The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine
- The Sainsbury Laboratory, United Kingdom
- University of Tübingen, Germany
- Biodiversity and Climate Research Centre, Germany
- Goethe University, Germany
- Senckenberg Gesellschaft für Naturforschung, Germany
- Max Planck Institute for Developmental Biology, Germany
- United States Department of Agriculture, United States
- Centre for Integrated Fungal Research, Germany
Figures
Figure 1
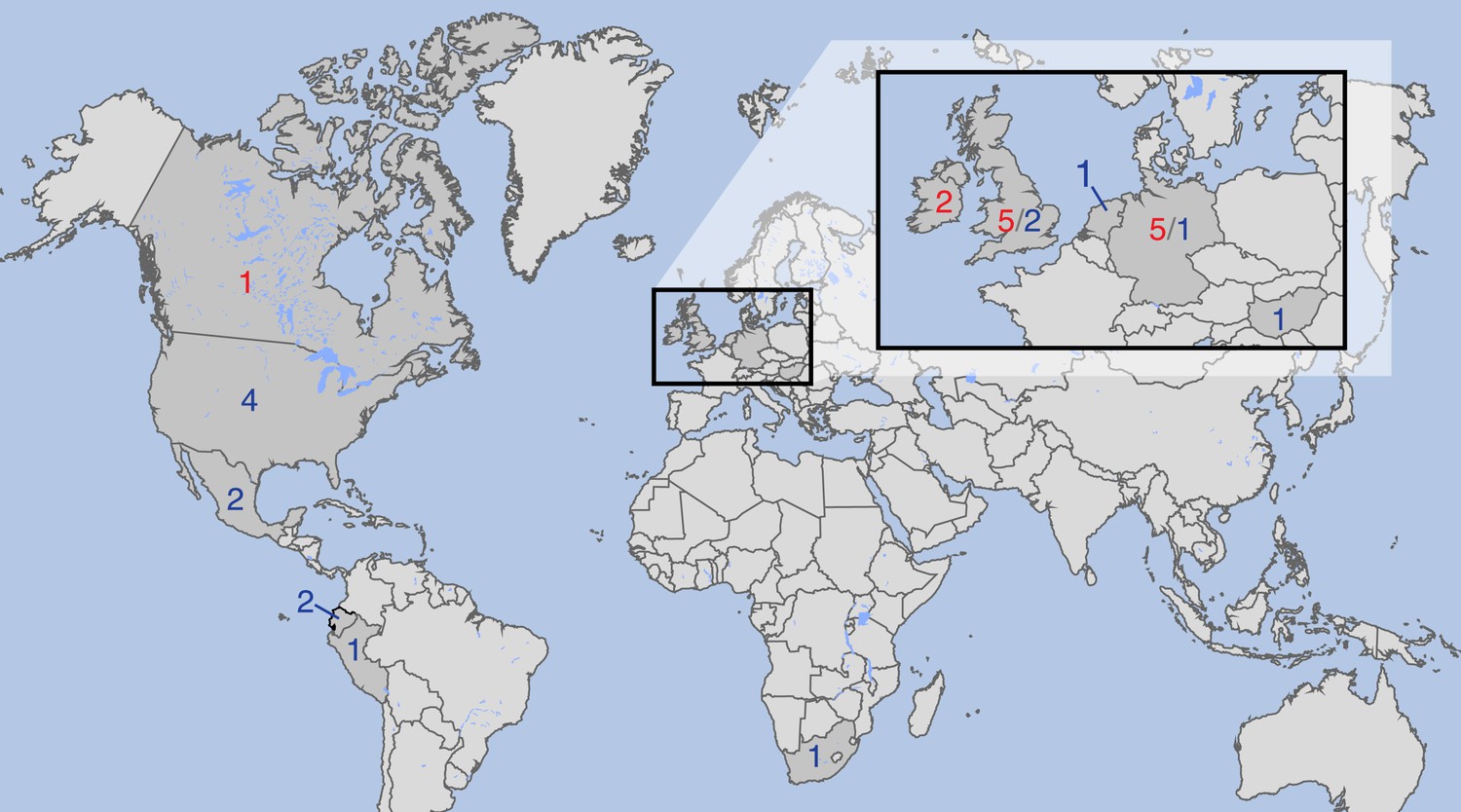
Countries of origin of samples used in whole-genome, mtDNA genome or both analyses.
Red indicates number of historic and blue of modern samples. More information on the samples is given in Tables 1 and 2.
Figure 2
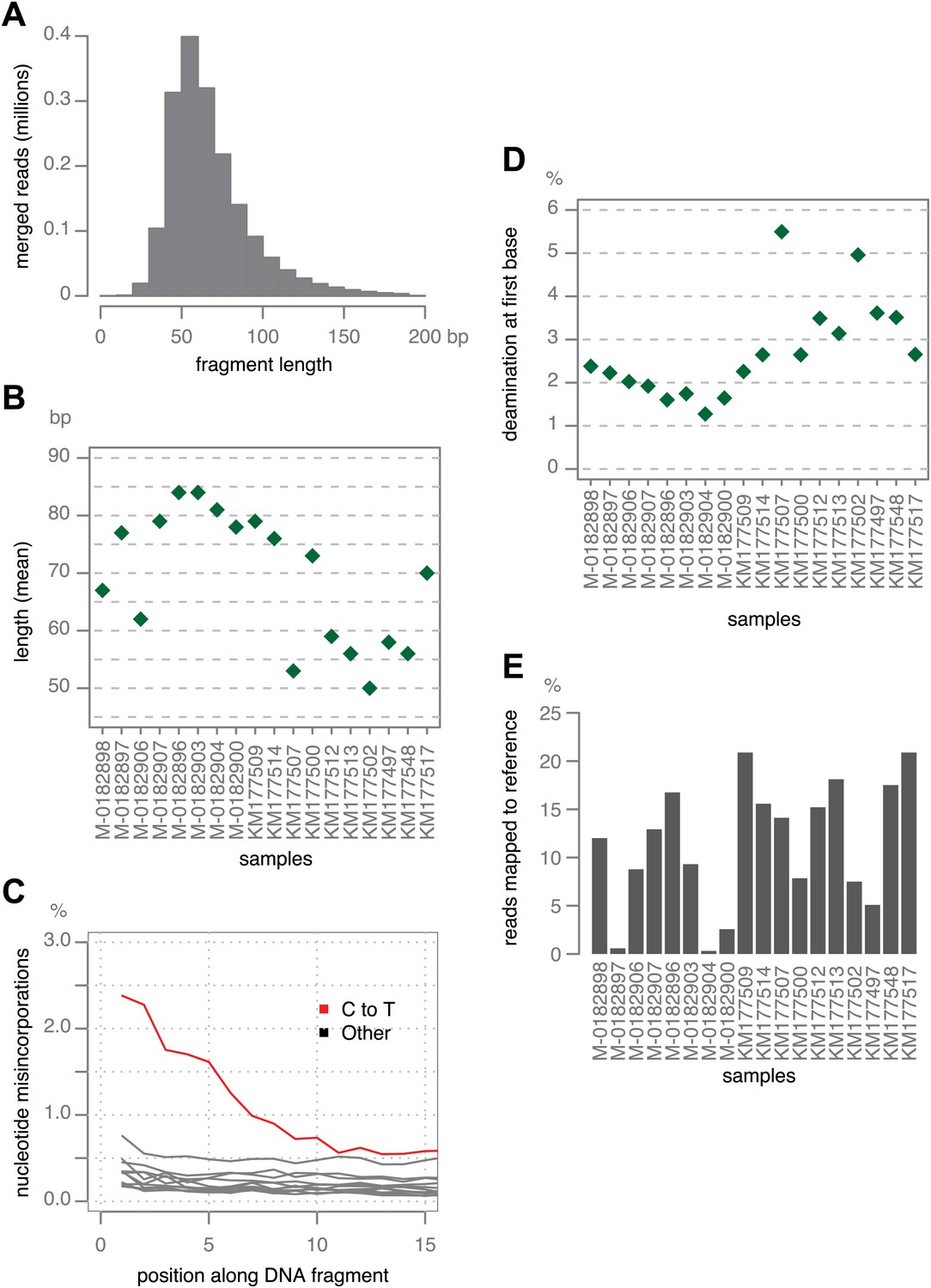
Ancient DNA-like characteristic of historic samples.
