Global cellular response to chemotherapy-induced apoptosis
- University of California, San Francisco, United States
- The College of New Jersey, United States
- Department of Pathology, University of California, San Francisco, United States
- Howard Hughes Medical Institute, University of California, San Francisco, United States
Figures
Figure 1 with 3 supplements
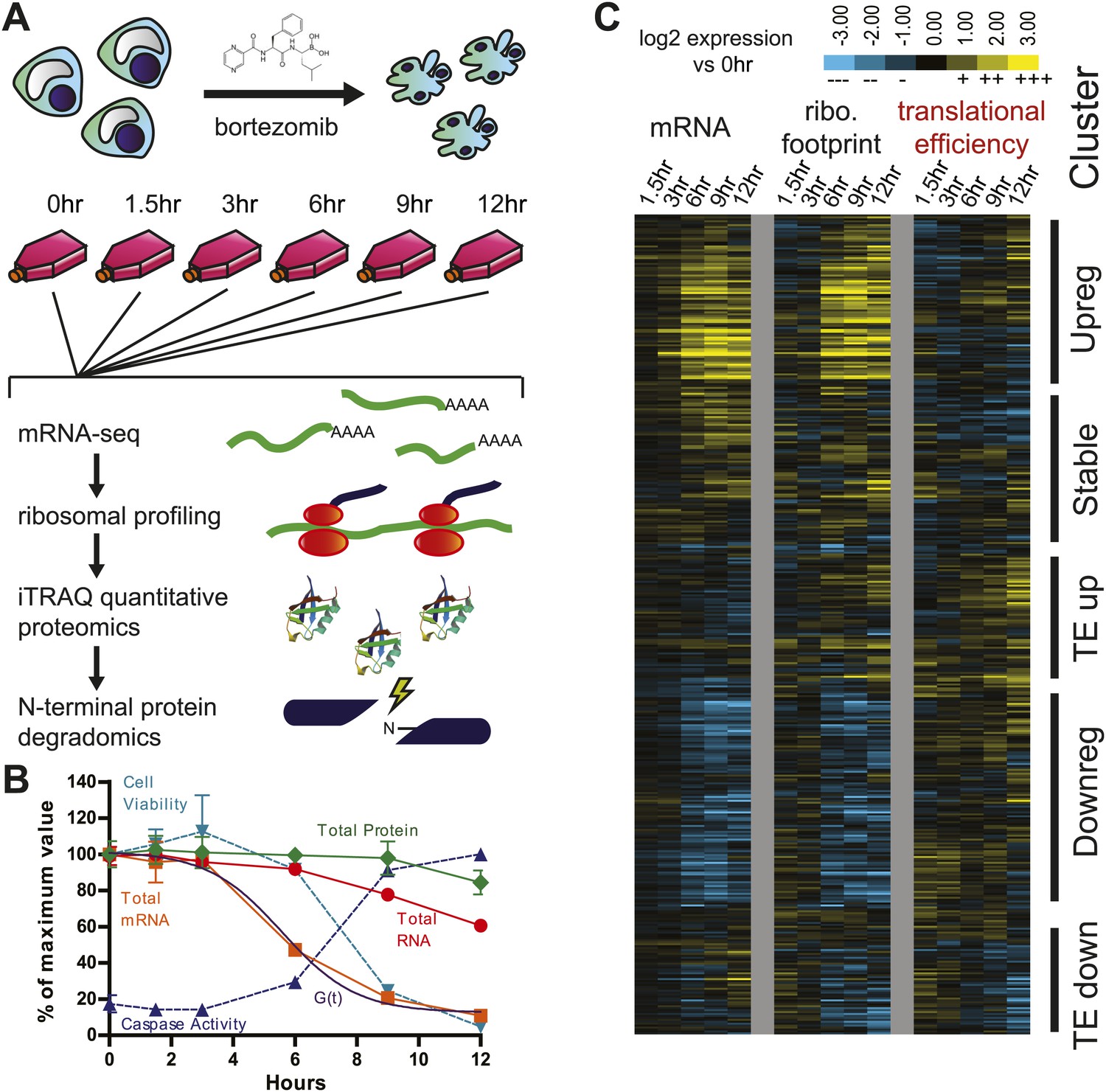
Experimental design and paired mRNA-seq/ribosome profiling data.
(A) MM1.S cells were prepared in six separate flasks each containing 3 × 108 cells. All time points past 0 hr were exposed to 20 nM bortezomib. Cells were harvested and analyzed using four systems-level technologies shown. (B) Cells undergo rapid apoptosis with initial increase in caspase activation seen at 6 hr and <10% cell viability by 12 hr (duplicate, mean ± SD). Total RNA and mRNA were isolated and measured by spectrophotometry; total protein was measured by BCA assay (measured in duplicate, isolated from 106 cells; mean ± SD). G(t), describing global mRNA degradation, is derived from a sigmoid fit to the total mRNA data. (C) Heat map showing log2 expression compared to 0 hr for normalized mRNA and ribosome footprint read density (reads per kilobase million, RPKM) for 5680 well-expressed transcripts (Figure 1—source data 1). Also shown is calculated translational efficiency (TE) relative to 0 hr, calculated as the ratio of (footprint RPKM)/(mRNA RPKM) per transcript at each time point. We used unsupervised hierarchical clustering to define five broad groups of transcripts with relation to mRNA, footprint, and translational efficiency changes.
-
Figure 1—source data 1
Deep sequencing data, translational efficiency, and gene clusters.
(A) mRNA-seq and (B) ribosome profiling deep sequencing data with assigned UCSC hg19 identifier, associated UniProt accession number, and gene identifier as defined by kgXref feature in UCSC genome browser. For samples at each time point, we display raw reads that uniquely mapped to canonical transcript as well as mapped reads normalized to total read number and transcript length (reads per kilobase million, RPKM) for comparison across samples. These 5680 tracked transcripts had RPKM ≥1 across all 12 samples (six mRNA-seq, six ribosome footprint). (C) Translational efficiency (TE) for all tracked transcripts as measured by (RPKM of footprint sample/RPKM of mRNA-seq sample) at each time point. Table is ordered by descending TE at 0 hr. The histone genes are artifactually elevated as these mRNA's do not contain poly (A) tails and are therefore under-represented in the mRNA-seq data, as previously noted in Ingolia et al. (2011). (D) List of transcripts included in each Cluster (as in Figure 1C) as uploaded to Ingenuity Pathway Analysis server for analysis (results in Table 1).
- https://doi.org/10.7554/eLife.01236.004
Figure 1—figure supplement 1
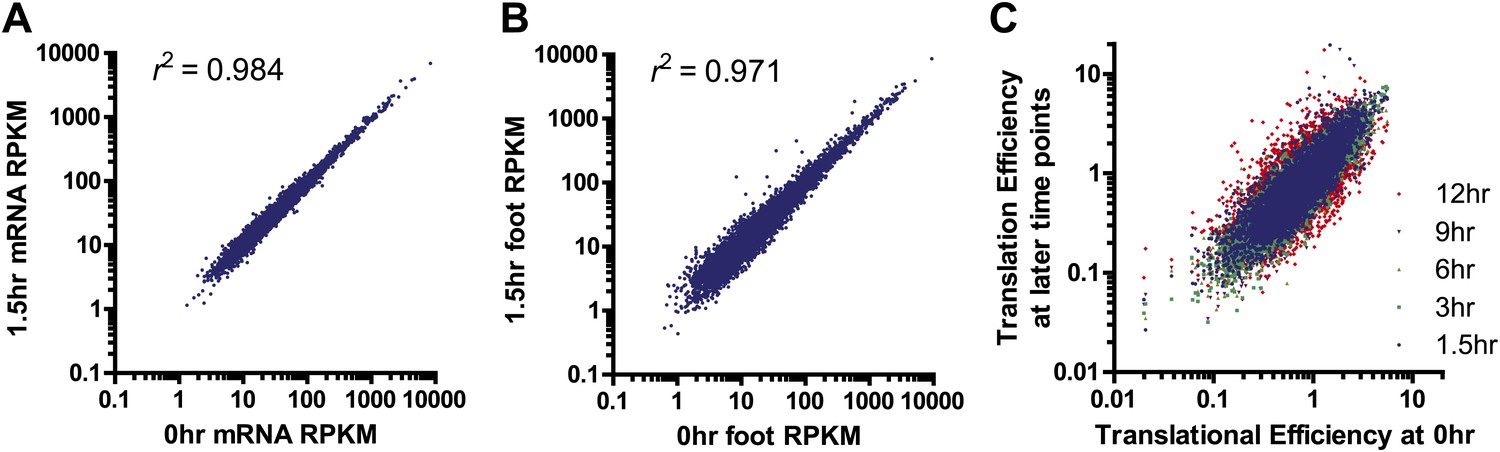
Sample comparison and biological subgroups.
(A) mRNA and (B) footprint samples prepared and sequenced independently at early time points show strong correlation in read density, demonstrating consistency in experimental methods. In footprint samples the mean difference between read density at 1.5 hr and 0 hr across transcripts = 28%, reflecting both experimental error and biological variation. (C) Translational efficiency spans >10-fold across all tracked transcripts without any large shifts across time points.
