Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis
- New York University School of Medicine and Hospital for Joint Diseases, United States
- The Kimmel Center for Biology and Medicine of the Skirball Institute, New York University School of Medicine, United States
- University of California, San Francisco, United States
- Weill Cornell Medical College, United States
- University of Trento, Italy
- Harvard School of Public Health, United States
- Memorial Sloan-Kettering Cancer Center, United States
- University of Valencia, Spain
- Weatherall Institute of Molecular Medicine, University of Oxford, United Kingdom
- Howard Hughes Medical Institute, New York University School of Medicine, United States
Figures
Figure 1 with 1 supplement
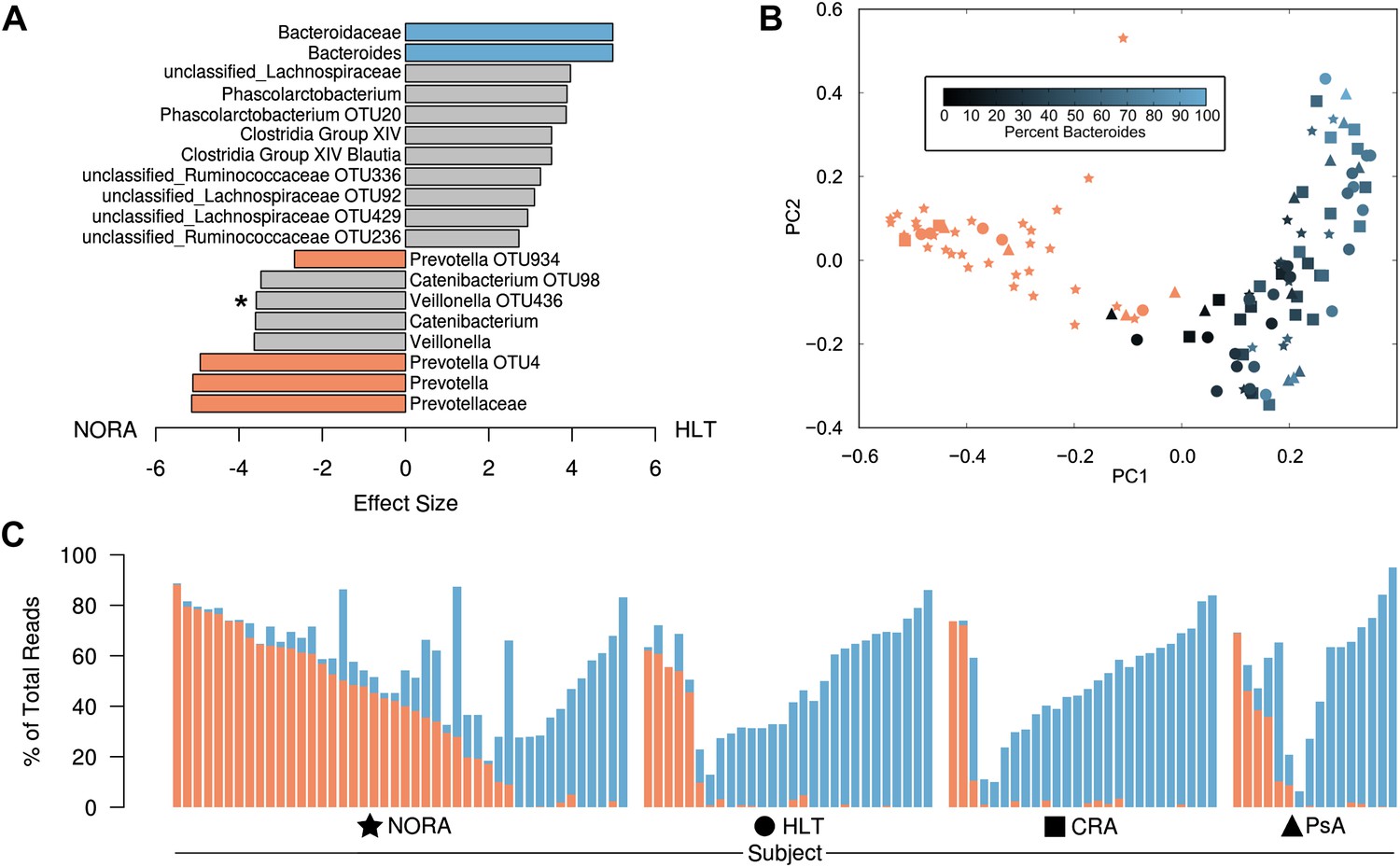
Differences in the relative abundance of Prevotella and Bacteroides in 114 subjects with and without arthritis, determined by 16S sequencing (regions V1–V2, 454 platform).
(A) LEfSe (Segata et al., 2011) was used to compare the abundances of all detected clades among all groups, producing an effect size for each comparison (‘Materials and methods’). All results shown are highly significant (q<0.01) by Kruskal-Wallis test adjusted with the Benjamini-Hochberg procedure for multiple testing, except that indicated with an asterisk, which is significant at q<0.05. Negative values (left) correspond to effect sizes representative of NORA groups, while positive values (right) correspond to effect sizes in HLT subjects. Prevotella was found to be over-represented in NORA patients, while Bacteroides was over-represented in all other groups. (B) The Bray-Curtis distance between all subjects was calculated and used to generate a principal coordinates plot in MOTHUR (Schloss et al., 2009). The first two components are shown. Subjects with an abundance of Prevotella greater than 10% were colored red. Other subjects were colored according to their Bacteroides abundance as shown. NORA subjects (stars) primarily cluster together according to their Prevotella abundance, and the x-axis is representative of differences in the relative abundance of Prevotella and Bacteroides. (C) The abundances of Prevotella (red) and Bacteroides (blue) are shown for all subjects, sorted in order of decreasing Prevotella abundance (>5%) and increasing Bacteroides abundance.
-
Figure 1—source data 1
Intermediate data and analysis tools for Figure 1.
- https://doi.org/10.7554/eLife.01202.005
-
Figure 1—source data 2
Intermediate data and analysis tools for Figure 1—figure supplement 1.