(A) Lengths of merged reads from historic sample M-0182898. (B) Mean lengths of merged reads from historic samples. (C) Nucleotide mis-incorporation in reads from the historic sample M-0182898. (D) Deamination at first 5′ end base in historic samples. (E) Percentage of merged reads that mapped to the P. infestans reference genome.
Figure 3 with 1 supplement
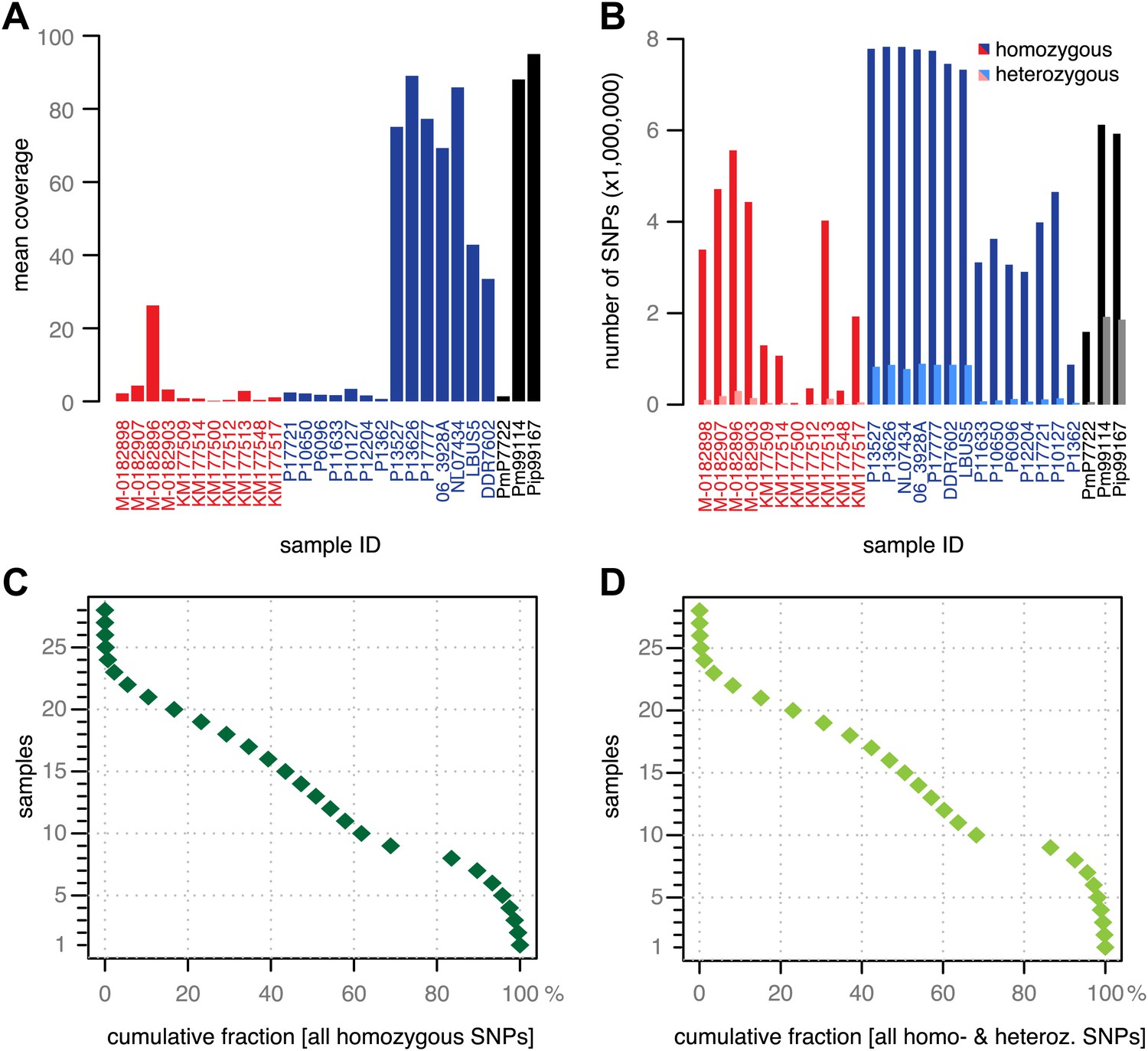
Coverage and SNP statistics.
(A) Mean nuclear genome coverage from historic (red) and modern (blue) samples. (B) Homo- and heterozygous SNPs in each sample. (C) Inverse cumulative coverage for all homozygous SNPs across all samples. (D) Same as (C) for homo- and heterozygous SNPs.
Figure 3—figure supplement 1
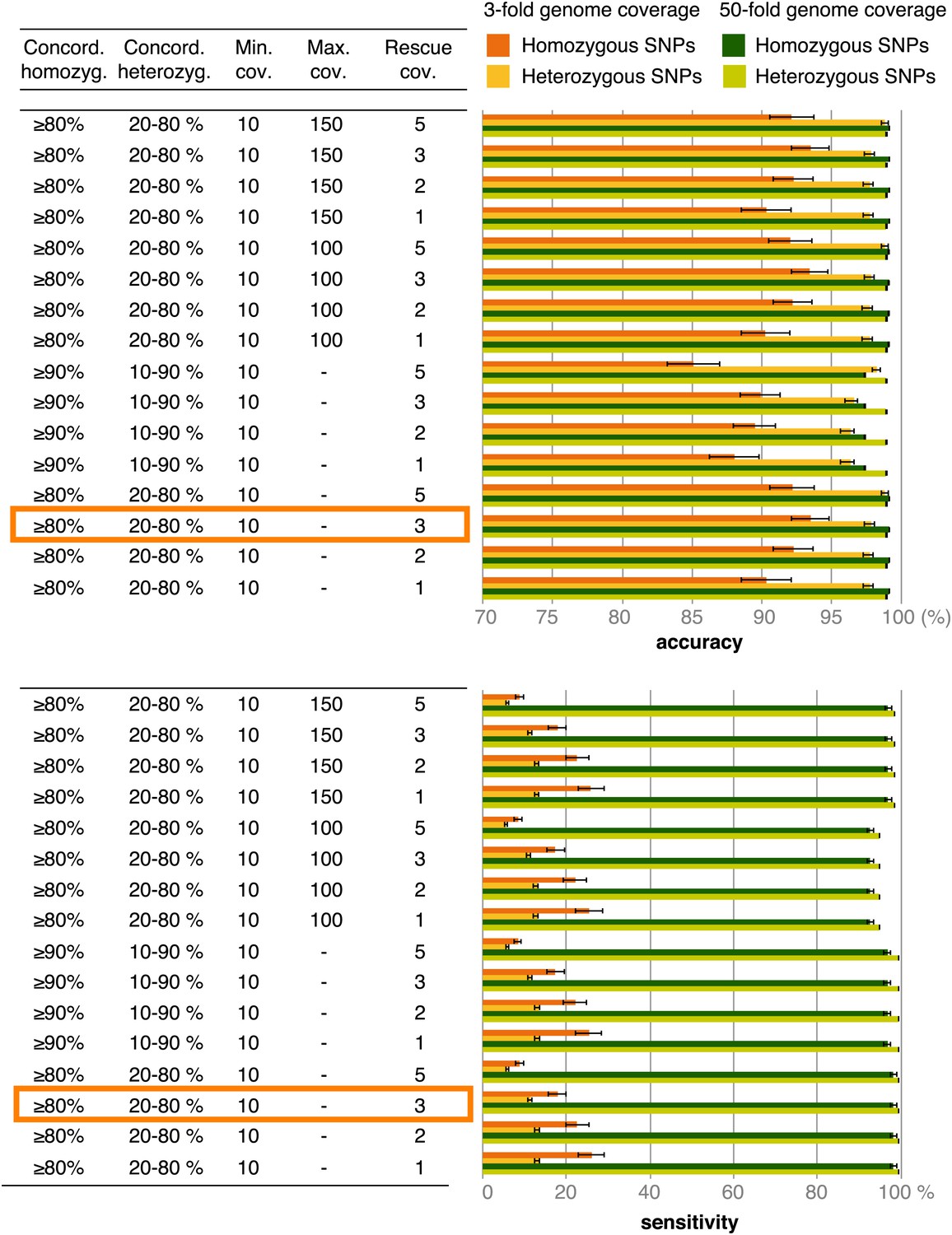
Accuracy and sensitivity of SNP calling at different cutoffs for SNP concordance based on 3- and 50-fold coverage of simulated data.
