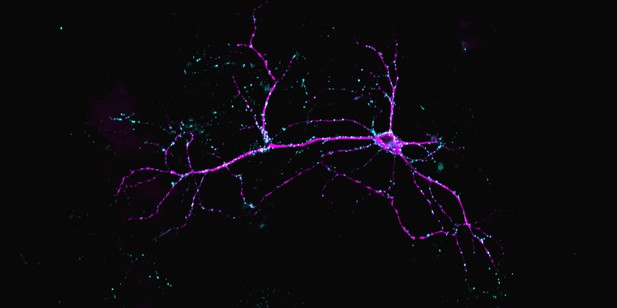
A neuron from the hippocampus (shown in pink) forming excitatory synapses (labelled in blue) which increase neuronal activity. Image credit: Maiken Østergaard (CC BY 4.0)
Neurons in the brain communicate with one another by passing molecules called neurotransmitters across the synapse connecting them together. Mutations in the machinery that controls neurotransmitter release can lead to epilepsy or developmental delays in early childhood, but how exactly is poorly understood.
Neurotransmitter release is primarily controlled by three proteins that join together to form the SNARE complex, and another protein called synaptotagmin-1. This assembly of proteins primes vesicles containing neurotransmitter molecules to be released from the neuron. When calcium ions bind to synaptotagmin-1, this triggers vesicles in this readily releasable pool to then fuse with the cell membrane and secrete their contents into the small gap between the communicating neurons.
Mutations associated with epilepsy and developmental delays have been found in all components of this release machinery. Here, Kádková, Murach, Østergaard et al. set out to find how three of these mutations, which are found in a protein in the SNARE complex called SNAP25, lead to aberrant neurotransmitter release.
Two of these mutations are located in the interface between the SNARE complex and synaptotagmin-1, while the other is found within the bundle of proteins that make up the SNARE complex. In vitro and ex vivo experiments in mice revealed that the two interface mutations led to defects in vesicle priming, while at the same time bypassing the control by synaptotagmin-1, resulting in vesicles spontaneously fusing with the cell membrane in an unregulated manner. These mutations therefore combine loss-of-function and gain-of-function features.
In contrast, the bundle mutation did not impact the number of vesicles in the releasable pool but reduced spontaneous and calcium ion evoked vesicle fusion. This was due to the mutation destabilizing the SNARE complex, which reduced the amount of energy available for merging vesicles to the membrane.
These findings reveal how SNAP25 mutations can have different effects on synapse activity, and how these defects disrupt the release of neurotransmitters. This experimental framework could be used to study how other synaptic mutations lead to diseases such as epilepsy. Applying this approach to human neurons and live model organisms may lead to the discovery of new therapeutic targets for epilepsy and delayed development.