- https://doi.org/10.7554/eLife.01202.006
Figure 1—figure supplement 1
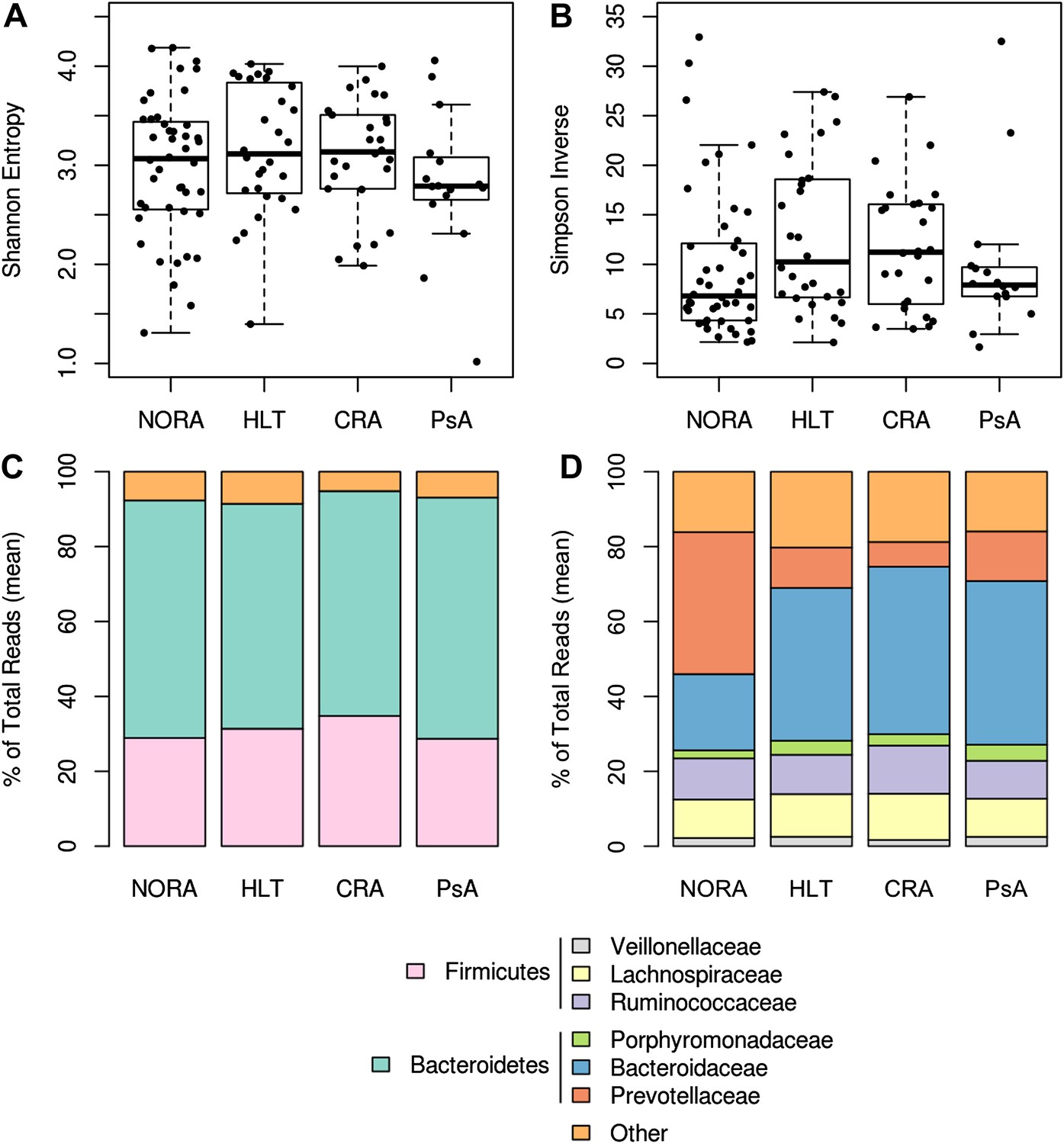
Gut microbiota richness, diversity and relative abundance in NORA patients and controls.
(A and B) Gut microbiota richness and diversity are similar among RA groups and healthy controls. (C) Phyla abundance by group. No significant differences were found at this taxonomic level. (D) Family abundance by group. NORA subjects have a significant increase in Prevotellaceae (red) and a concomitant decrease in Bacteroidaceae (blue) by FDR-adjusted Kruskal-Wallis test (q<0.01).
Figure 2 with 1 supplement
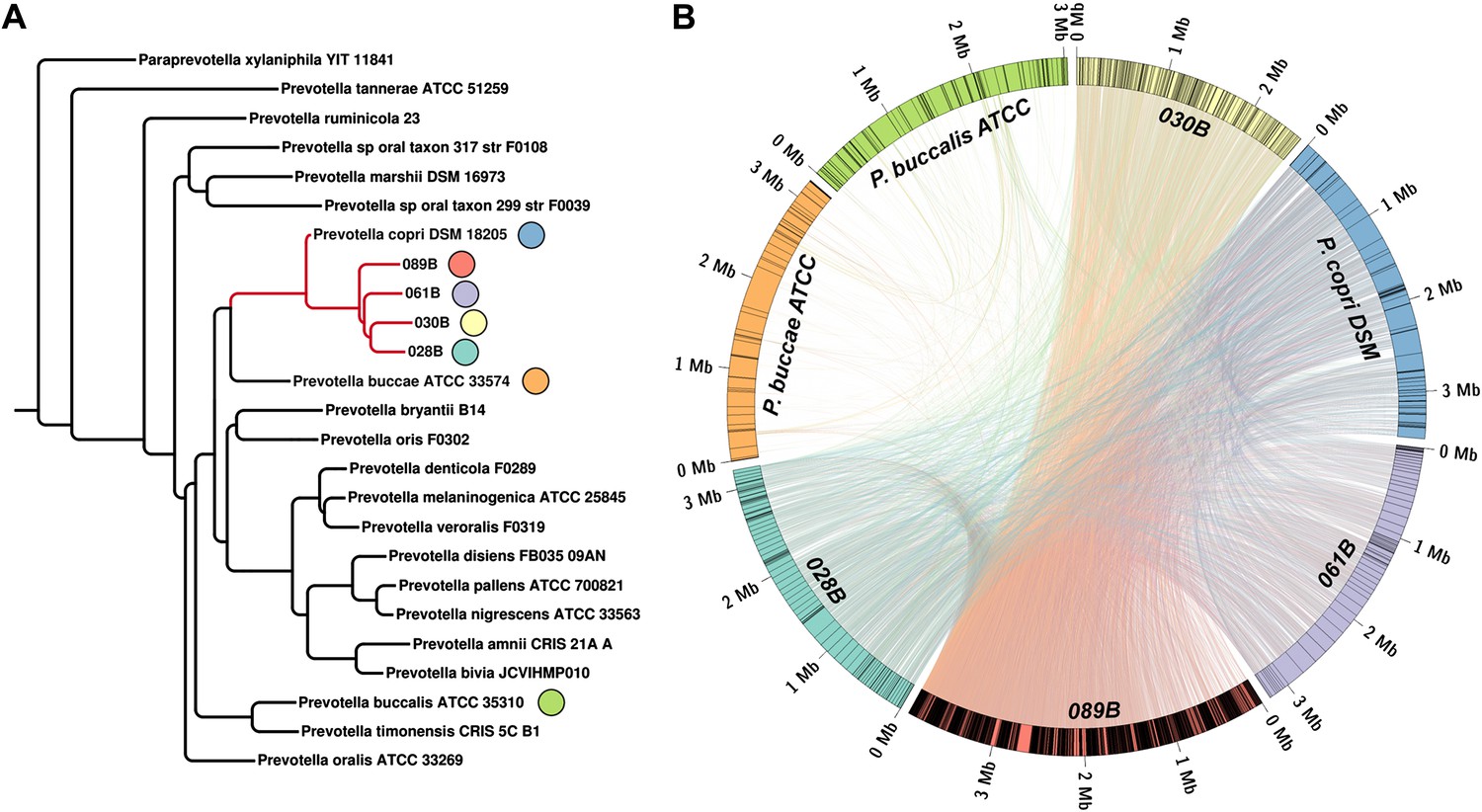
Homology-based classification of patient-associated Prevotella.