Rescue cov.—minimum coverage required to accept SNP calls in low-coverage genomes based on these SNPs having been found in high-coverage genomes. The cutoffs enclosed in orange rectangles were used for the final analysis.
Figure 4 with 2 supplements
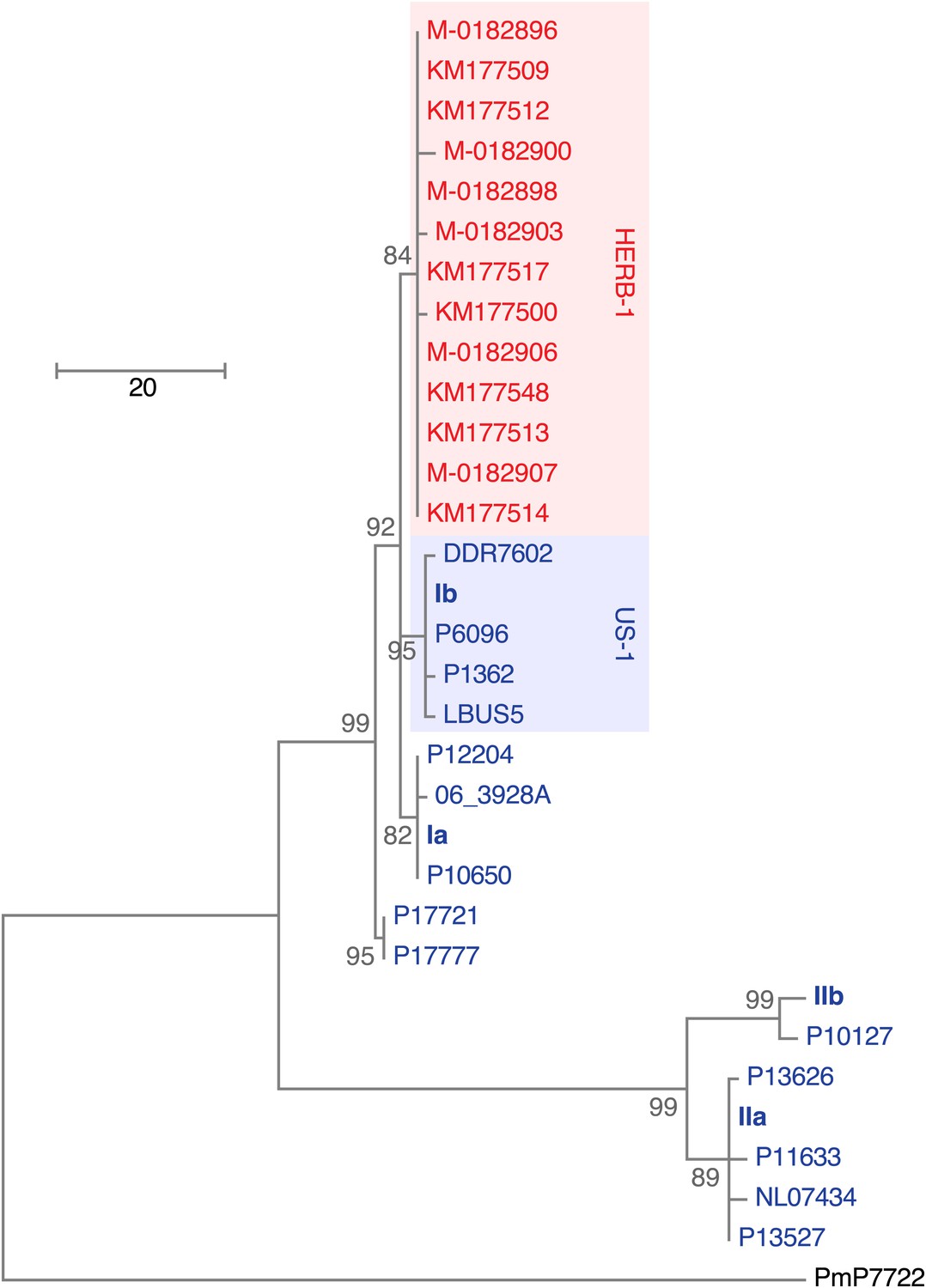
Maximum-parsimony phylogenetic tree of complete mtDNA genomes.
Sites with less than 90% information were not considered, leaving 24,560 sites in the final dataset. Numbers at branches indicate bootstrap support (100 replicates), and scale indicates changes.
Figure 4—figure supplement 1
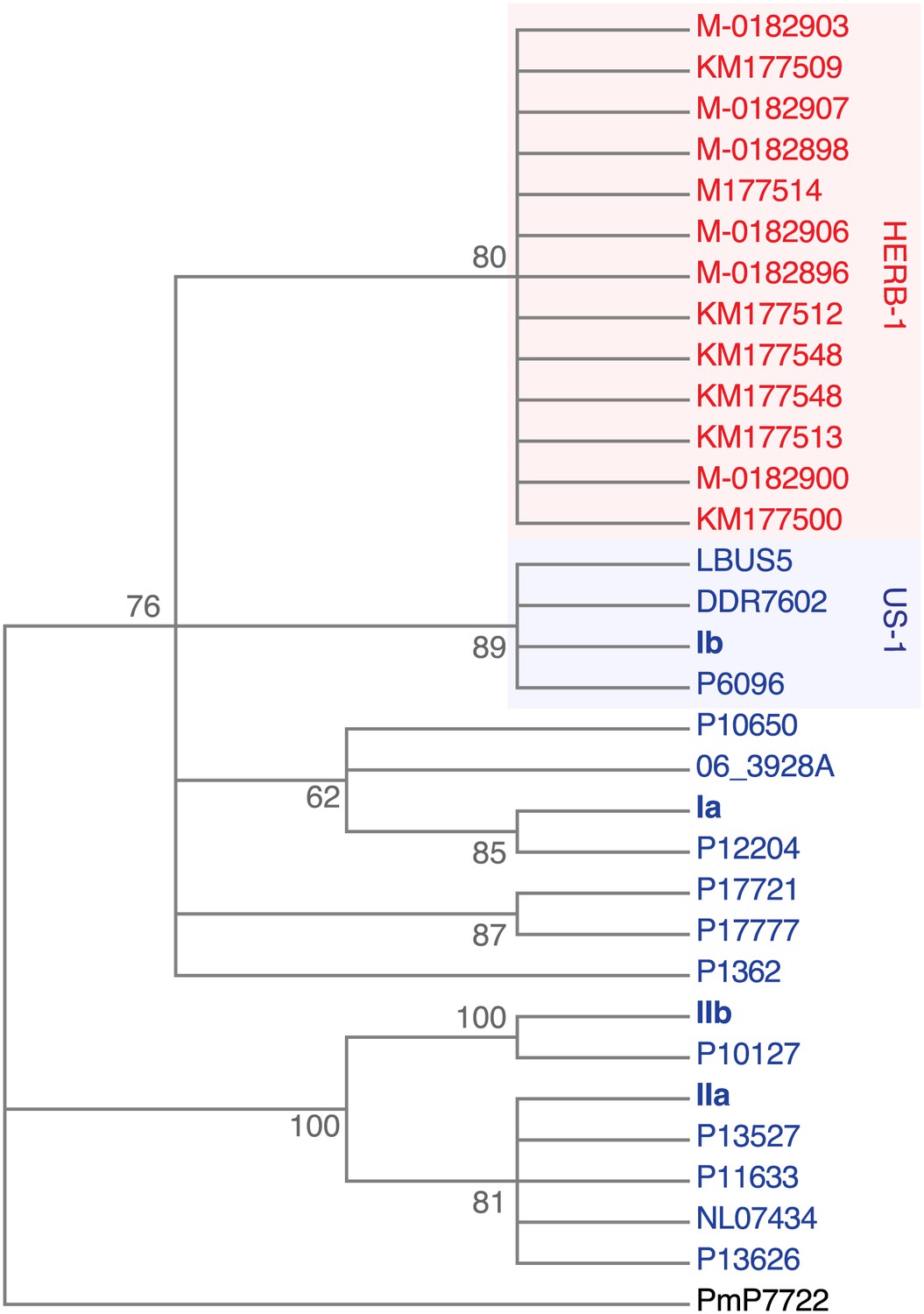
Maximum-likelihood phylogenetic tree of complete mtDNA genomes.
