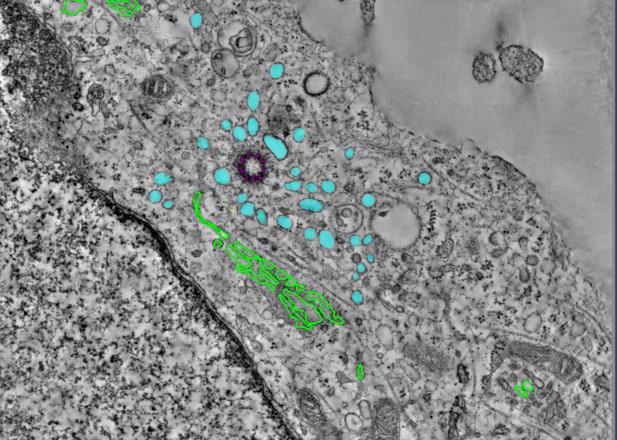
Accumulation of coatless vesicles (cyan) around a rudimentary cilium (purple) in a cell lacking WDR35; the Golgi organelle is outlined in green. Image credit: Tooba Quidwai (CC BY 4.0)
Most human cells have at least one small hair-like structure on their surface called a cilium. These structures can act as antennae and allow the cell to sense signals from the rest of the body. To do this, they contain proteins that differ from the rest of the cell. The content of cilia depends on regulated delivery of these proteins in and out of cilia by a process called the intraflagellar transport or IFT, which involves a large complex made of several proteins. This complex shuttles the cargo proteins back and forth between the base and the tip of the cilia.
However, ciliary proteins are not produced in the cilia; instead, they are made in a different part of the cell and then they are transported to the ciliary base. At the point where they enter the cilia, they were thought to bind to the assembling IFT ‘trains’ and be transported across the ciliary gate to the positions where they are needed in cilia. One of the components of the IFT machinery is a protein called WDR35, also known as IFT121. If the gene that codes for this protein is faulty or missing, it results in severe disorders in both humans and mice including a range of potentially lethal skeletal dysplasias.
Interestingly, without WDR35, cells cannot build functional cilia. The absence of this protein not only disrupts IFT, stopping certain ciliary proteins and their associated membranes from entering cilia; it also causes a ‘traffic jam’ with a pile-up of transport intermediates from the place in cell where they are made to the cilia. It is unclear why a mutation in one of the components of the IFT would have this effect, raising the question of whether WDR35, or IFTs a whole, has another role in bringing the cargo proteins into the cilia.
To understand this phenomenon, Quidwai et al. analysed the structure of WDR35 and other IFT proteins and found that they are very similar to a protein complex called COPI, which is involved in transporting membrane proteins around the cell. When certain proteins are newly made, they are stored in small lipid bubbles – called vesicles – that then selectively move to where the proteins are needed. COPI coats these vesicles, helping them get to where they need to go in a process called vesicular transport. Quidwai et al. found that WDR35 and other IFT proteins are able to bind to specific types of lipid molecules, suggesting that they might be assisting in a form of vesicle transport too. Indeed, when mouse cells grown in the lab were genetically engineered so they could not produce WDR35, coatless vesicles accumulated around the base of the cilia. Adding back WDR35 to these mutant cells rescued these defects in vesicle transport to cilia as well as allowed functional cilia to be formed.
These results provide evidence that WDR35, likely with other IFT proteins, acts as a COPI-like complex to deliver proteins to growing cilia. Further research will investigate the composition of these vesicles that transport proteins to cilia, and help pinpoint where they originate. Quidwai et al.’s findings not only shed light on how different genetic mutations found in patients with cilia dysfunction affect different steps of transporting proteins to and within cilia. They also increase our understanding of the cellular roadmap by which cells shuttle building blocks around in order to assemble these important ‘antennae’.