Four NORA subjects with a high abundance of Prevotella OTU4 were selected for shotgun sequencing and metagenome assembly. (A) The resulting metagenomic contigs were used to generate a phylogenomic tree with PhyloPhlAn (Segata et al., 2013). (B) Assemblies were filtered by alignment to the reference Prevotella copri DSM 18205 genome, keeping contigs with at least one 300 bp region aligned at 97% identity or greater. The resulting draft patient-derived P. copri assemblies were aligned to one another, the reference P. copri genome, and two distinct Prevotella taxa (Prevotella buccae and Prevotella buccalis). Colored arcs represent assemblies as labeled, lines connecting arcs represent regions of >97% identity >1 kb in length, and gray lines dividing colored arcs represent boundaries between contigs. These results demonstrate that Prevotella OTU4, OTU12, and OTU934 form a clade with P. copri (left, red highlighted subtree) that is genetically distinct from more distant Prevotella taxa.
-
Figure 2—source data 1
Intermediate data and analysis tools for Figure 2.
- https://doi.org/10.7554/eLife.01202.010
-
Figure 2—source data 2
Intermediate data and analysis tools for Figure 2—figure supplement 1.
- https://doi.org/10.7554/eLife.01202.011
Figure 2—figure supplement 1
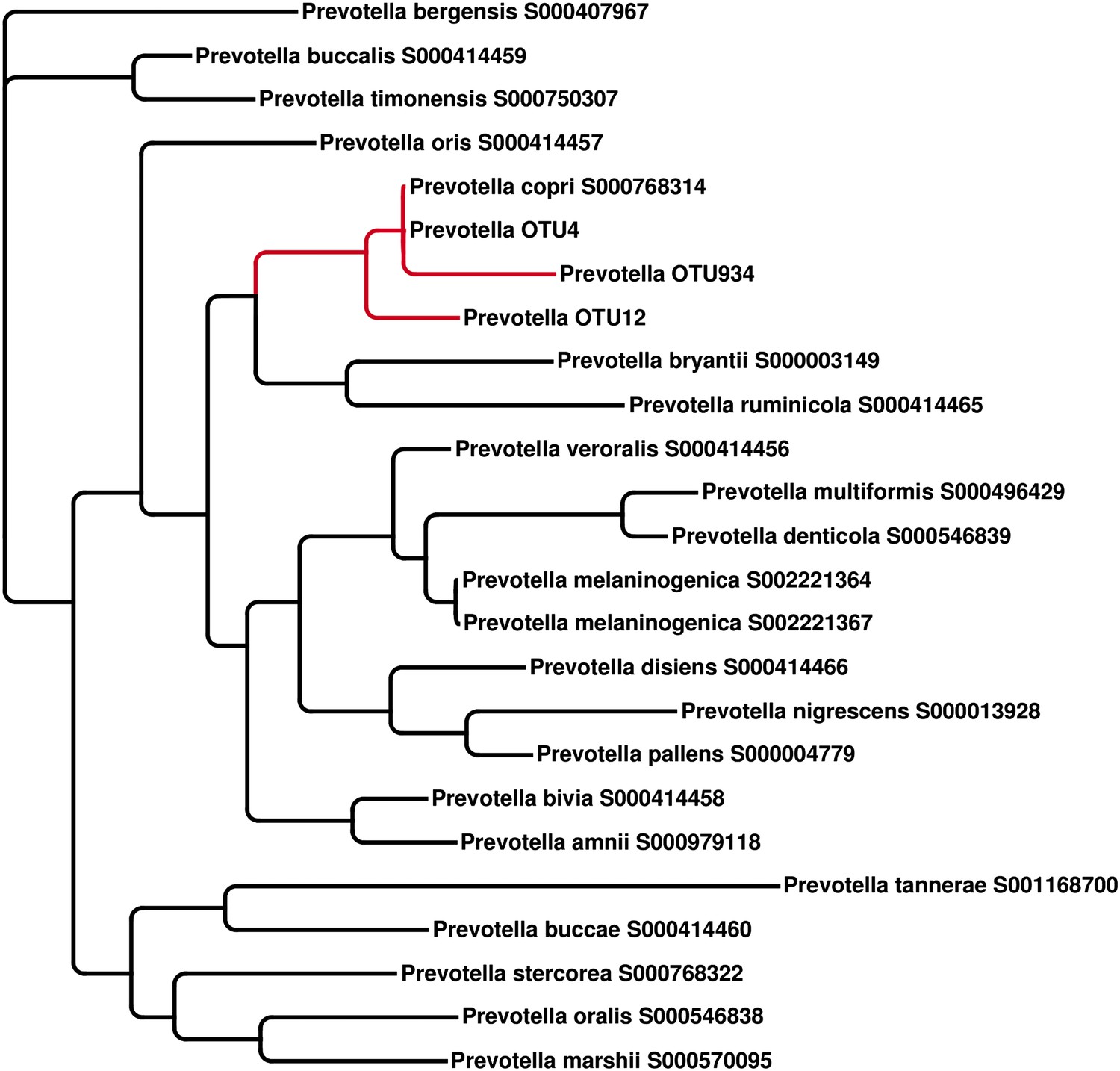
The representative 16S sequenced reads for Prevotella OTU4, OTU12, and OTU934 were aligned with MUSCLE (Edgar, 2004) and clustered with FastTree (Price et al., 2010)
All three Prevotella OTUs cluster with the full-length reference 16S sequence of P. copri.