Figure 1—figure supplement 2
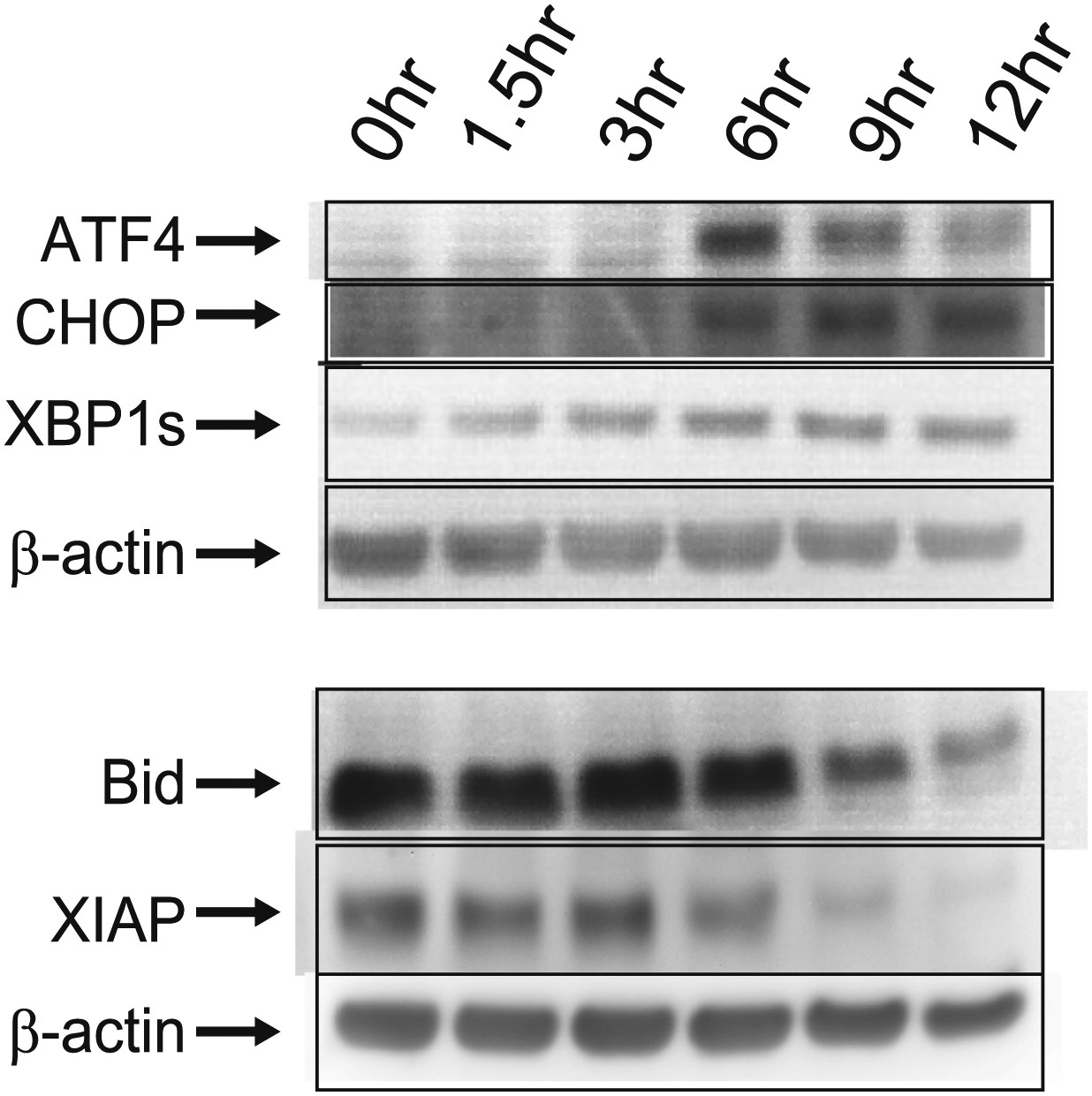
Immunoblots.
Western blotting for proteins related to ER stress/UPR (ATF4, CHOP, XBP1s [‘s’ for spliced, active form]) and apoptosis (Bid, XIAP).
Figure 1—figure supplement 3

Biological subgroups.
Genes curated from the literature related to ER stress (left) and apoptosis (right) that were found in among the ∼5700 tracked transcripts. Genes highlighted with ribosome footprint analysis are highlighted with red arrows (DDIT3 = CHOP; DDIT4 = REDD1; PPP1R15A = GADD34). All data are shown as Log2 expression ratio vs 0 hr.
Figure 2 with 1 supplement
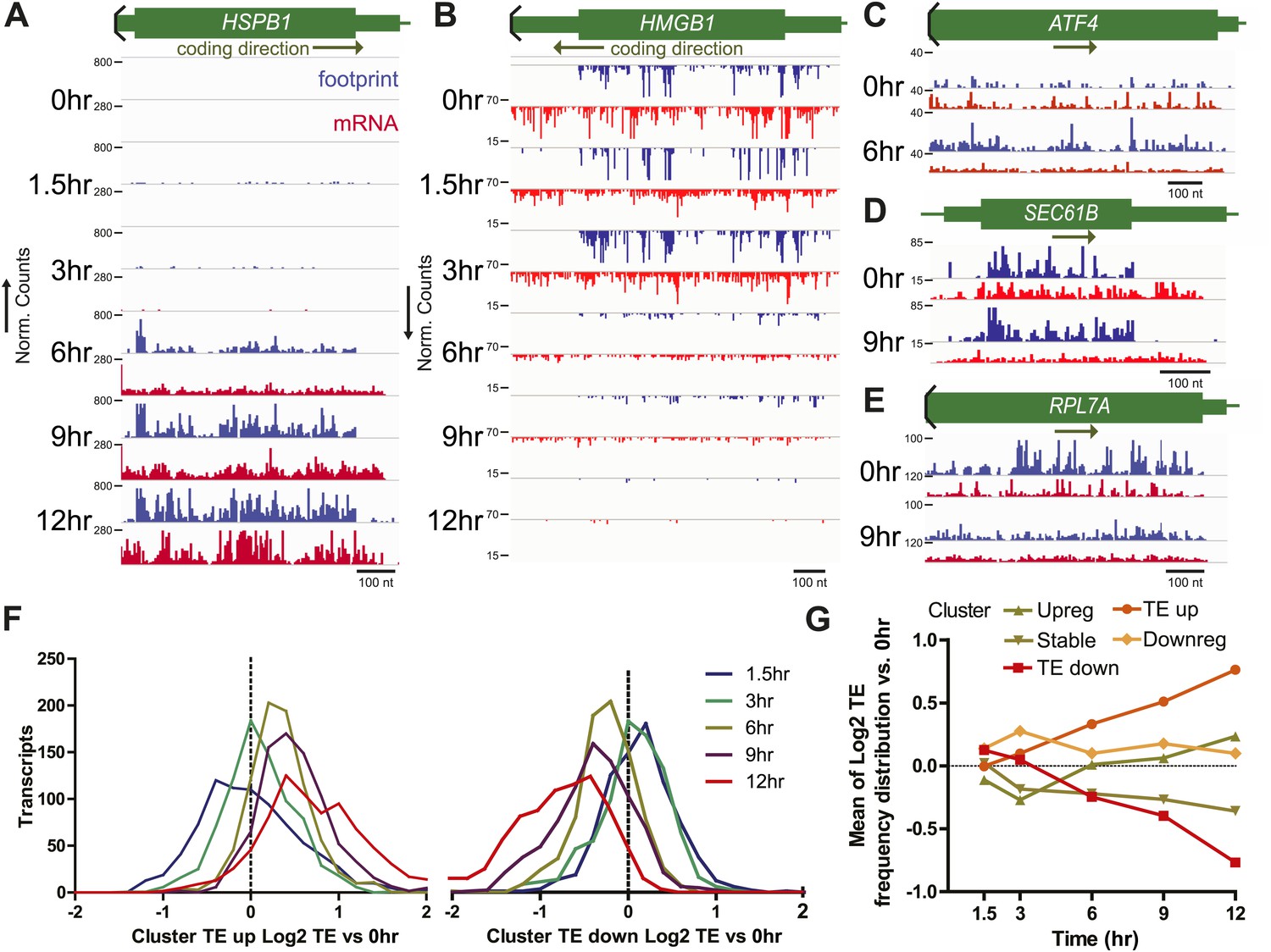
Translational efficiency (TE) during bortezomib-induced apoptosis.
(A and B) Paired mRNA and ribosome footprint reads for HSPB1 (Cluster Upreg) and HMGB1 (Cluster Downreg) showing that protein translation generally tracks with transcript abundance. Displayed read counts are median-normalized to total aligned reads in 0 hr sample for comparison across the time course. Thick green bars = protein coding sequence (CDS); medium bars = untranslated regions (UTR); thin lines = intronic or intergenic regions. Read counts are inverted depending on coding direction. (C) ATF4 shows increased footprint read density in the absence of mRNA increase at 6 hr. (D) SEC61B, a downstream target of XBP1, shows increased footprints and decreased mRNA reads, leading to increased TE. (E) Many ribosome structural proteins, including RPL7A, are found in Cluster TE down. (F) Log2 changes in transcript TE from Cluster TE up and Cluster TE down at 12 hr vs 0 hr demonstrate statistically significant changes in the overall TE distribution (p<0.0001, Mann-Whitney test). (G) Mean (±SEM) changes in TE during the apoptotic time course generally do not exceed twofold even at later time points.
Figure 2—figure supplement 1

Subgroup analysis from biological pathways showing changes in translation efficiency from Ingenuity Pathway Analysis.
Genes included in IPA as XBP1 downstream targets (left) and Genes included as part of the eIF2 signaling pathway in IPA (right). Green boxed area indicates numerous ribosome structural proteins with decreased footprint reads relative to mRNA. SEC61B and RPL7A (shown in Figure 2) are highlighted with red arrows, as is PERK (EIF2AK3). All data are shown as Log2 expression ratio vs 0 hr.
Figure 3 with 3 supplements

Nucleotide resolution of ribosome profiling reveals changes in 5′ UTR translation.
