizing harmful reactive molecules, while the zinc ion stabilizes the protein against aggregation. Sannigrahi et al. found that when the zinc ion was removed, the SOD1 protein attached to a structure inside the cell called the mitochondria and formed toxic aggregates.
Sannigrahi et al. then used these observations to build a computational model that incorporated different mutations that have been previously associated with ALS. The model suggests that mutations close to the site where zinc binds to the SOD1 protein increase disease severity and shorten survival time after diagnosis. This model was then experimentally validated using two disease variants of ALS that have mutations close to the sites where zinc or copper binds.
These findings still need to be tested in animals and humans to see if these mechanisms hold true in a multicellular organism. This discovery could help design new ALS treatments that target the zinc binding site on SOD1 or disrupt the protein’s interactions with the mitochondria.