Sites with less than 90% information were not considered, leaving 24,560 sites in the final dataset. Numbers at branches indicate bootstrap support (100 replicates).
Figure 4—figure supplement 2

mtDNA sequences around diagnostic Msp1 restriction site (grey) for reference haplotype modern strains (blue) and historic strains (red).
The Msp1 (CCGG) restriction site is only present in the Ib haplotype; all other strains have a C-to-T substitution (CTGG).
Figure 5

Correlation between nucleotide distance of mtDNA genomes of HERB-1/haplotype Ia/haplotype Ib clade to the outgroup P17777 and sample age in calendar years before present.
https://doi.org/10.7554/eLife.00731.012
Figure 6
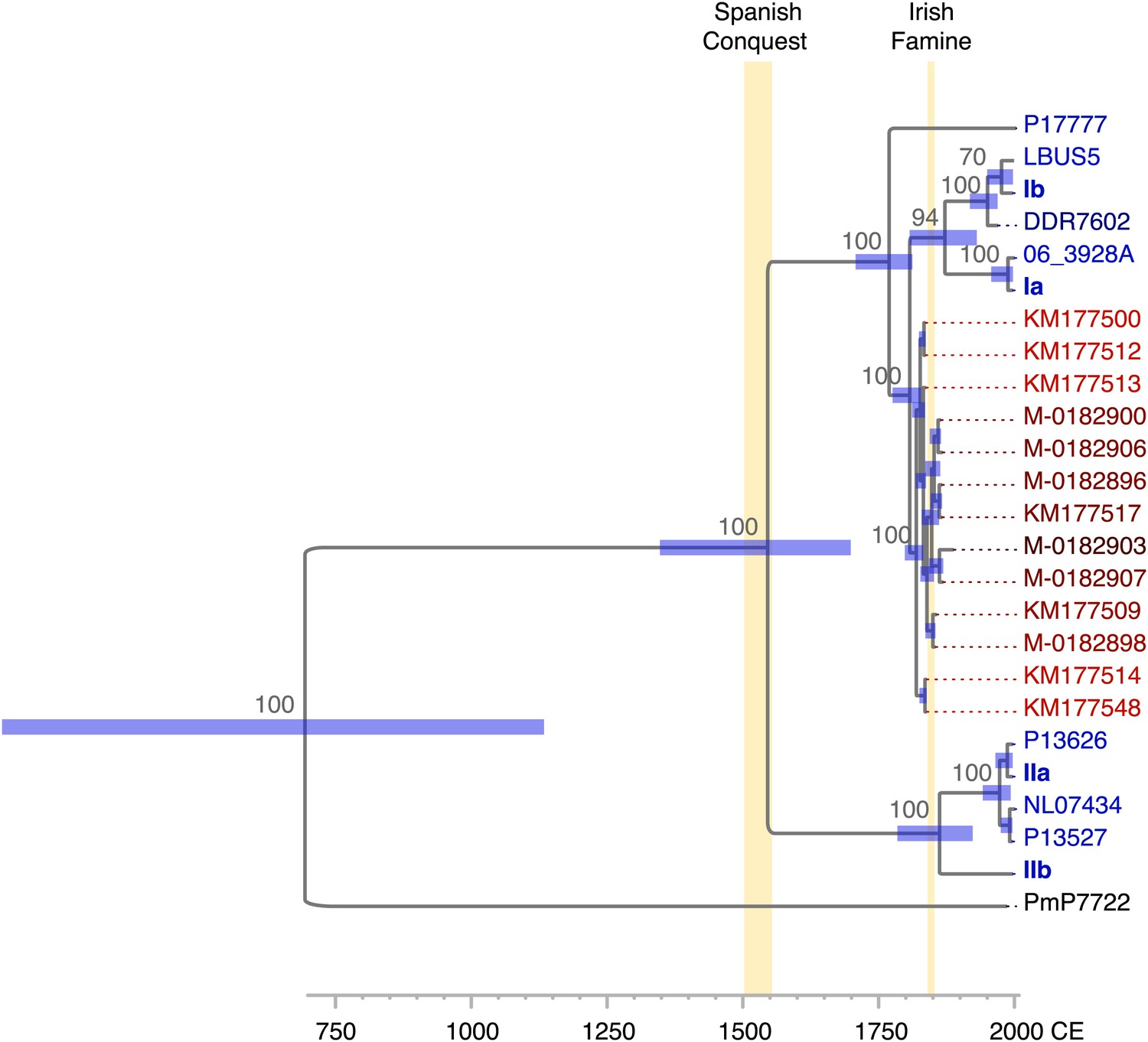
Divergence estimates of mtDNA genomes.
Bayesian consensus tree from 147,000 inferred trees. Posterior probability support above 50% is shown next to each node. Blue horizontal bars represent the 95% HPD interval for the node height. Light yellow bars indicate major historical events discussed in the text. See Figure 5 and Table 3 for detailed estimates at the four main nodes in P. infestans.
Figure 7 with 1 supplement
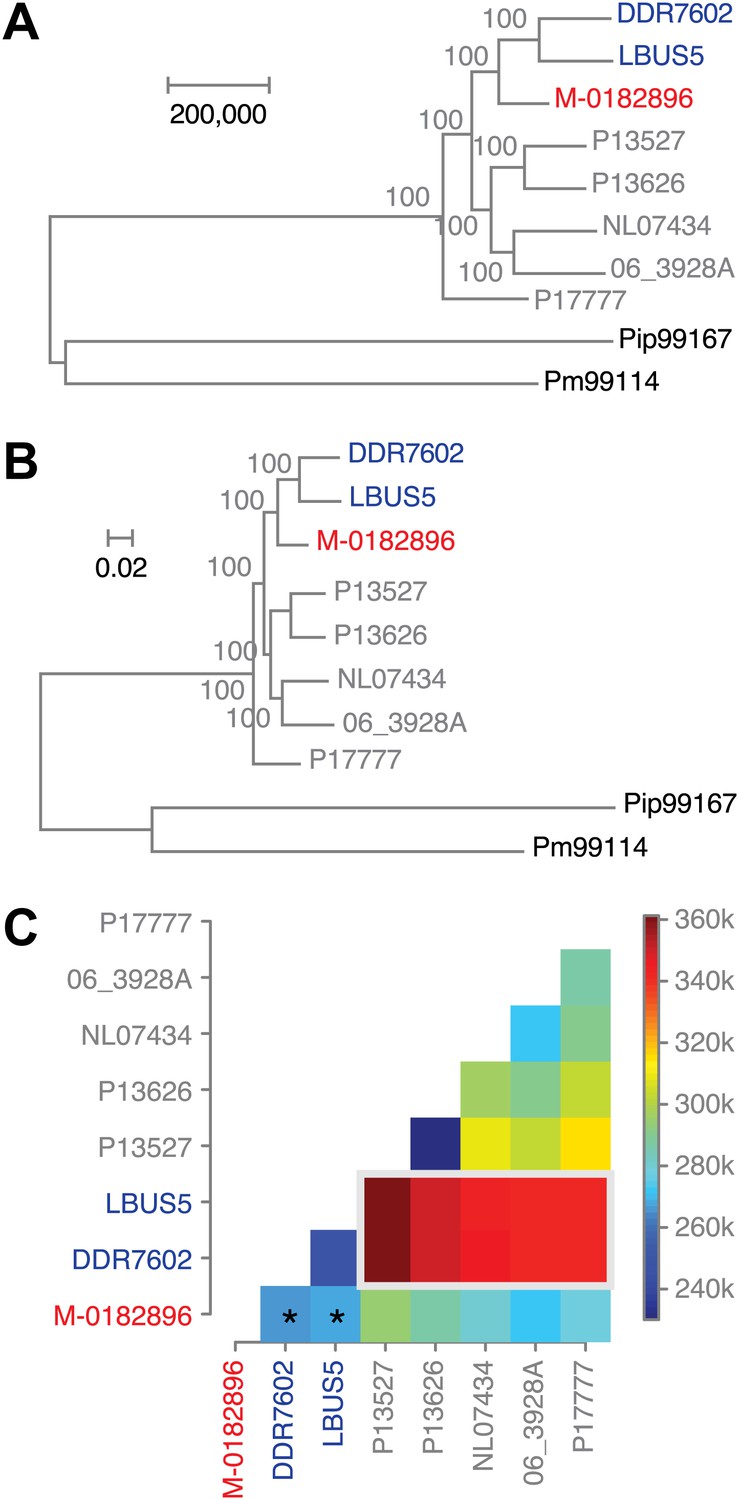
Phylogenetic trees of high-coverage nuclear genomes using both homozygous and heterozygous SNPs.