Figure 3 with 2 supplements
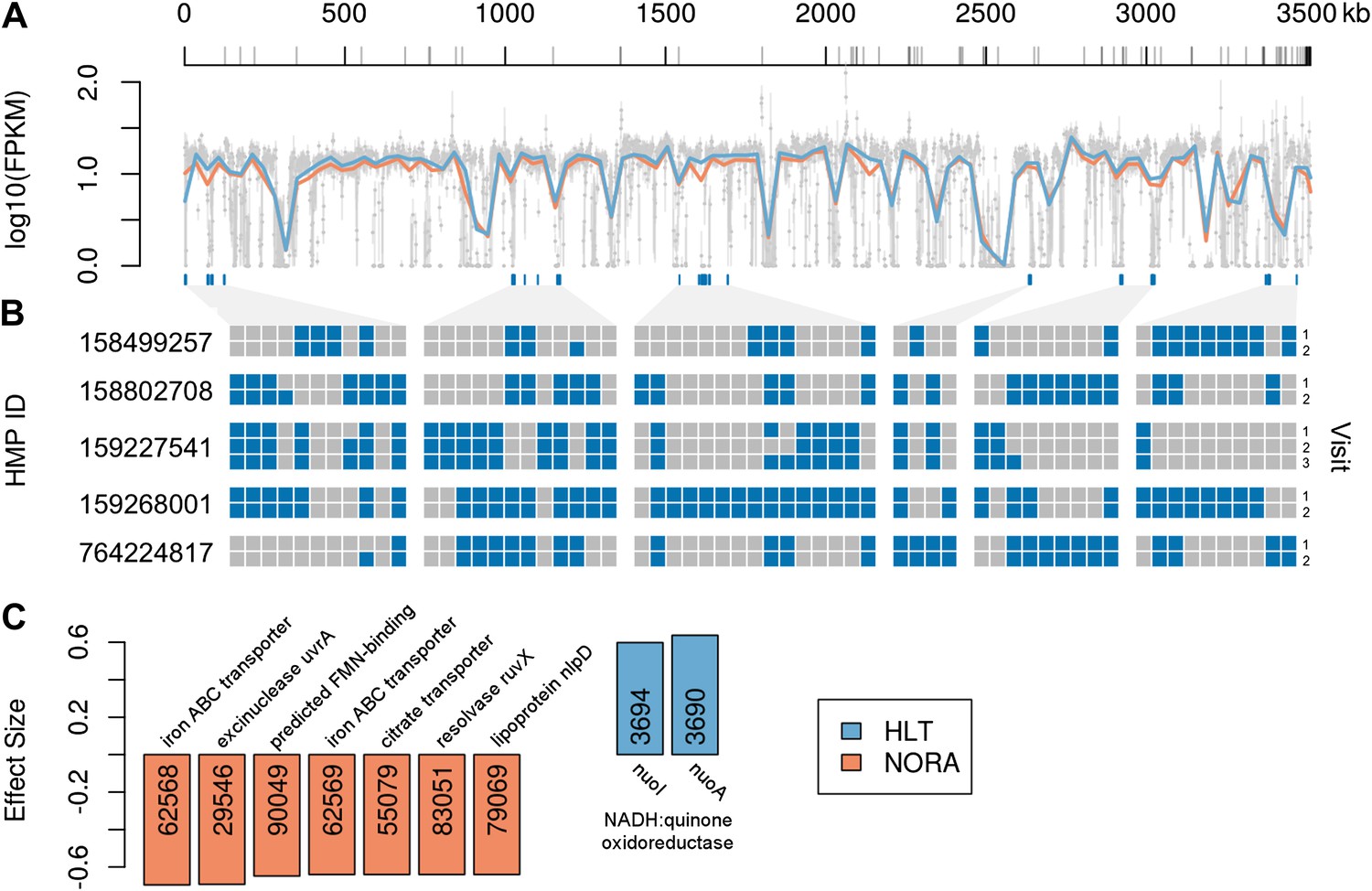
Comparison of P. copri genomes from healthy and NORA subjects.
(A) Comparative coverage of the draft P. copri DSM 18205 genome between individuals and within healthy and NORA groups. Gray points are median fragments per kilobase per million (FPKM) for 1-kb windows, gray lines within the plot are the interquartile range for each window, red and blue lines the LOWESS-smoothed average for NORA and healthy groups, respectively. Gray lines on the horizontal axis represent boundaries between assembled contigs. Regions are variably covered between subjects and groups, with several genomic islands lacking overall or especially variable (dark blue lines below the plot). (B) The presence (blue) or absence (gray) of previously-reported P. copri-unique marker genes (Segata et al., 2012) in 11 stool samples from five subjects of the Human Microbiome Project (HMP) are shown as a heatmap. We report, in columns, only those P. copri-specific markers showing variable presence/absence patterns across the considered HMP samples. Each row represents a different sample collection date, groups of rows represent subjects, and groups of columns correspond to different variably covered genomic islands. Strains of P. copri are defined by the presence and absence of particular genes, which remain stable for at least 6 months in these individuals. All inter- and intra-individual comparisons between rows are highly statistically significant (p<<0.001, ‘ Materials and methods’). (C) The P. copri pangenome was identified by finding P. copri ORFs in all HMP and NORA cohort subjects, and the presence or absence of these ORFs was calculated for each subject (‘Materials and methods’, Figure 3—figure supplement 1). Several ORFs are statistically significant biomarkers between healthy and NORA status (q<0.25) (Supplementary file 1B, ‘Materials and methods’).
-
Figure 3—source data 1
Intermediate data and analysis tools for Figure 3.
- https://doi.org/10.7554/eLife.01202.015
-
Figure 3—source data 2
Intermediate data and analysis tools for Figure 3—figure supplement 1.
- https://doi.org/10.7554/eLife.01202.016
Figure 3—figure supplement 1
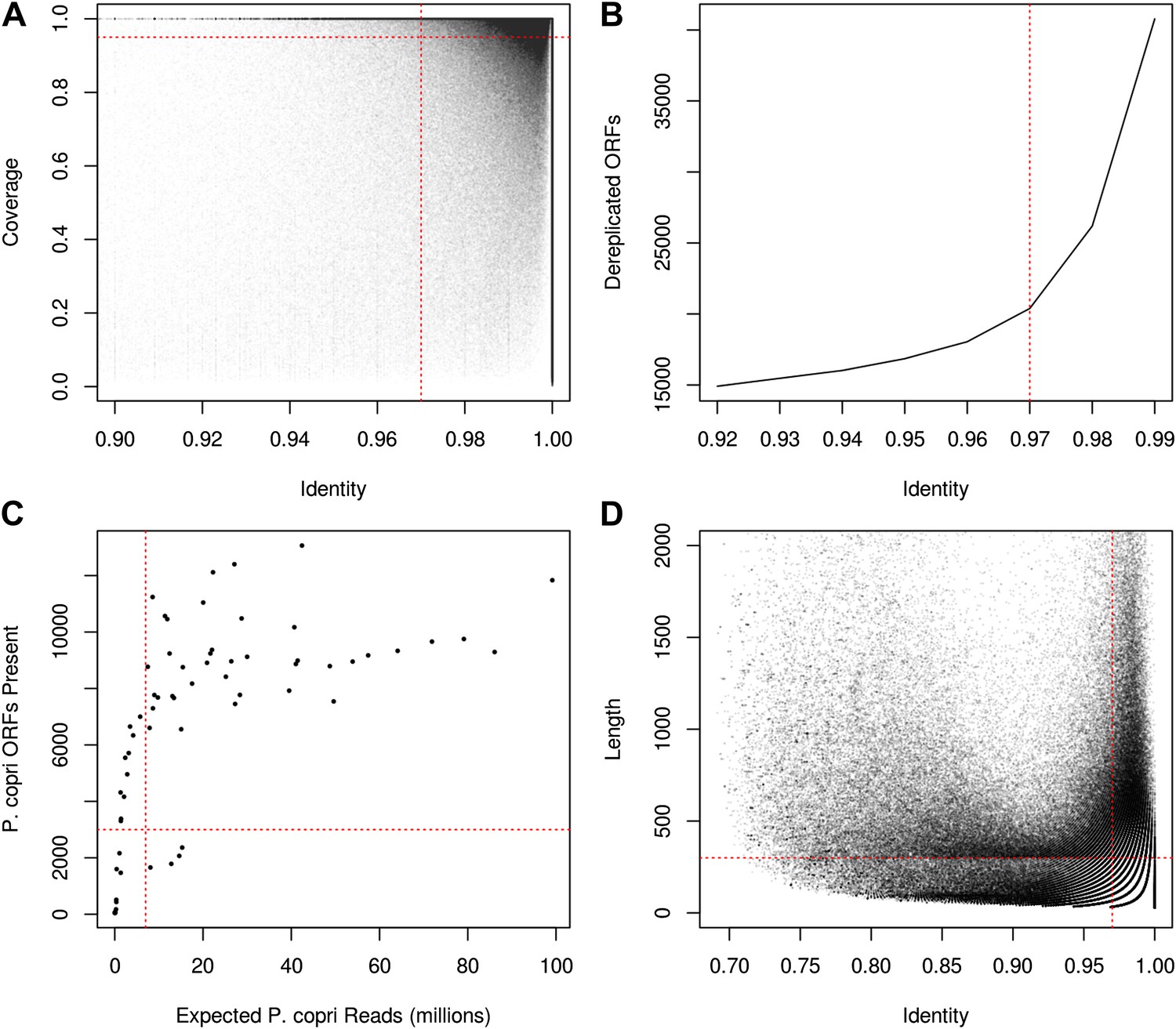
Recovery of P. copri pangenome from HMP/RA shotgun reads and determination of presence/absence of P. copri ORFs by alignment of reads to pangenome gene catalog.