(A) Metagene analysis, the alignment of footprint reads across all transcripts, to both the 5′ CDS start site and 3′ termination site (Ingolia et al., 2009). Reads counts are median-normalized to total aligned reads per sample. No significant changes are noted in overall read alignments during apoptosis. (B) Relative translation of 5′ UTR measured by the ratio of 5′ UTR reads to CDS reads per transcript. The log2 change in this ratio is compared to the 5′ UTR/CDS ratio at 0 hr. (C and D) Examples of genes with most increased or decreased relative 5′ UTR translation include HSPA8 (Hsc70 protein) and CYCS (cytochrome c), respectively. (E–H) Genes known to be involved in ER stress response show increased read density at 5′ UTR upstream open reading frames (uORFs). ATF4 shows density at known uORFs as well as a single upstream AUG codon. Putative uORF translation with noted initiation codon is also identified based on areas of increased read density for DDIT4 (REDD1), PPP1R15B, and TXNIP (see detailed sequence information in Figure 3—figure supplement 2).
-
Figure 3—source data 1
5′ UTR translation.
(A) RPKM values of reads uniquely mapped to either 5′ UTR or CDS of transcripts (same list of transcripts as in Figure 1—source data 1). Sequence and length of 5′ UTR and CDS (for RPKM calculation) are defined based on the canonical transcript isoform annotated in hg19. (B) Transcripts with 5′ UTR/CDS translation ratio increased ≥twofold at a minimum of three time points compared to transcript 5′ UTR/CDS translation ratio at 0 hr. (C) as in B but for transcripts with decreased 5′ UTR to CDS translation ratio.
- https://doi.org/10.7554/eLife.01236.012
Figure 3—figure supplement 1
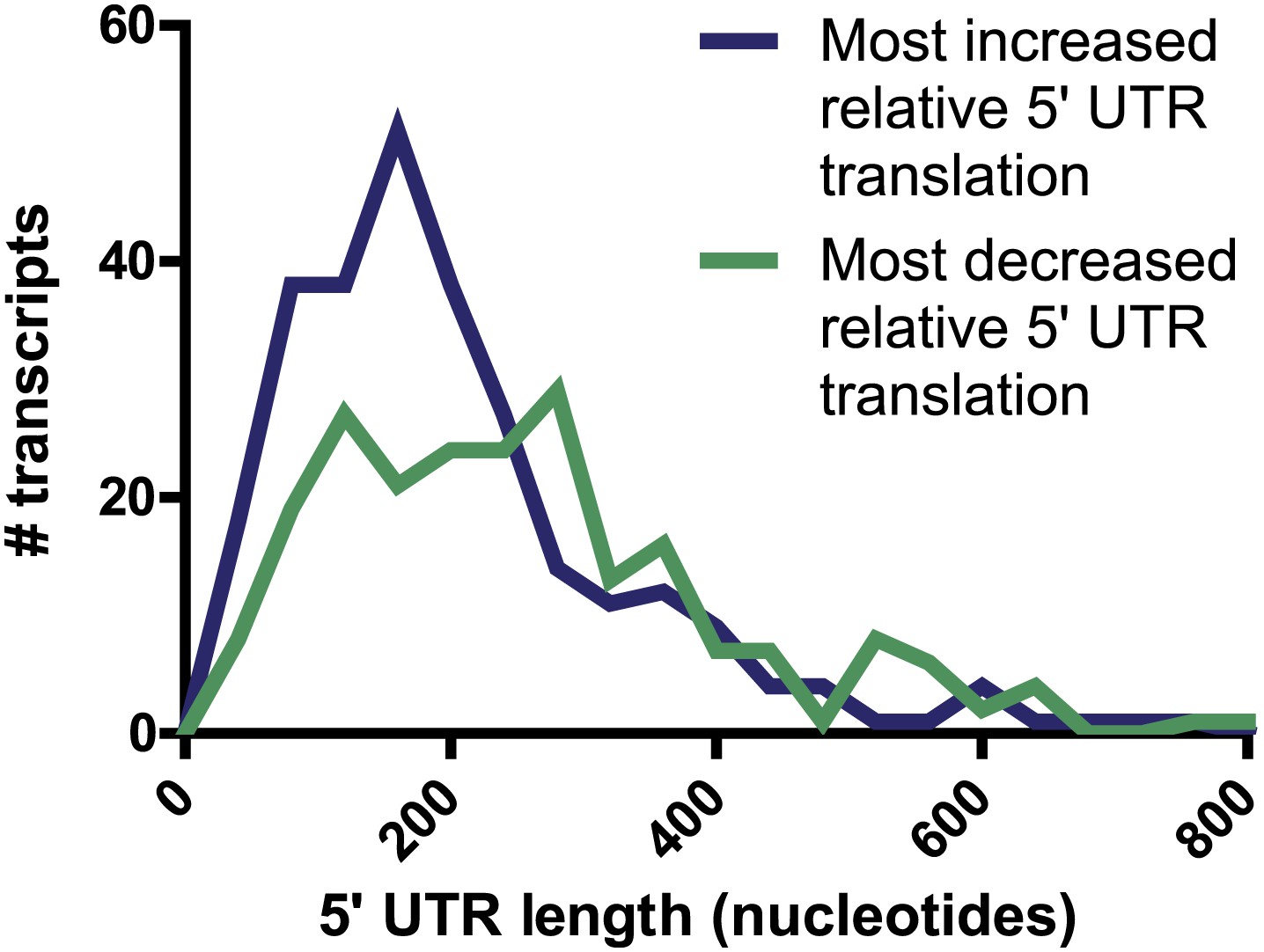
5′ UTR length.
For genes found to have most increased (274 genes) and most decreased (219 genes) relative 5′ UTR translation during the time course, there is no significant difference in the distribution of annotated 5′ UTR length (p=0.94, Mann-Whitney U test).
Figure 3—figure supplement 2
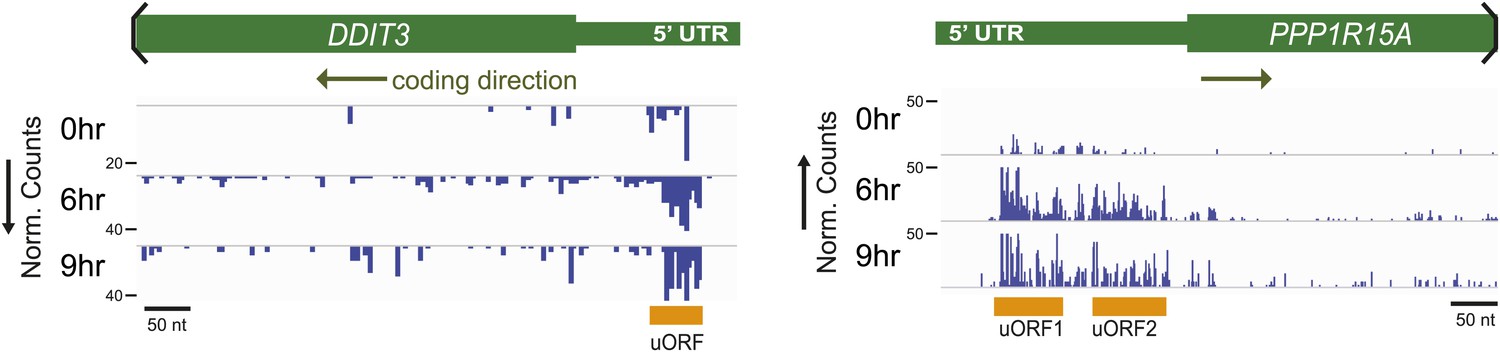
Ribosome profiling demonstrates strong footprint read density in known uORFs of DDIT3 (CHOP) and PPP1R15A (GADD34).
https://doi.org/10.7554/eLife.01236.014
Figure 3—figure supplement 3
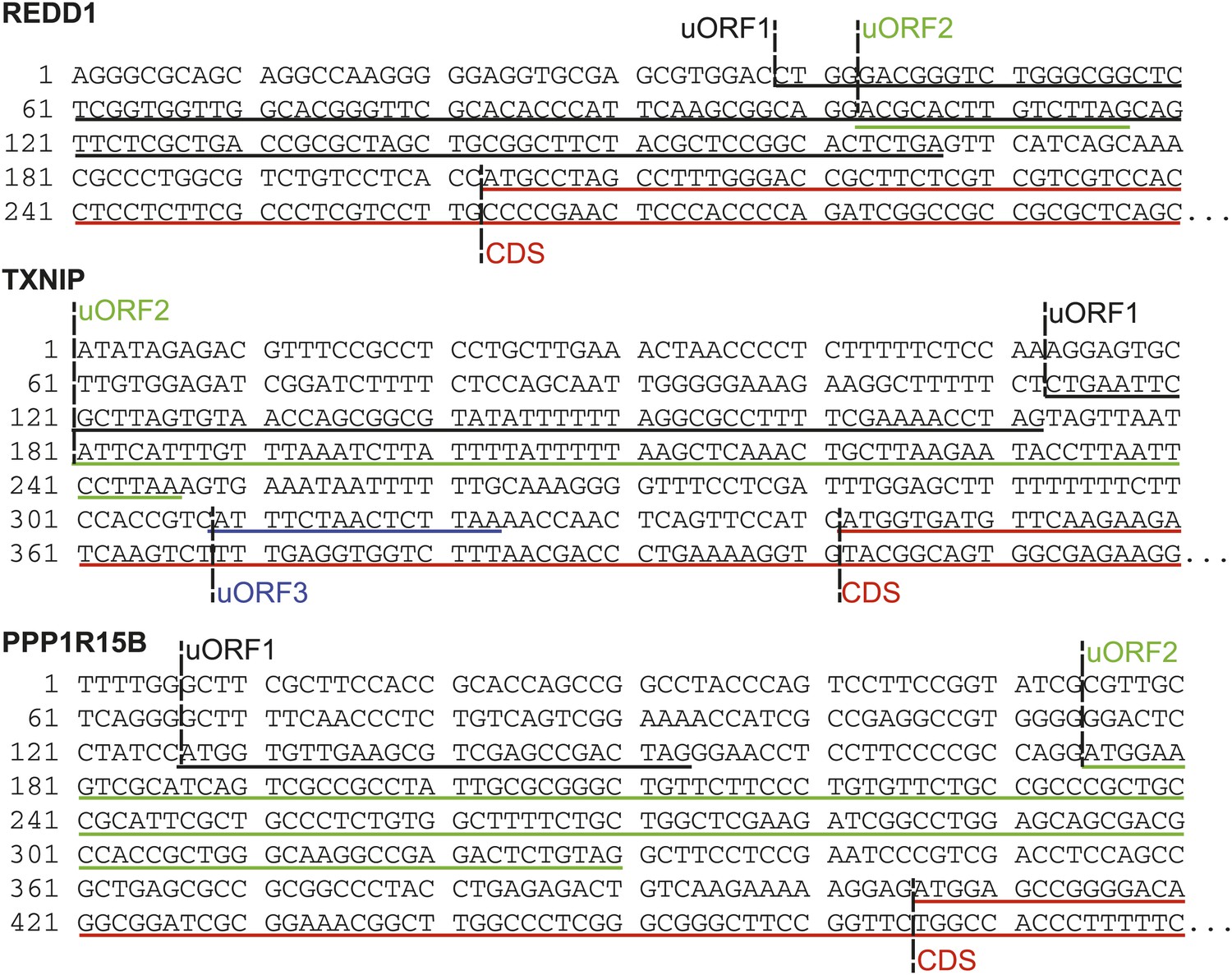
Sequence data indicating uORFs suggested by increased footprint read density in REDD1, TXNIP, and PPP1R15B 5′ UTRs.