(A) Maximum-parsimony tree, considering only sites with at least 95% information, leaving 4,498,351 sites in the final dataset. Numbers at branches indicate bootstrap support (100 replicates), and scale indicates genetic distance. (B) Maximum-likelihood tree. (C) Heat map of genetic differentiation (color scale indicates SNP differences). US-1 strains DDR7062 and LBUS5 have the genomes sequences closest to M-0182896 (asterisks). The two US-1 isolates in turn are outliers compared to all other modern strains (highlighted by a gray box).
Figure 7—figure supplement 1

Phylogenetic trees of high- and low-coverage nuclear genomes.
(A) Neighbor-joining tree of high-coverage genomes using 4,595,012 homo- and heterozygous SNPs. Numbers at branches indicate bootstrap support (100 replicates), and scale indicates genetic distance. (B) Neighbor-joining tree of high- and low-coverage genomes using 2,101,039 homozygous and heterozygous SNPs. Numbers at branches indicate bootstrap support above 50, from 100 replicates. Scale indicates genetic distance. (C) Maximum parsimony tree of high- and low-coverage genomes using 315,394 SNPs homozygous and heterozygous SNPs (using only sites with at least 80% information).
Figure 8 with 1 supplement
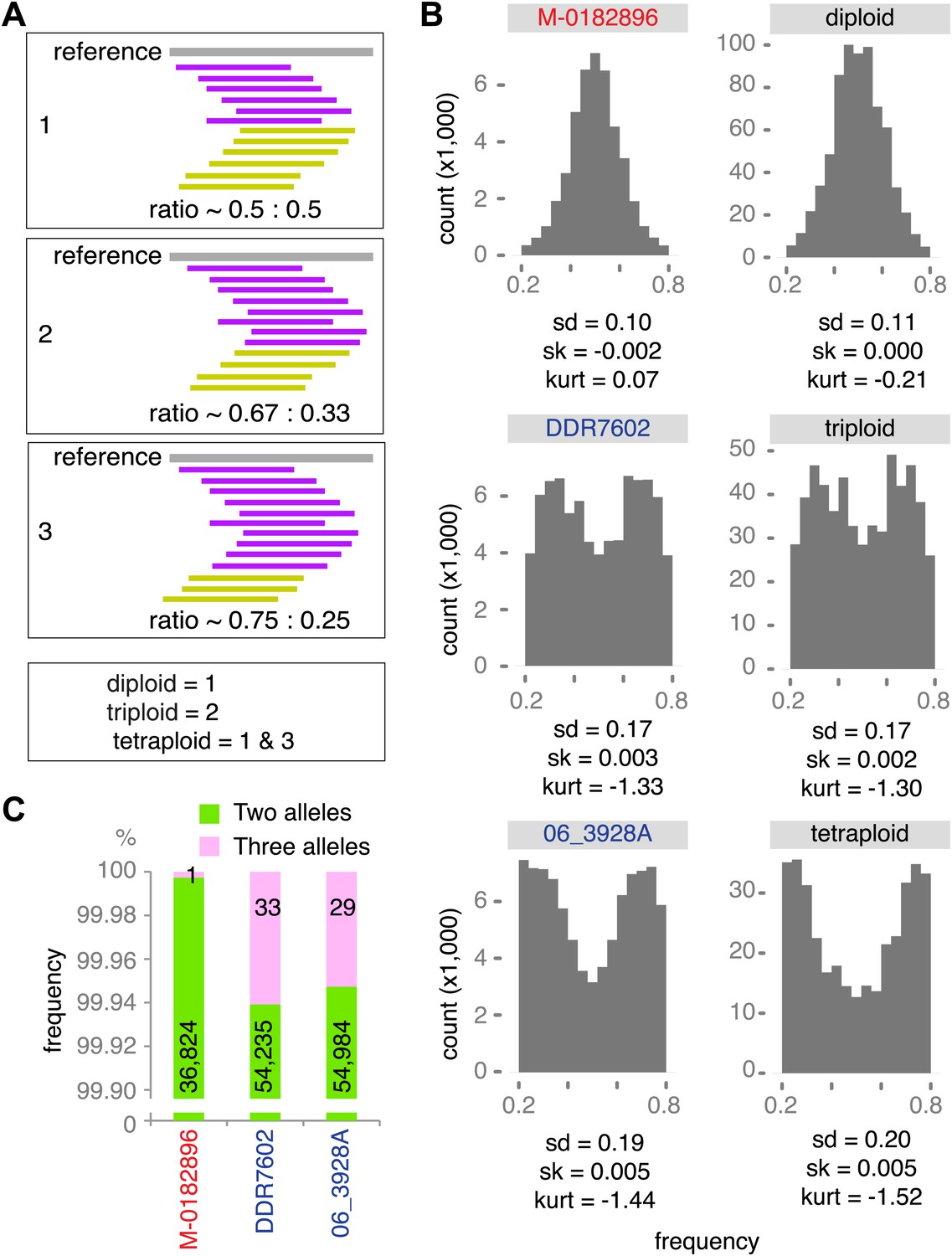
Ploidy analysis.
(A) Diagram of expected read frequencies of reads at biallelic SNPs for diploid, triploid and tetraploid genomes. (B) Reference read frequency at biallelic SNPs in gene dense regions (GDRs) for the historic sample M-0182896, two modern samples, and simulated diploid, triploid and tetraploid genomes. The simulated tetraploid genome is assumed to have 20% of pattern 1 and 80% of pattern 3 shown in (A). The shape and kurtosis of the observed distributions are similar to the corresponding simulated ones. (C) Polymorphic positions with more than one allele in the GDR.