(A) Genes were called present in a sample if they were covered by aligned reads at an identity threshold of >97% over >97% of their length (red lines). (B) ORFs were called on contigs using MetaGeneMark (Zhu et al., 2010) and were dereplicated with UCLUST (Edgar, 2010) at an identity threshold of 97% (red line). (C) Recovery of a sample's P. copri pangenome saturated at approximately 7 million reads (red line). We therefore excluded samples with less than 7 million P. copri reads, defined as P. copri abundance determined by MetaPhlAn (Segata et al., 2012) multiplied by the total number of quality-filtered reads. Samples with P. copri abundance likely misestimated (i.e., those with <3000 ORFs present) were also excluded (Supplementary file 1A). (D) Contigs were said to have originated from P. copri if they had at least one hit >97% identity over >300 bp (red lines).
Figure 3—figure supplement 2
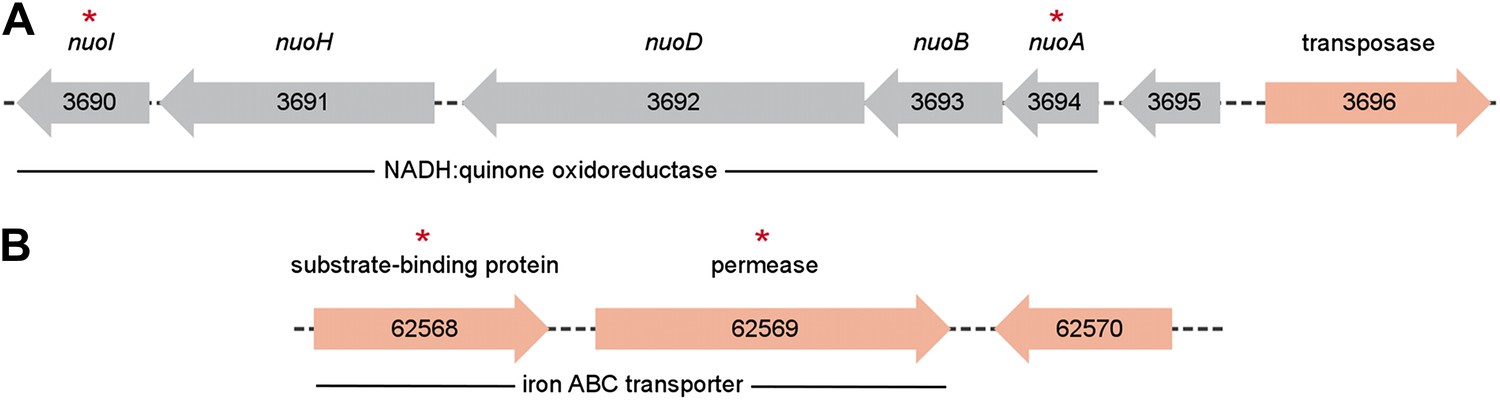
Metagenomic context of discriminative biomarker ORFs.
ORFs found in the P. copri DSM 18205 reference genome are colored red, while those identified as differentially present in healthy and NORA groups are indicated with red asterisks. (A) Two ORFs, 3690 and 3694, are healthy-specific, occur on the same contig, and encode different components of the same NADH:quinone oxidoreductase. (B) Similarly, ORFs 62,568 and 62,569 occur on the same contig, are NORA-specific, and encode components of the same iron ABC transporter.