Putative REDD1 uORFs are translated out of frame with the CDS; TXNIP uORFs 1 and 2 are out of frame, while uORF3 is in frame; both PPP1R15B uORFs are in frame with the CDS.
Figure 4 with 4 supplements
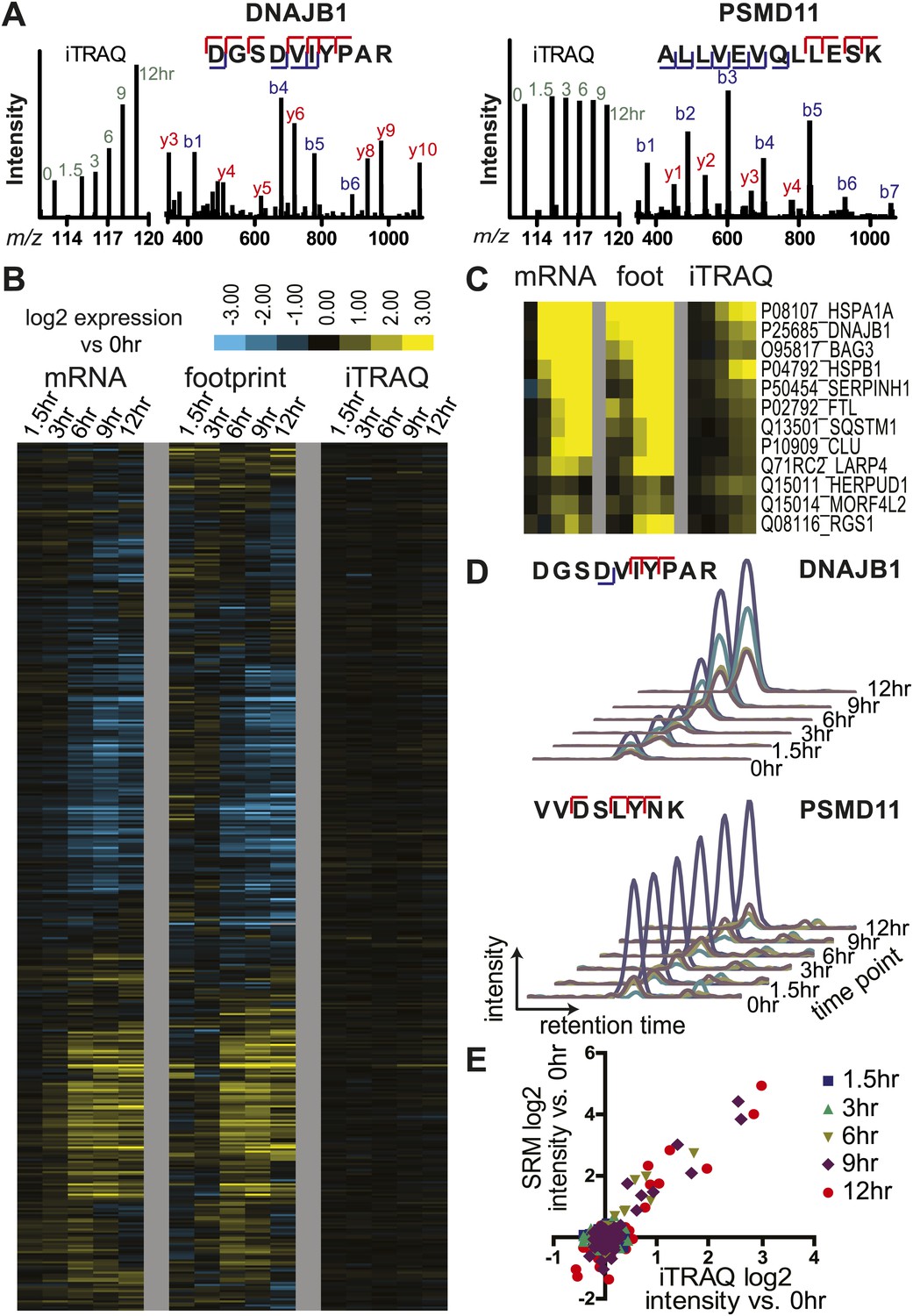
Unbiased and targeted quantitative proteomics reveals abundance changes in a small subset of proteins.
(A) Example mass spectra demonstrating both m/z peaks used for peptide sequencing and iTRAQ reporter ion signal to measure relative abundance across time points. (B) Hierarchical clustering heat map of paired mRNA and ribosome footprint relative read density vs 0 hr with relative iTRAQ protein abundance for 2572 proteins. (C) Inset of heat map for proteins increased in relative abundance by >50% at ≥2 time points shows few proteins are measurably produced during bortezomib-induced apoptosis. (D) Targeted selected reaction monitoring (SRM) assays orthogonally validate iTRAQ data for 152 proteins. In this representative data, each colored trace monitors the intensity of a given parent and fragment ion pair, as demarcated in the peptide sequence; multiple co-eluting peaks positively identify a targeted peptide. (E) Protein abundance measured by both SRM and iTRAQ demonstrate strong correlation across time points (r = 0.80).
-
Figure 4—source data 1
Proteomic data.
iTRAQ proteomic data organized at the protein (A) and peptide (B) level for 2686 proteins. All proteins included had a minimum of two unique peptides mapping to the protein; all peptides showed minimum iTRAQ reporter ion intensity of 300 cps at 0 hr. (C) iTRAQ proteomic data matched by UniProt Accession number to genes tracked in mRNA and footprint data across the time course. (D) List of proteins identified by iTRAQ proteomics but not present in analyzed genes from RNA deep sequencing. (E) List of parent and fragment ion transitions used for all peptides in SRM validation of iTRAQ data. (F) Protein and (G) peptide intensity data for targets tracked in SRM assay.
- https://doi.org/10.7554/eLife.01236.017
Figure 4—figure supplement 1
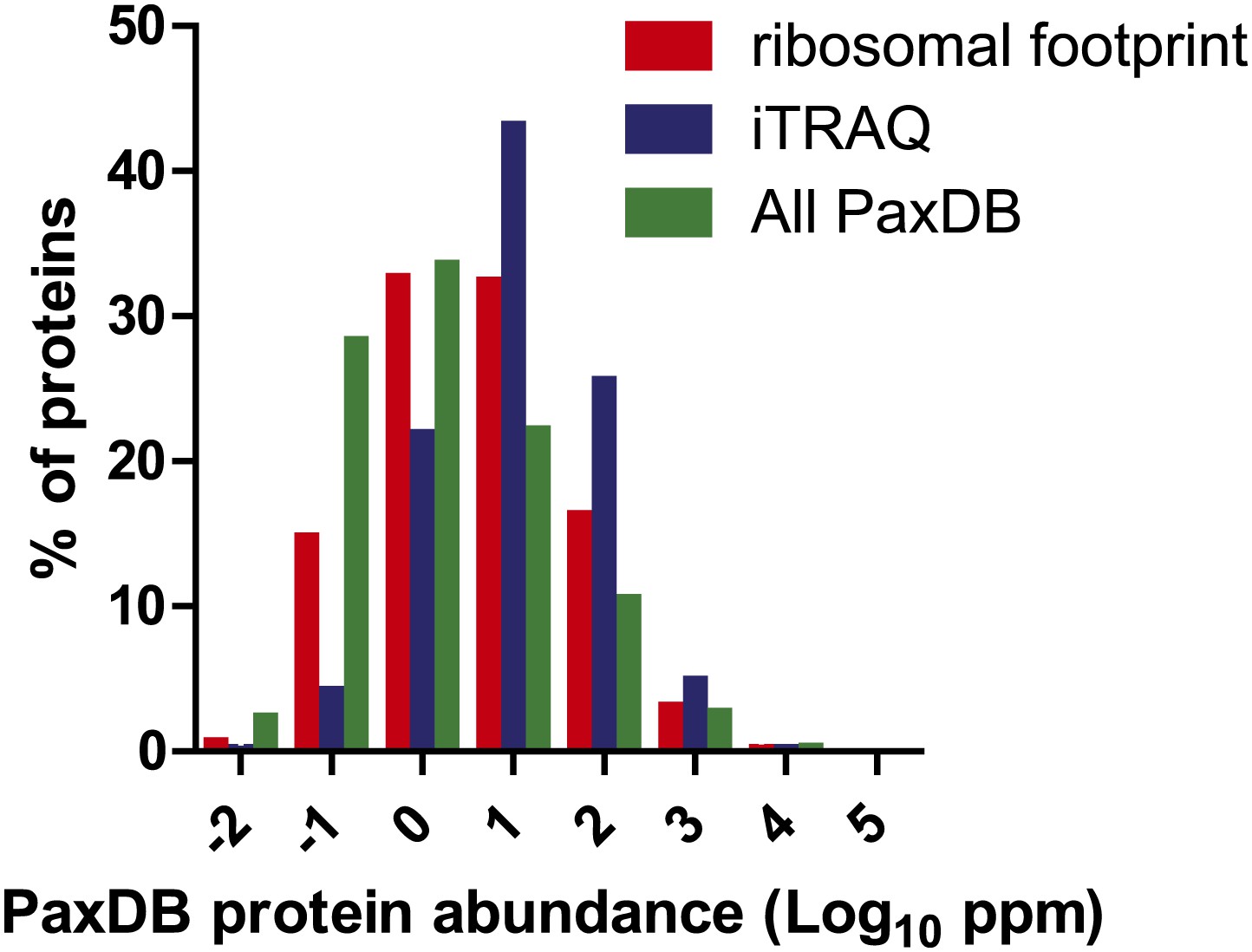
Protein abundance comparison.