Figure 8—figure supplement 1
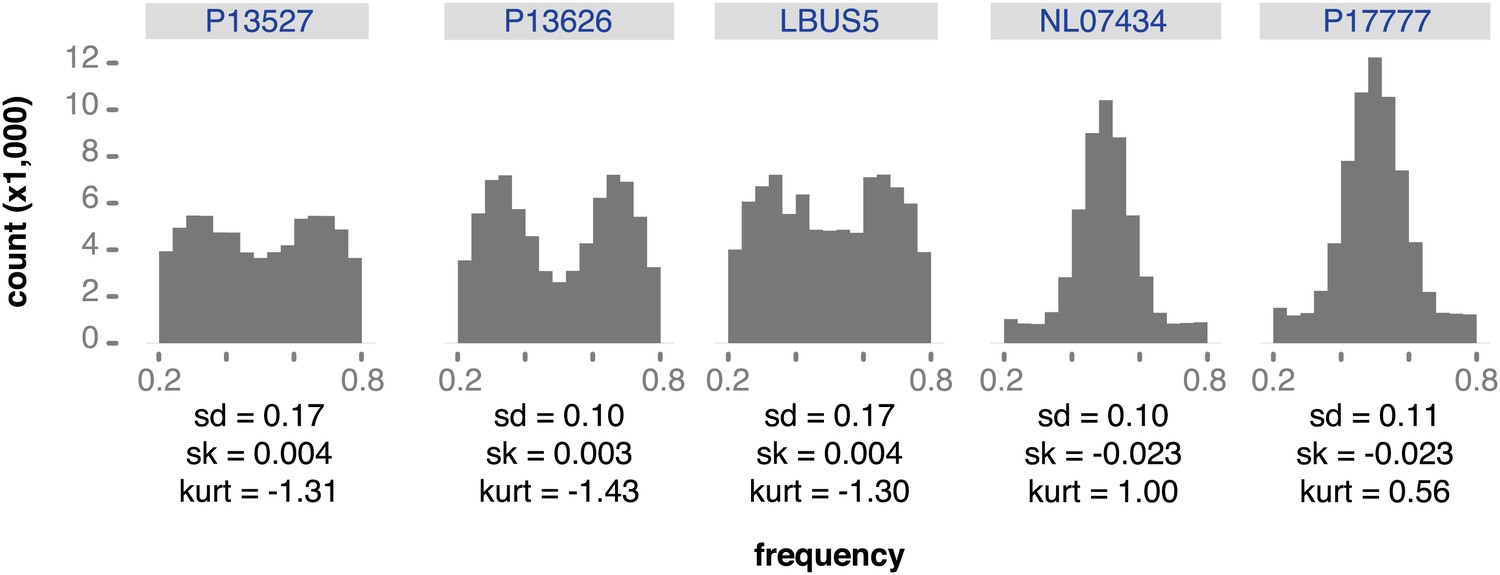
Reference read frequency at biallelic SNPs in gene dense regions (GDRs) for five modern high-coverage samples.
https://doi.org/10.7554/eLife.00731.018
Figure 9
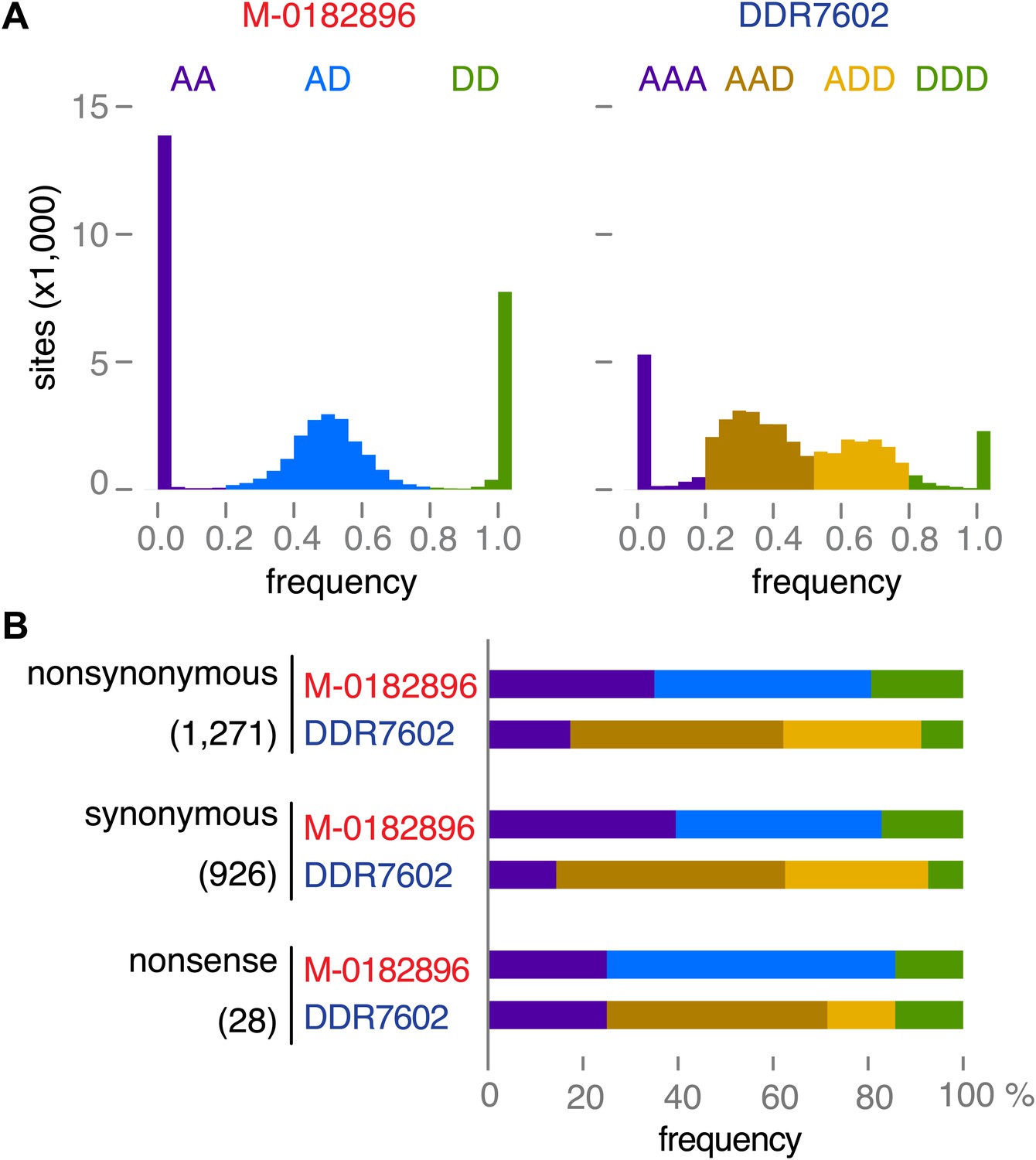
Read allele frequencies of historic genome M-0182896 and US-1 isolate DDR7602.
Alleles were classified as ancestral or derived using outgroup species P. mirabilis and P. ipomoeae. There were 40,532 segregating sites. (A) Distributions of derived alleles at sites segregating between M-0182896 and DDR7602. (B) Annotation of the different site classes.