Figure 4
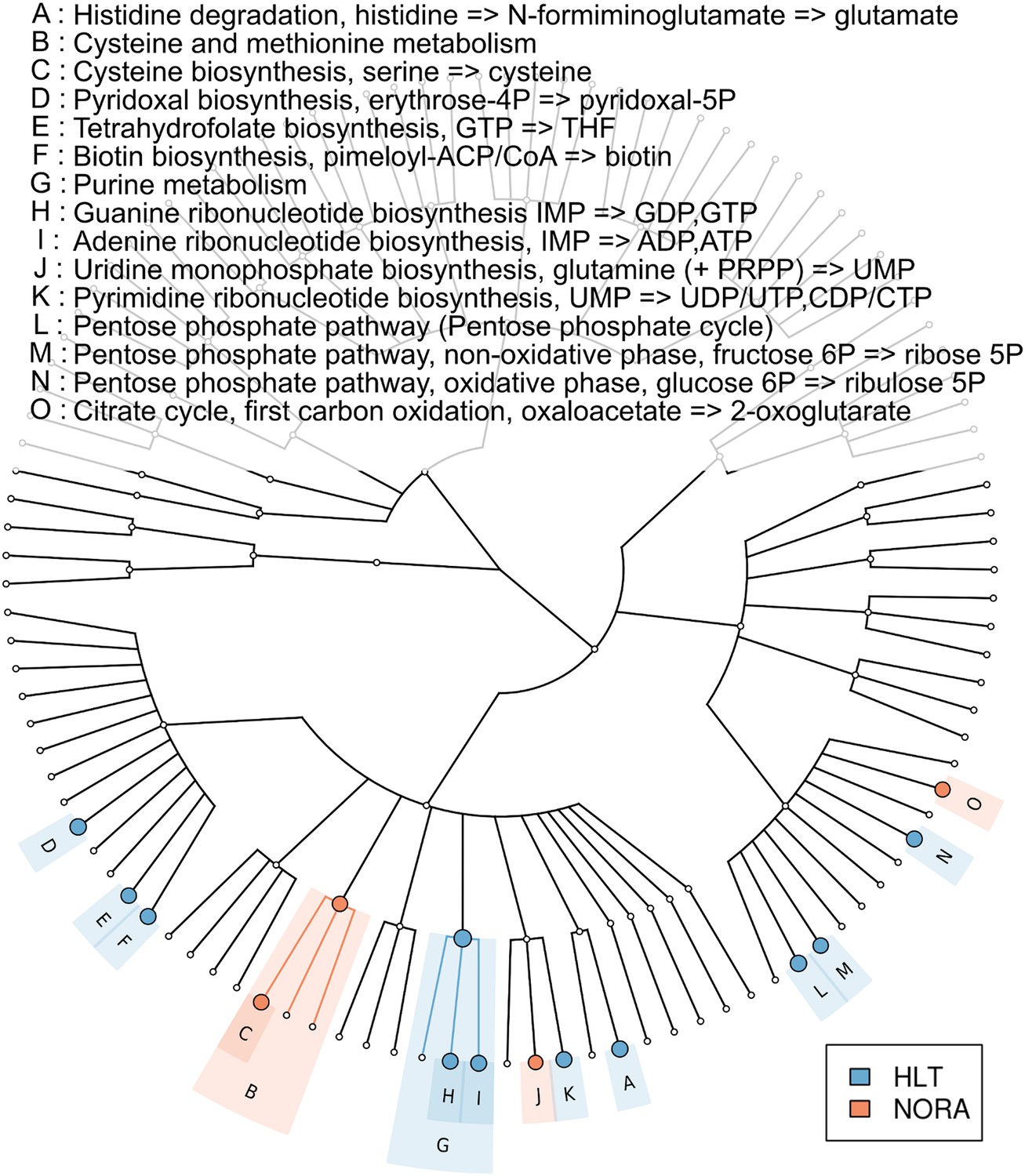
Metabolic pathway representation in the microbiome of healthy and NORA subjects.
HUMAnN (Abubucker et al., 2012) was applied to metagenomic reads (paired-end, 100 nt, Illumina platform) from NORA subjects (n = 14) and healthy controls (n = 5) to quantitate the abundances of hierarchically related KEGG modules in these samples (‘Materials and methods’ and Supplementary file 1A). LEfSe (Segata et al., 2011) was used to find statistically significant differences between groups at an alpha cutoff of 0.001 and an effect size cutoff of 2.0. Results shown here are highly significant (p<0.001) and represent large differences between groups. Modules highlighted in red are over-abundant in NORA samples while modules highlighted in blue are over-abundant in healthy samples. Prevotella-dominated NORA metagenomes have a dearth of genes encoding vitamin and purine metabolizing enzymes, and an excess of cysteine metabolizing enzymes.
-
Figure 4—source data 1
Intermediate data and analysis tools for Figure 4.
- https://doi.org/10.7554/eLife.01202.020
Figure 5
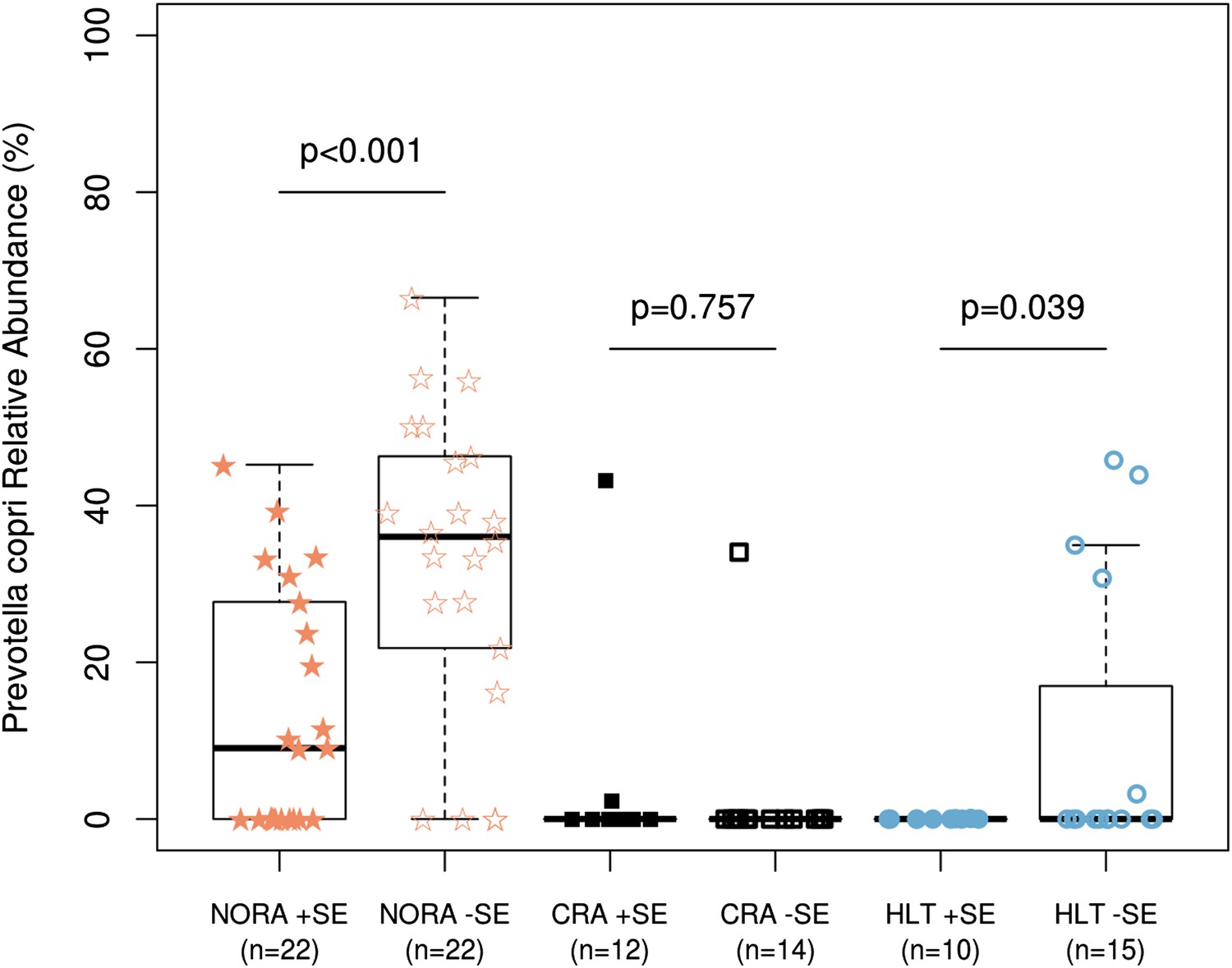
Relationship of host HLA genotype to abundance of P. copri (OTU4, OTU12, and OTU934 combined relative abundance).