Frequency distribution of protein abundance monitored by iTRAQ proteomics (∼2700 proteins) and ribosome profiling (∼5700 proteins) compared to average cellular protein abundance as estimated in PaxDB (www.pax-db.org). We note that both distributions examine similar portions of the proteome, though the iTRAQ demonstrates greater coverage of higher abundance proteins.
Figure 4—figure supplement 2

Comparison of baseline (0 hr) read density of mRNA transcripts vs number of identified peptides mapping to each protein in iTRAQ proteomics.
We find a weak but positive correlation (R = 0.27, p<0.0001). This distribution is comparable to the relationship between absolute transcript and protein copies at baseline in Figure 5—figure supplement 2.
Figure 4—figure supplement 3
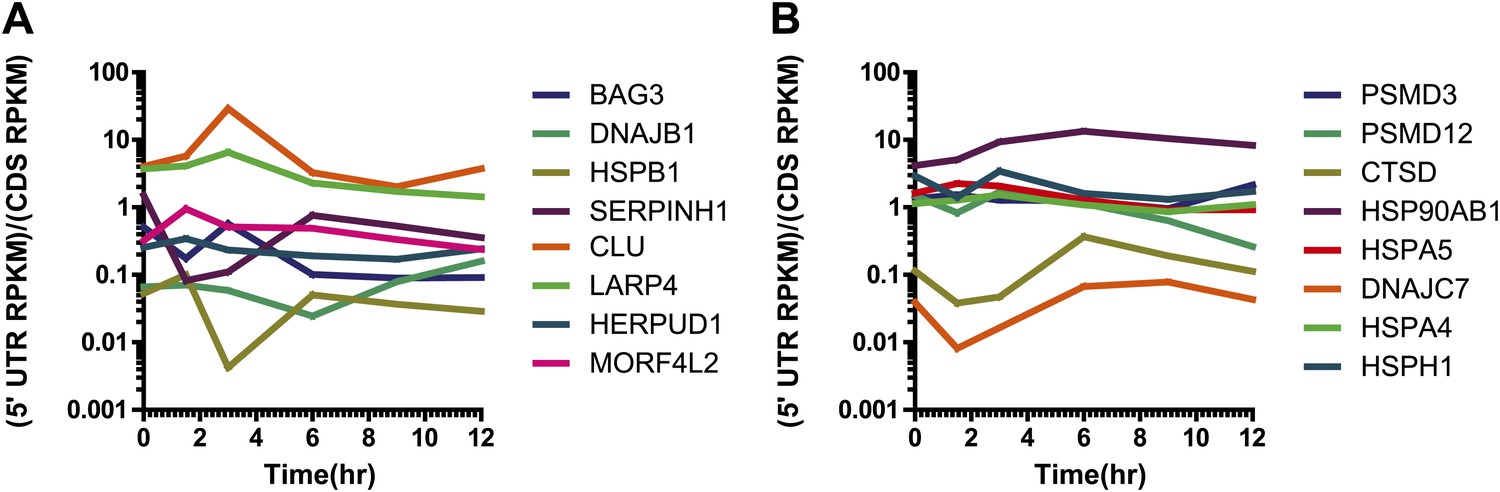
Relative 5′ UTR translation across the time course for upregulated genes.
Relative 5′ UTR translation (measured as normalized footprint read density in 5′ UTR vs normalized footprint read density in coding sequence) over the time course for selected genes (A) upregulated at the mRNA and footprint level and also increased by iTRAQ proteomics and (B) upregulated at the mRNA and footprint level with no detected change by iTRAQ. We note that for both sets of genes relative 5′ UTR translation stays relatively constant during bortezomib-induced apoptosis, consistent with genome-wide analysis in Figure 3B.
Figure 4—figure supplement 4

Unbiased and targeted proteomics comparison to deep sequencing data.
Data from 150 proteins targeted in SRM assay to validate iTRAQ data. Comparison heat map of mRNA and footprint read density with iTRAQ and SRM relative protein abundance.
Figure 5 with 5 supplements
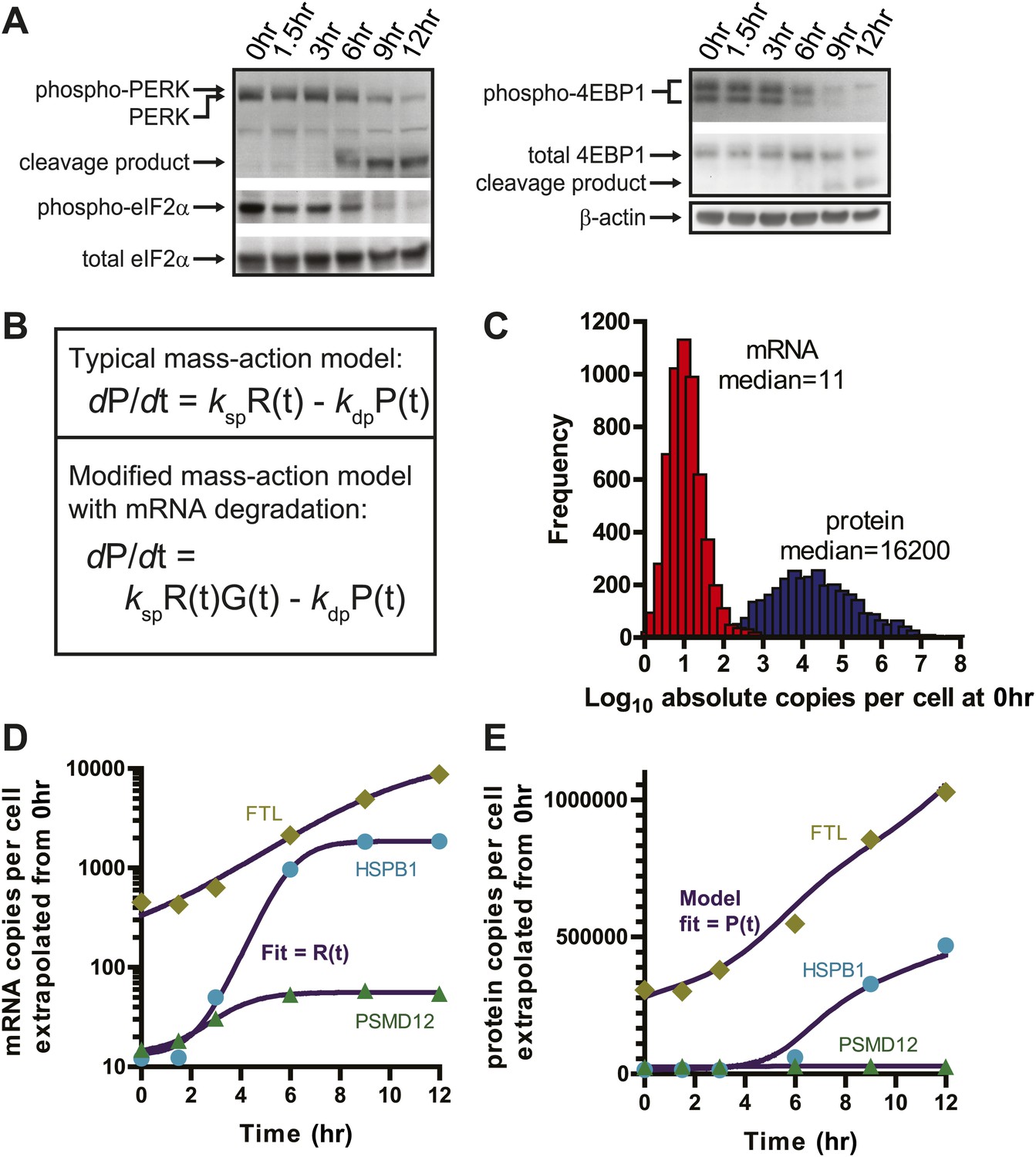
Translational shutdown during apoptosis and quantitative modeling reconciles deep sequencing and proteomic data.
(A) Western blotting tracked UPR proteins important in modulating translation via ER stress. We detect the known caspase cleavage of PERK and 4E-BP1. (B) Differential rate equations describing protein production. R(t) = mRNA abundance; P(t) = protein abundance; ksp = protein translation rate constant; kdp = protein degradation rate constant. G(t) describes global mRNA degradation (Figure 1B). (C) Absolute copies per cell at 0 hr of both mRNA (∼5700 transcripts) and protein measured by iBAQ (∼3400 proteins) demonstrates distributions of same order of magnitude as those seen in mouse NIH3T3 cells (Schwanhausser et al., 2011). (D) Extrapolation of absolute mRNA copy numbers per cell based on RPKM, not accounting for mRNA degradation, is well-fit by either a sigmoid or quadratic function to define R(t). (E) Absolute protein copies per cell were extrapolated from iBAQ results based on SRM assay intensity. The modified mass-action model incorporating mRNA degradation was able to well-fit the subset of proteins that were detectably increased and also explained why others increased at the transcriptional level did not show protein increases.