Figure 10 with 1 supplement
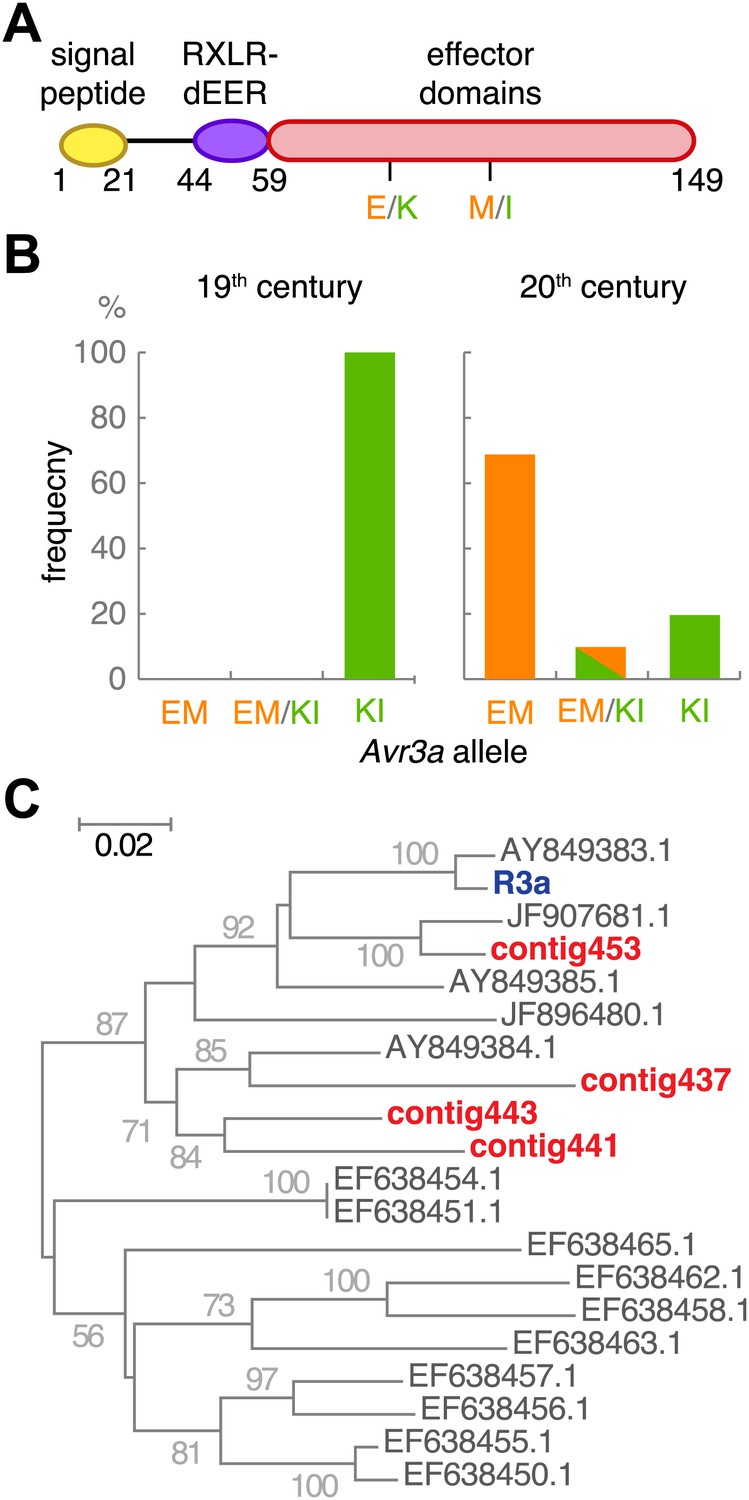
The effector gene Avr3a and its cognate resistance gene R3a.
(A) Diagram of AVR3A effector protein. (B) Frequency of Avr3a alleles in historic and modern P. infestans strains. (C) Neighbor-joining tree of R3a homologs from potato, based on 0.67 kb partial nucleotide sequences of S. tuberosum R3a (blue, accession number AY849382.1) and homologs (dark grey) in GenBank, and de novo assembled contigs from M-0182896 (red). Numbers at branches indicate bootstrap support with 500 replicates. Scale indicates changes.
Figure 10—figure supplement 1
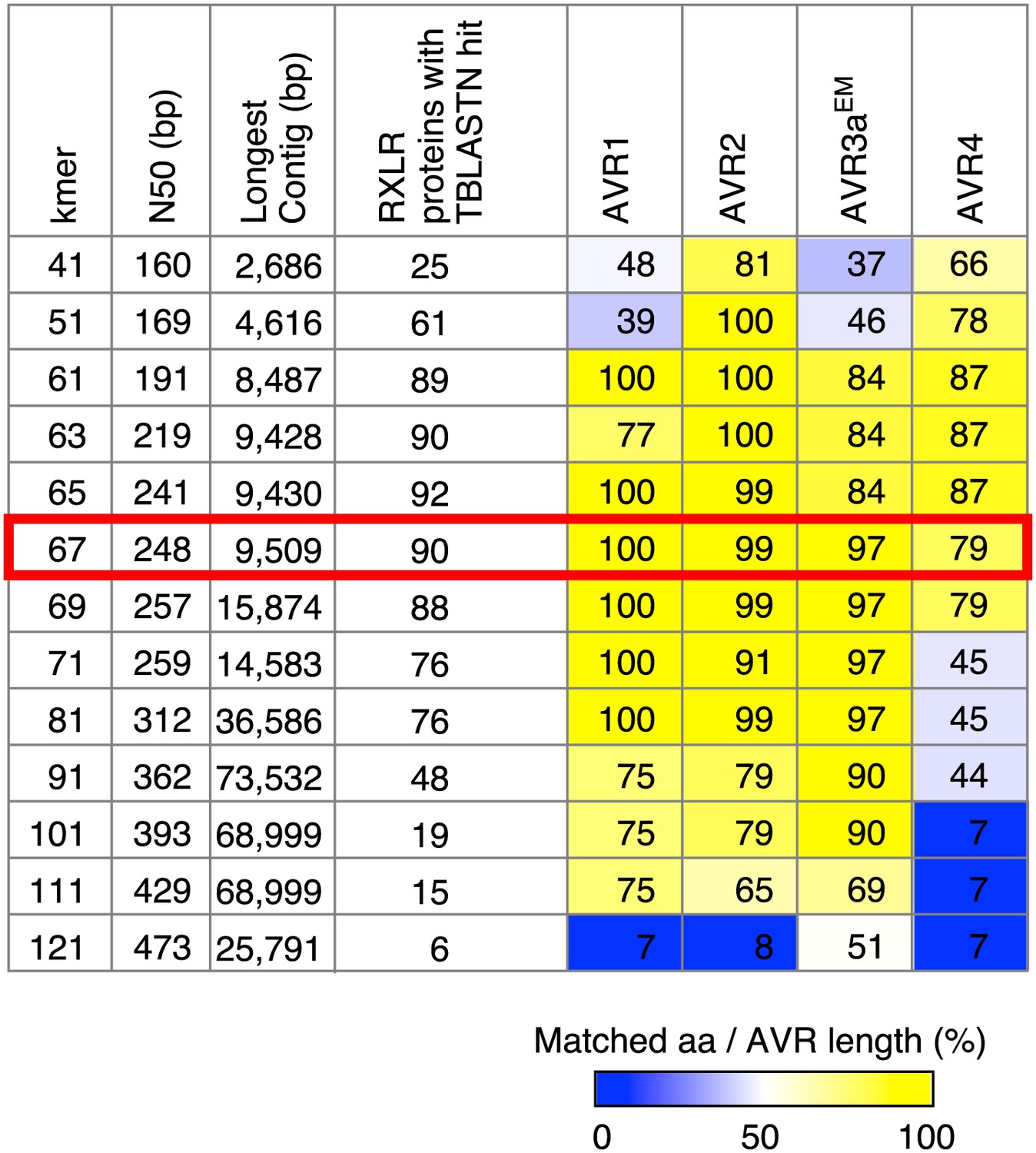
Summary of de novo assembly of RXLR effector genes.
TBLASTN query was performed with 549 RXLR proteins as a query and contigs as a database. When the High-scoring Segment Pair (HSP) and matched amino acids both covered ≥99% of the query length, we recorded a hit. Results with the optimal k-mer size are highlighted.
Figure 11

Suggested paths of migration and diversification of P. infestans lineages HERB-1 and US-1.
The location of the metapopulation that gave rise to HERB-1 and US-1 remains uncertain; here it is proposed to have been in North America.