The HLA-class II genotype of all subjects was determined by sequence-based typing methodology (‘Materials and methods’). Groups were subdivided by the presence or absence of shared-epitope RA risk alleles (+/− SE as indicated above) and correlated with relative abundance of intestinal P. copri. A statistically significant correlation is seen between P. copri abundance and the genetic risk for rheumatoid arthritis in NORA (red stars) and healthy (blue circles) subjects by Welch’s two-tailed t test.
-
Figure 5—source data 1
Intermediate data and analysis tools for Figure 5.
- https://doi.org/10.7554/eLife.01202.022
Figure 6 with 1 supplement
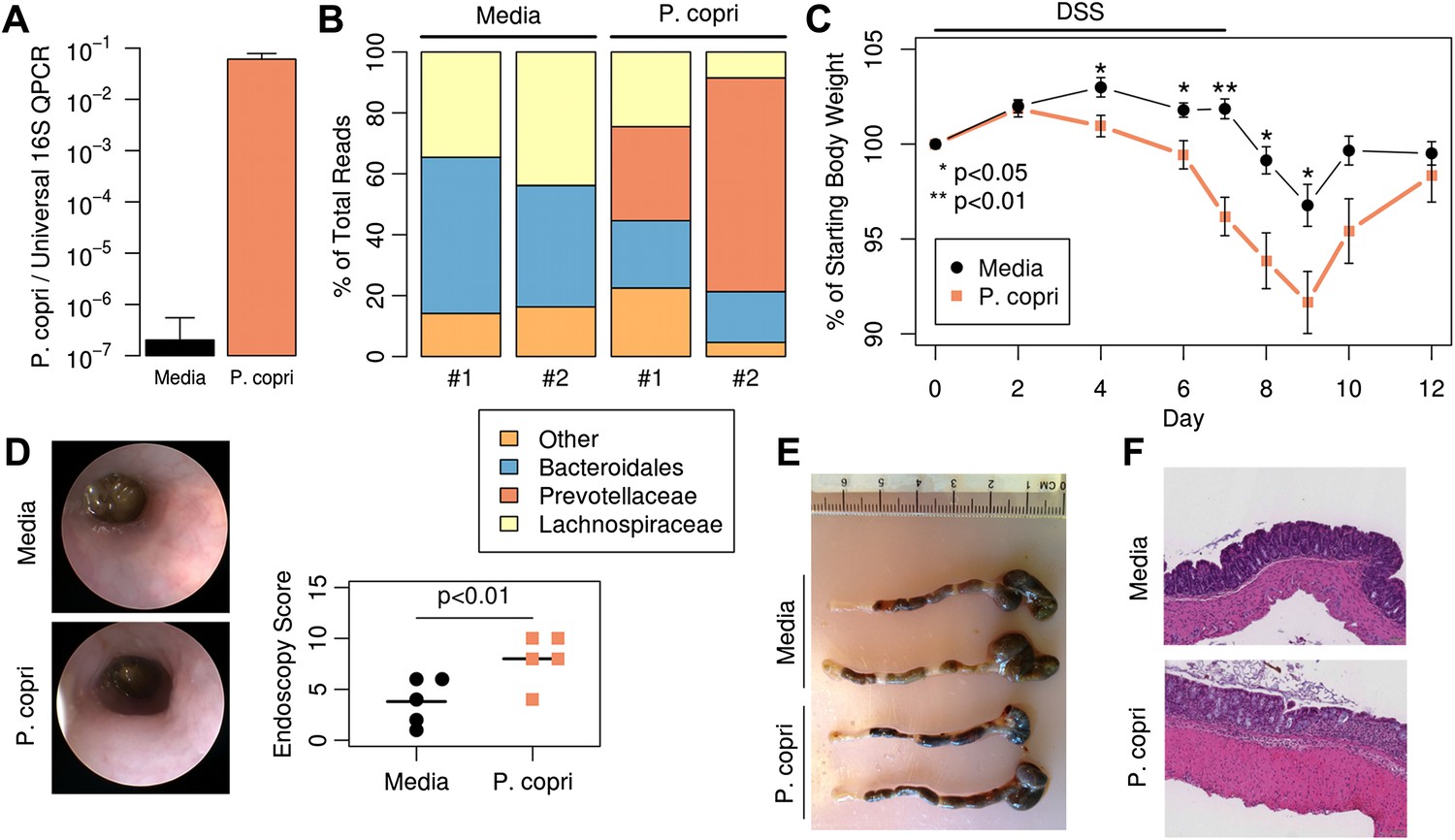
Colonization with P. copri dominates the colonic microbiome and exacerbates local inflammatory responses.
(A) DNA was extracted from fecal pellets of media-gavaged mice and P. copri-gavaged mice 2 weeks after colonization and assayed by QPCR with P. copri specific primers compared to universal 16S. (B) Relative abundance of bacterial families in fecal DNA from media-gavaged and P. copri-colonized mice (shown in duplicate) by high-throughput 16S sequencing (regions V1–V2, 454 platform). (C) C57BL/6 mice colonized with P. copri (n = 15) or media alone (n = 13) controls were exposed to DSS for seven days and percent of starting body weight is shown. Composite data from three representative experiments are shown. (D) Representative colonoscopic images of mice colonized with P. copri or media gavage following DSS-induced colitis. Endoscopic colitis score for five individual animals is displayed. (E and F) Gross pathology (E) and histology (F) of colons from mice colonized with P. copri or media gavage following DSS-induced colitis.
-
Figure 6—source data 1
Intermediate data and analysis tools for Figure 6.