-
Figure 5—source data 1
mRNA and protein absolute abundance.
(A) Absolute copies of mRNA and (B) protein per MM1.S cell at 0 hr as determined using mRNA read density and iBAQ calculation, respectively, as described in the ‘Materials and methods’. (C) Relative 5′ UTR translation at baseline vs absolute protein copies per cell by iBAQ. (D) Extrapolated mRNA and protein copies per cell for 13 genes as used for model fitting as shown in Figure 5—figure supplement 4.
- https://doi.org/10.7554/eLife.01236.023
Figure 5—figure supplement 1
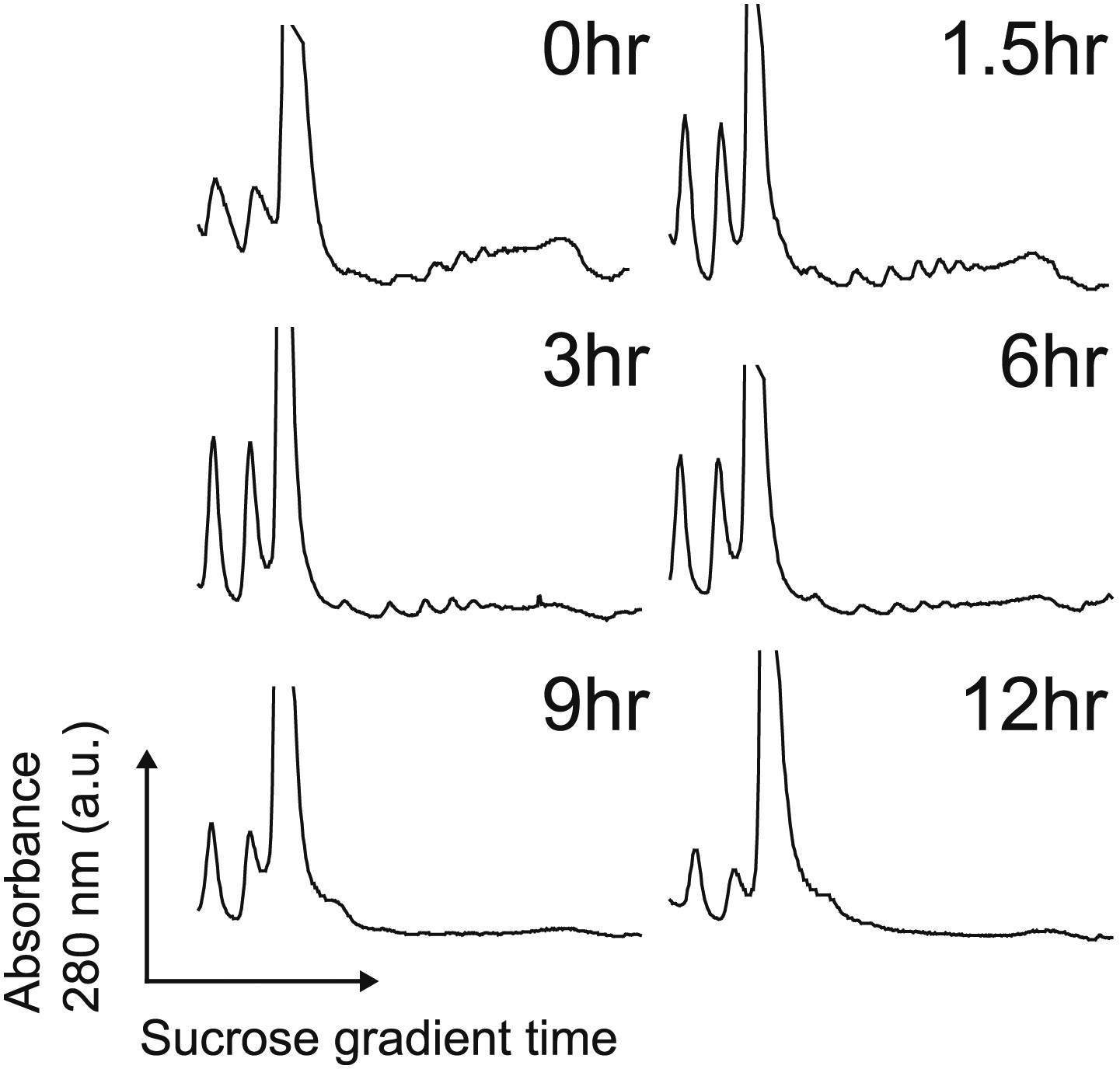
Polysome profiles.
Polysome profiles by sucrose gradient during the apoptotic time course show decrease in polysome peaks over time during during apoptosis. Translation is largely suppressed well before any loss in cell viability.
Figure 5—figure supplement 2
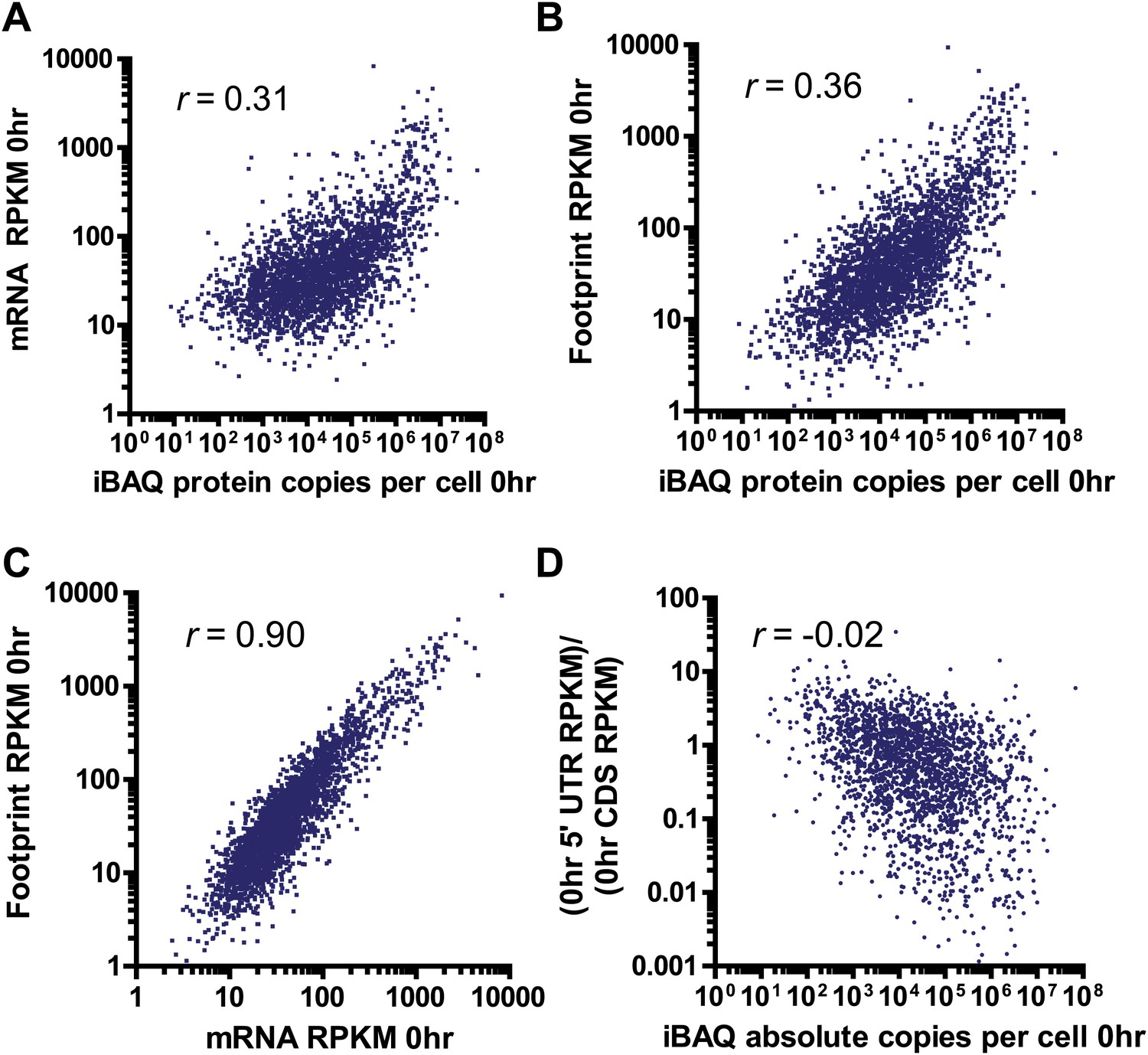
Comparison between deep sequencing and proteomic data.
Correlation of mRNA amount (A) and ribosome footprint (B) read density (in RPKM) with absolute protein abundance by iBAQ MS analysis at 0 hr in MM1.S cells demonstrates limited correlation. (C) Strong correlation of ribosome footprint and mRNA read density at 0 hr. All correlations in A–C with p<0.0001. (D) Relative 5′ UTR translation at baseline vs protein abundance. We note a weak but non-significant correlation (p=0.35) suggesting that increased 5′ UTR translation may relate to lower steady-state protein abundance.