Tables
Table 1
Provenance of P. infestans samples
| ID | Country of origin | Collection year | Host species | Reference* | |
| Herbarium samples | KM177500 | England | 1845 | Solanum tuberosum | 1 |
| KM177513 | Ireland | 1846 | Solanum tuberosum | 1 | |
| KM177502 | England | 1846 | Solanum tuberosum | 1 | |
| KM177497 | England | 1846 | Solanum tuberosum | 1 | |
| KM177514 | Ireland | 1847 | Solanum tuberosum | 1 | |
| KM177548 | England | 1847 | Solanum tuberosum | 1 | |
| KM177507 | England | 1856 | Petunia hybrida | 1 | |
| M-0182898 | Germany | Before 1863 | Solanum tuberosum | 2 | |
| KM177509 | England | 1865 | Solanum tuberosum | 1 | |
| M-0182900 | Germany | 1873 | Solanum lycopersicum | 2 | |
| M-0182907 | Germany | 1875 | Solanum tuberosum | 1 | |
| KM177517 | Wales | 1875 | Solanum tuberosum | 1 | |
| M-0182897 | USA | 1876 | Solanum lycopersicum | 2 | |
| M-0182906 | Germany | 1877 | Solanum tuberosum | 2 | |
| M-0182896 | Germany | 1877 | Solanum tuberosum | 2 | |
| M-0182904 | Austria | 1879 | Solanum tuberosum | 2 | |
| M-0182903 | Canada | 1896 | Solanum tuberosum | 2 | |
| KM177512 | England | NA | Solanum tuberosum | 1 | |
| Modern samples | 06_3928A | England | 2006 | Solanum tuberosum | 3 |
| DDR7602 | Germany | 1976 | Solanum tuberosum | 4 | |
| P1362 | Mexico | 1979 | Solanum tuberosum | 5 | |
| P6096 | Peru | 1984 | Solanum tuberosum | 5 | |
| P7722 (P. mirabilis) | USA | 1992 | Solanum lycopersicum | 5 | |
| P9464 | USA | 1996 | Solanum tuberosum | 5 | |
| P12204 | Scotland | 1996 | Solanum tuberosum | 5 | |
| P13527 | Ecuador | 2002 | Solanum andreanum | 5 | |
| P10127 | USA | 2002 | Solanum lycopersicum | 5 | |
| P13626 | Ecuador | 2003 | Solanum tuberosum | 5 | |
| P10650 | Mexico | 2004 | Solanum tuberosum | 5 | |
| LBUS5 | South Africa | 2005 | Petunia hybrida | 6 | |
| P11633 | Hungary | 2005 | Solanum lycopersicum | 5 | |
| NL07434 | Netherlands | 2007 | Solanum tuberosum | 3 | |
| P17777 | USA | 2009 | Solanum lycopersicum | 5 | |
| P17721 | USA | 2009 | Solanum tuberosum | 5 |
-
*
1, Kew Royal Botanical Gardens; 2, Botanische Staatssammlung München; 3, Cooke et al. (2012); 4, Kamoun et al. (1999); 5, World Oomycete Genetic Resource Collection at UC Riverside, CA; 6, Dr Adele McLeod, Univ. of Stellenbosch, South Africa.
Table 2
Sequencing strategy
| ID | Instrument and read type | Sequencing center | Coverage |
| M-0182896 | HiSeq 2000 (2 × 101 bp) | MPI | High |
| M-0182897 | HiSeq 2000 (2 × 101 bp) | MPI | Low* |
| M-0182898 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| M-0182900 | HiSeq 2000 (2 × 101 bp) | MPI | Low† |
| M-0182903 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| M-0182904 | HiSeq 2000 (2 × 101 bp) | MPI | Low* |
| M-0182906 | HiSeq 2000 (2 × 101 bp) | MPI | Low† |
| M-0182907 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177497 | MiSeq (2 × 150 bp) | MPI | Low |
| KM177500 | MiSeq (2 × 150 bp) | MPI | Low* |
| KM177502A | MiSeq (2 × 150 bp) | MPI | Low* |
| KM177507 | MiSeq (2 × 150 bp) | MPI | Low* |
| KM177509 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177512 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177513 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177514 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177517 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| KM177548 | MiSeq (2 × 150 bp) and HiSeq 2000 (2 × 101 bp) | MPI | Low |
| 06_3928A | GAIIX (2 × 76 bp) | TSL | High |
| DDR7602 | GAIIX (2 × 76 bp) | TSL | High |
| LBUS5 | GAIIX (2 × 76 bp) | TSL | High |
| NL07434 | GAIIX (2 × 76 bp) | TSL | High |
| P10127 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P10650 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P12204 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P13527 | GAIIX (2 × 76 bp) | TSL | High |
| P1362 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P13626 | GAIIX (2 × 76 bp) | TSL | High |
| P11633 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P17721 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P17777 | GAIIX (2 × 76 bp) | TSL | High |
| P6096 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P7722 | HiSeq 2000 (2 × 101 bp) | MPI | Low |
| P9464 | HiSeq 2000 (2 × 101 bp) | MPI | Low* |
| PIC99114 | GAIIX (2 × 76 bp) | TSL | High |
| PIC99167 | GAIIX (2 × 76 bp) | TSL | High |
-
*
Samples not included in any analysis due to extremely low coverage.
-
†
Samples used only in mtDNA analysis.
Table 3
Inferred time to most recent common ancestor (TMRCA) for different splits in the mtDNA tree
| Node | TMRCA (ya) | ||
| Best estimate | Lower 2.5% | Upper 2.5% | |
| I/HERB-1, II | 460 | 300 | 643 |
| Ia/Ib, HERB-1 | 234 | 187 | 290 |
| HERB-1 strains | 182 | 168 | 201 |
| IIa, IIb | 142 | 78 | 214 |
Table 4
Presence or absence of avirulence effector genes in historic and modern samples, expressed as percentages of effector genes covered by reads
| Avr gene | R gene | HERB-1* | US-1† | 20th century non-US-1 | Outgroups | ||||||
| EC3527 | EC3626 | P17777 | 06_3928A | NL07434 | Merged | Pm PIC99114 | Pip PIC99167 | ||||
| Avr1 | R1 | 100 | 100 | 0 | 0 | 100 | 0 | 0 | 100 | 98 | 100 |
| Avr2 | R2 | 100 | 100 | 100 | 100 | 100 | 81 | 100 | 77 | 97 | 100 |
| Avr3a | R3a | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 28 |
| Avr3b | R3b | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 100 | 100 | 100 |
| Avr4 | R4 | 100 | 100 | 100 | 100 | 95 | 89 | 100 | 99 | 85 | 92 |
| Avrblb1 | Rpi-blb1 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 |
| Avrblb2 | Rpi-blb2 | 100 | 100 | 100 | 100 | 92 | 100 | 100 | 89 | 88 | 0 |
| Avrvnt1 | Rpi-vnt1 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| AvrSmira1 | Rpi-Smira1 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 97 | 100 |
| AvrSmira2 | Rpi-Smira2 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 0 |
-
Sequences and polymorphisms are shown in Table 5 and Table 5—source data 1.
-
*
Same sequences obtained for M-0182896 and merged sequences.
-
†
Same sequences obtained for DDR7602 and LBUS5.
Table 5
Amino acid differences in the avirulence effectors AVR1, AVR2, AVR3a and AVR4 encoded by the T30-4 reference genome, HERB-1 and DDR7602 (US-1)
| Position | Substitution | Note | ||
| T30-4 | HERB1 | DDR7602 | ||
| AVR1 (PITG_16,663) | ||||
| 80 | T | T | T, S | HERB-1 polymorphisms shared with T30-4 and DDR7602. |
| 142 | I | I, T | T | |
| 154 | V | V, A | A | |
| 185 | I | I | I, V | |
| AVR2 (PITG_22,870) | ||||
| 31 | N | K | K | HERB-1 identical to DDR7602. |
| AVR3a (PITG_14,371) | ||||
| 19 | S | C | C | HERB-1 identical to DDR7602; both correspond to AVR3aKI isoform. |
| 80 | E | K | K | |
| 103 | M | I | I | |
| 139 | M | L | L | |
| AVR4 (PITG_07,387) | ||||
| 19 | T | T, I | T | HERB-1 polymorphisms shared with T30-4 and DDR7602. |
| 139 | L | S | L, S | |
| 221 | L | V | L, V | |
| 271 | V | F | V, F | |
-
IDs in parentheses refer to gene models in reference genome. Full-length sequences of deduced amino acid sequences of HERB-1 AVR1, AVR2, AVR3a and AVR4 are provided in Table 5—source data 1.
-
Table 5—source data 1
Full-length sequences of deduced amino acid sequences of HERB-1 AVR1, AVR2, AVR3a and AVR4.
- https://doi.org/10.7554/eLife.00731.022
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine
eLife 2:e00731.
https://doi.org/10.7554/eLife.00731




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}