- https://doi.org/10.7554/eLife.01202.024
Figure 6—figure supplement 1
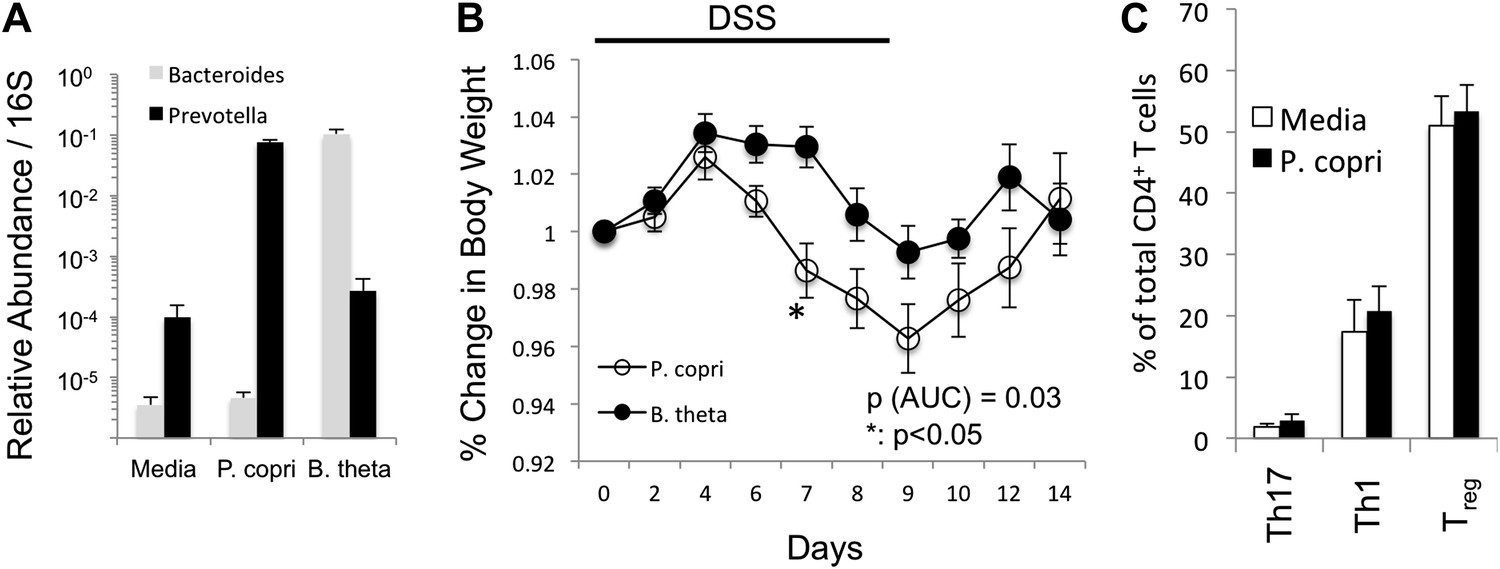
P. copri colonization exacerbates chemically induced colitis.
(A) DNA was extracted from fecal pellets of media, P. copri, and B. thetaiotamicron gavaged mice 2 weeks after colonization and assayed by QPCR with P. copri or Bacteroides specific primers compared to universal 16S amplicon. (B) C57BL/6 mice colonized with P. copri (n = 10) or B. theta (n = 10) were exposed to DSS for seven days and percent of starting body weight is shown. (C) Percent of total CD4+ T-cells in the colonic lamina propria expressing IL-17 (Th17) or IFNγ (Th1) following PMA/ionomycin stimulation or expressing Foxp3 (Treg).
Tables
Table 1
Demographic and clinical data among subjects with new-onset rheumatoid arthritis (NORA), chronic, treated rheumatoid arthritis (CRA), psoriatic arthritis (PsA), and healthy controls (HLT)
https://doi.org/10.7554/eLife.01202.003| NORA | CRA | PsA | Healthy | |
|---|---|---|---|---|
| (n = 44) | (n = 26) | (n = 16) | (n = 28) | |
| Age, years, mean (median) | 42.4 (40.0) | 50.0 (49.0) | 46.3 (46.0) | 42.8 (40.0) |
| Female, % | 75 | 88 | 56 | 75 |
| Disease duration, months, mean (median) | 5.4 (2.0) | 72.3 (48.0) | 0.8 (0.0) | N/A |
| Disease activity parameters | ||||
| ESR, mm/h, mean | 34.6 | 33.5 | 19.7 | 10.2 |
| CRP, mg/l, mean | 20.6 | 8.2 | 7.6 | 1.1 |
| DAS28, mean (median) | 5.4 (5.7) | 4.7 (5.0) | 4.8 (4.7) | N/A |
| Patient VAS pain, mm, mean (median) | 61.4 (57.5) | 51.5 (62.5) | 50.6 (45.0) | N/A |
| TJC-28, mean (median) | 11.2 (8.5) | 7.6 (7.0) | 8.8 (6.5) | N/A |
| SJC-28, mean (median) | 8.3 (8.0) | 4.6 (3.0) | 4.8 (3.0) | N/A |
| Autoantibody status | ||||
| IgM-RF positive, % | 95 | 81 | 13 | 11 |
| ACPA positive, % | 100 | 85 | 6 | 7 |
| IgM-RF and/or ACPA positive, % | 100 | 96 | 13 | 14 |
| IgM-RF titer, kU/l, mean (median) | 341.3(157.0) | 178.2 (89.0) | 3.6 (0.0) | 20.5 (0.0) |
| ACPA titer, kAU/l, mean (median) | 117.6 (114.0) | 90.8 (57.0) | 1.6 (0.0) | 9.6 (0.0) |
| Medication use | ||||
| Methotrexate, % | 0 | 42 | 6 | 0 |
| Prednisone, % | 0 | 15 | 6 | 0 |
| Biological agent, % | 0 | 12 | 0 | 0 |
Table 2
Draft genome assembly statistics of four subjects with a high abundance of Prevotella OTU4
https://doi.org/10.7554/eLife.01202.008| Total | P. copri aligned | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject ID | Group | Prevotella OTU4 abundance (%) | # reads | # of contigs | Size (Mb) | N50 (kb) | Mean depth | # of contigs | Size (Mb) | N50 (kb) | Mean depth |
| 028B | NORA | 27.7 | 1,240,515 | 19,988 | 23.24 | 1.45 | 6.13 | 115 | 3.21 | 59.84 | 36.76 |
| 030B | NORA | 50.9 | 1,041,546 | 21,579 | 17.35 | 1.01 | 6.97 | 232 | 2.60 | 16.18 | 44.14 |
| 061B | NORA | 66.5 | 1,209,392 | 9,241 | 12.8 | 1.58 | 9.88 | 74 | 3.23 | 79.98 | 172.64 |
| 089B | NORA | 56.3 | 1,395,872 | 12,112 | 23.47 | 4.64 | 23.12 | 1,963 | 3.96 | 3.19 | 30.39 |
| Ref. genome | – | – | – | – | – | – | – | 83 | 3.51 | 131.4 | – |
Table 3
Statistical comparisons of Prevotella copri prevalence between cohort groups
https://doi.org/10.7554/eLife.01202.013Additional files
-
Supplementary file 1
(A) Read statistics of sequenced samples included in and excluded from biomarker analyses. (B) Presence/absence, p-values and FDR statistics for differentially represented ORFs in the P. copri pangenome biomarker analysis, with annotations. (C) KOs present in P. copri DSM 18205 but not in any Bacteroides accounting for at least 5% of the total microbiota in any subject of the Human Microbiome Project. (D) KOs present in all genomes available for Bacteroides accounting for at least 5% of the total microbiota in any subject of the Human Microbiome Project and not present in P. copri DSM 18205. (E) HLA-DRB1 alleles were determined for subjects in the cohort. Counts of RA risk alleles (shared epitope) are indicated as 0 for homozygotes not at risk, one for heterozygotes, and two for homozygotes at risk (‘Materials and methods). Shared epitope alleles appear in bold.
- https://doi.org/10.7554/eLife.01202.026
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis
eLife 2:e01202.
https://doi.org/10.7554/eLife.01202




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}