Figure 5—figure supplement 3
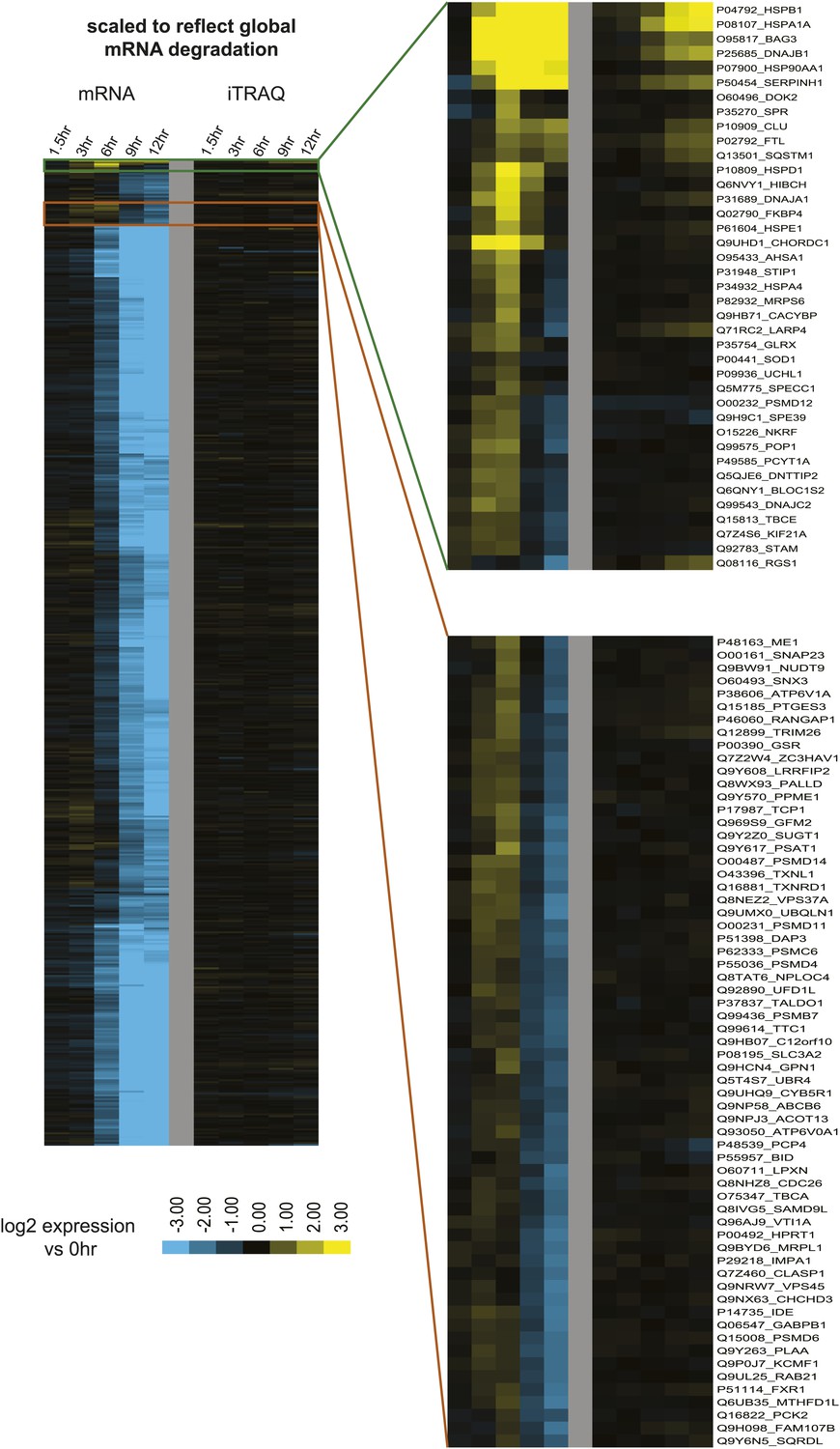
mRNA-seq data scaled by global mRNA degradation.
Relative mRNA expression data from Figure 4B scaled to reflect global mRNA degradation at later time points during apoptosis. By this scaling (scaling factors for mRNA based on fit G(t) function = 1.0 at 0 hr, 0.93 at 1.5 hr, 0.91 at 3 hr, 0.48 at 6 hr, 0.17 at 9 hr, 0.12 at 12 hr), assuming that all mRNA are degraded at the same rate, the absolute mRNA abundance can be estimated across all cells in the population. Upper inset demonstrate that transcripts with absolute mRNA abundance changes across the entire time course are more likely to have increases in protein level, whereas those with transient increases in mRNA generally do not result in detectable protein production.
Figure 5—figure supplement 4
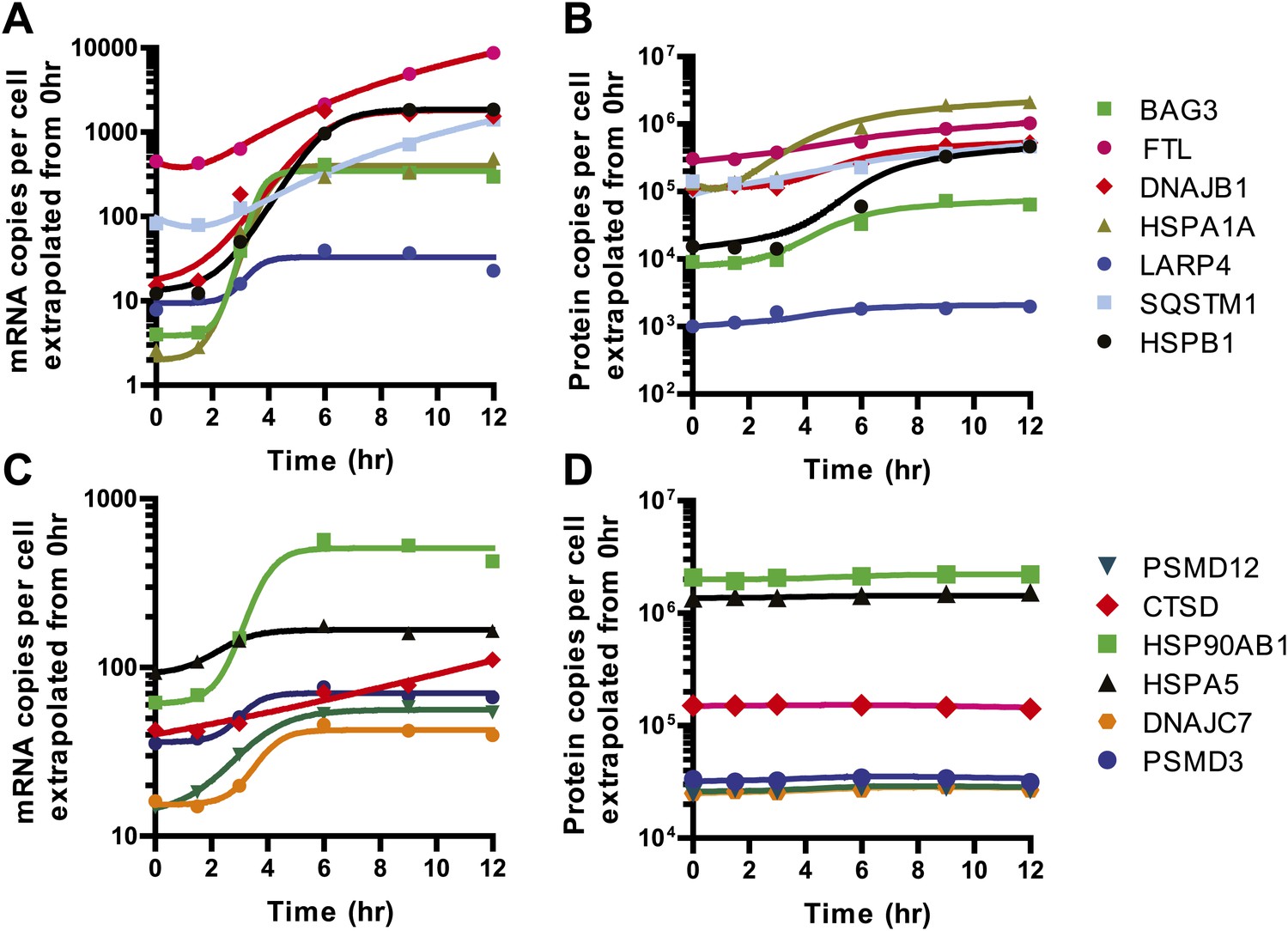
Model fits to 13 selected genes.
We used our quantitative model to describe a subset of genes increased at both the transcript and protein level (A and B) and those where relative transcript levels increased but protein levels were unchanged (C and D). Solid lines in (A and C) represent quadratic fits (FTL, SQSTM1, CTSD) or four-parameter sigmoid fits (all others) to relative mRNA abundance data to describe R(t), as in Figure 5E. Solid lines in (B and D) represent fits to modified mass-action to describe P(t) as shown in Figure 5F.
Figure 5—figure supplement 5

Predicted protein changes from mRNA changes.
Model predictions demonstrating that even during apoptosis, for the same increase in relative transcript levels low-abundance proteins (500 copies per cell at baseline) will have substantial relative protein increases, whereas high abundance proteins (100,000 copies per cell at baseline) will have much smaller changes in relative protein abundance. This is even true when the absolute transcript abundance change is much greater for the high abundance protein and including ksp, the rate constant describing proteins produced per mRNA per hour, as greater for the high abundance protein based on the results of Schwanhausser et al. 2011.
Figure 6 with 2 supplements
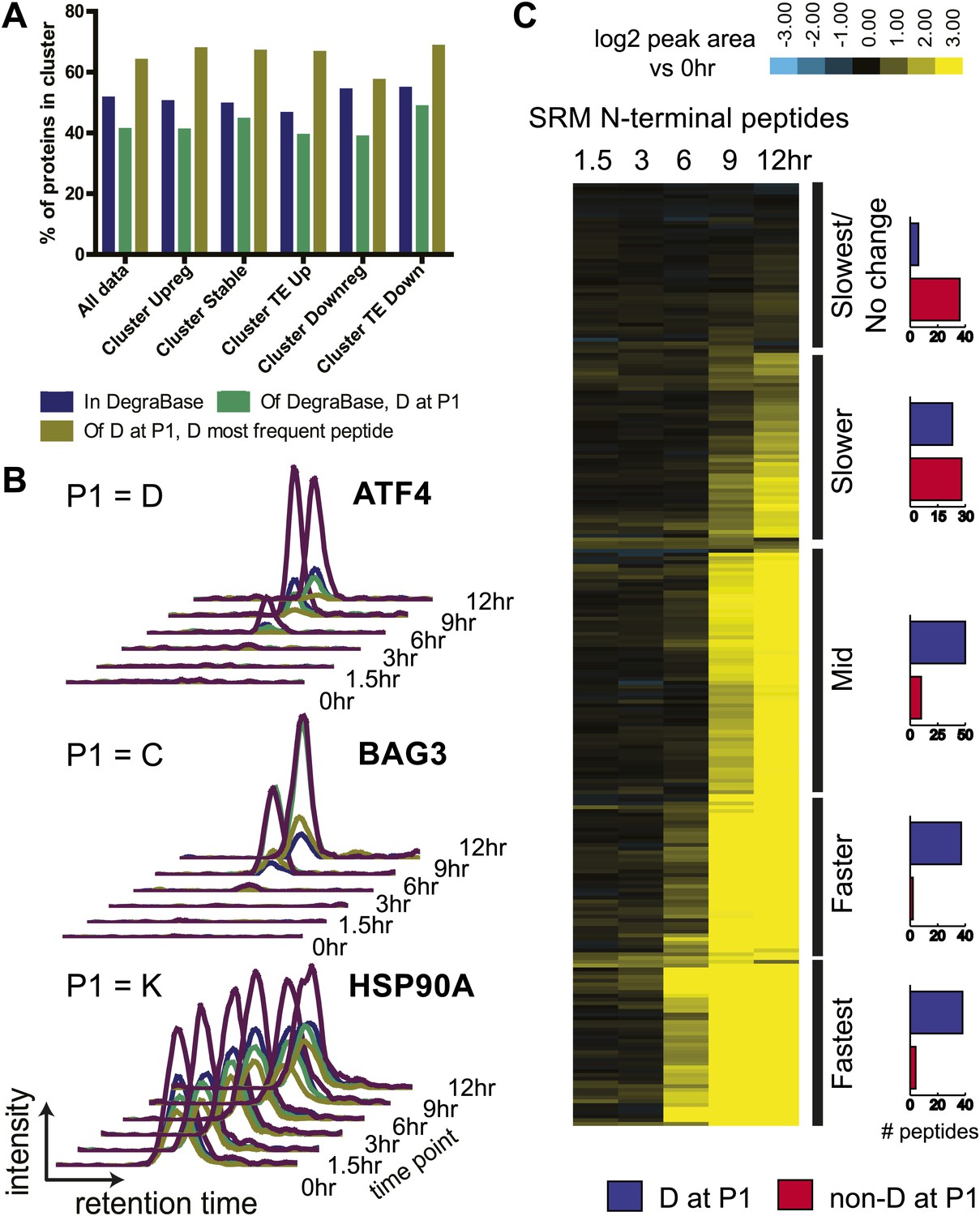
N-terminomics tracks proteolytic cleavage substrates.
(A) We compared defined hierarchical clusters (Figure 1C) to all proteolytic cleavage substrates present in the DegraBase, a database of over 8000 cleavage events (Crawford et al., 2013). D at P1 suggests a putative caspase substrate. We find no significant variation in levels of proteolysis by χ2 analysis of each cluster compared to the set of all substrates. (B) Representative SRM intensity for appearance of proteolytically-processed peptides, both caspase- and non-caspase-cleaved (Figure 6—source data 1). Each trace represents a single parent ion/fragment ion transition; co-elution of multiple targeted transitions confirms peptide. (C) Heat map showing relative SRM intensity of 252 proteolytic peptides monitored in this system compared to 0 hr baseline. Five groups defined by unsupervised hierarchical clustering representing relative cleavage kinetics show caspase proteolysis generally precedes other apoptotic proteolytic events.
-
Figure 6—source data 1
Degradomics SRM assay.
(A) SRM intensity data for peptides included in targeted degradomics assay. P1 residue refers to residue immediately N-terminal to identified proteolytic cleavage site based on protein sequence. (B) List of parent and fragment ion transitions tracked for targeted degradomic SRM assay.
- https://doi.org/10.7554/eLife.01236.030
Figure 6—figure supplement 1

N-terminomics biological data subgroups.
Log2 expression of N-terminomics SRM data across apoptotic time course. N-terminomics data divided by biological function show that caspase proteolysis proceeds with similar rates across biological subdivisions. Proteins increased at the transcriptional and ribosome footprint level after bortezomib treatment are primarily included in the ‘chaperones and cell stress’ and ‘proteasome and ubiquitination’ groups. The ‘translation’ group demonstrates cleavage of many translation initiation factors.
Figure 6—figure supplement 2

N-terminomics data with caspase and non-caspase cleavage sites in same substrate.
These data demonstrate that even in the same protein substrates, caspase cleavage tends to proceed more rapidly during apoptosis than non-caspase cleavage.
Figure 7

Systems-level analysis suggests potential chemotherapeutic combinations with bortezomib in myeloma.
(A) Bort = bortezomib. HSF1 inhibitor used was KRIBB11 (Yoon et al., 2011) and shows additive effect with bortezomib. (B) PERK inhibitor was GSK2606414 and shows additive effect with bortezomib (Axten et al., 2012). All measurements in 24-well plates in triplicate; mean ± SD shown.
Tables
Table 1
Biological relevance of findings from Ingenuity Pathway Analysis (IPA)
https://doi.org/10.7554/eLife.01236.010| Cluster | Molecular and cellular function | p value | Canonical pathway | p value | Upstream regulator | p value |
|---|---|---|---|---|---|---|
| Cluster Upreg | Post-translational modification | 4.15 e-11 | Protein ubiquitination pathway | 1.50e-34 | NFE2L2 (NRF2) | 7.77e-16 |
| Protein Folding | 4.15 e-11 | NRF2-mediated oxidative stress response | 2.90e-10 | HSF1 | 1.70e-14 | |
| Cell death and survival | 2.47 e-10 | EIF2AK3 (PERK) | 6.47e-10 | |||
| Protein synthesis | 8.82 e-09 | |||||
| Protein degradation | 9.63 e-09 | |||||
| Cluster Stable | RNA post-transcriptional modification | 1.44e-09 | – | – | ||
| Molecular transport | 8.87 e-09 | |||||
| Protein trafficking | 8.87 e-09 | |||||
| Cluster TE Up | – | Mitochondrial dysfunction | 6.04e-10 | XBP1 | 8.79e-10 | |
| Cluster Downreg | Cell death and survival | 7.56e-12 | – | TP53 | 2.67e-15 | |
| Cellular growth and proliferation | 3.61e-11 | MYC | 1.53e-10 | |||
| DNA replication, recombination and repair | 3.81e-09 | XBP1 | 1.65e-10 | |||
| RNA post-transcriptional modification | 8.91e-08 | INSR | 5.94e-09 | |||
| E2F4 | 6.20e-09 | |||||
| Cluster TE Down | RNA post-transcriptional modification | 8.88e-11 | EIF2 signaling | 2.35e-17 | – | |
| Gene expression | 3.24e-09 | Regulation of eIF4 and p70S6K signaling | 1.40e-08 | |||
| 5% transcripts highest TE at 0 hr | – | Mitchondrial dysfunction | 1.52e-26 | – | ||
| Increased 5′ UTR translation | – | EIF2 signaling | 2.45 e-17 | MYC | 1.09e-08 | |
| HSF1 | 8.11e-08 | |||||
| Decreased 5′ UTR translation | – | – | – |
-
Clusters defined by hierarchical clustering of mRNA, footprint, and translational efficiency data as shown in Figure 1B (included genes listed in Figure 1—source data 1). We also analyzed the top 5% of transcripts by translational efficiency at 0 hr as calculated by (footprint RPKM)/(mRNA RPKM) per transcript. p values are as calculated using Fisher′s exact test by Ingenuity Pathway Analysis software. We report findings here with a p<10−7. Results from each category (Molecular and cellular function; Canonical pathway; Upstream regulator) are listed in order of decreasing p, independent of the other categories.
Table 2
Comparison of caspase- and non-caspase cleavages during apoptotic time course as monitored by N-terminomics and SRM
https://doi.org/10.7554/eLife.01236.033| Total D = P1 peptides | 163 | |
| Total non-D = P1 peptides | 89 | |
| Tryptic-like (K or R at P1) | 24 | |
| Non-tryptic (all other residues at P1) | 65 |
| Cluster | D at P1 | Non-D at P1 | |
|---|---|---|---|
| Slowest/no change | 6 | 36 | |
| Slow | 23 | 28 | |
| Mid | 50 | 10 | |
| Faster | 37 | 2 | |
| Fastest | 38 | 4 |
| Non-D peptides in fastest three clusters | UniProt | Gene | P1 | Sequence Position | MEROPS annotated? | |
|---|---|---|---|---|---|---|
| Fastest | (C)GQVAAAAAAQPPASHGPER | O95817 | BAG3 | C | 151 | N |
| (S)AVGFNEMEAPTTAYK | P14317 | HCLS1 | S | 208 | N | |
| (P)GHGSGWAETPR | O75533 | SF3B1 | P | 304 | N | |
| (Q)VLTVPATDIAEETVISEEPPAKR | Q06547 | GABP1 | Q | 306 | N | |
| Fast | (Q)ALKEEPQTVPEMPGETPPLSPIDMESQER | P05412 | JUN | Q | 223 | N |
| (P)AVNGATGHSSSLDAR | Q07817 | BCL2L1 | P | 63 | N | |
| Mid | (E)AAGATGDAIEPAPPSQGAEAK | P49006 | MARCKSL1 | E | 56 | N |
| (Q)AASGDVQTYQIR | P16220 | CREB1 | Q | 243 | N | |
| (E)GGIDMDAFQER | P29083 | GTF2E1 | E | 297 | N | |
| (A)SIFGGAKPVDTAAR | P23588 | EIF4B | A | 358 | Y–meprin alpha (Becker-Pauly et al., 2011) | |
| (E)AIQNFSFR | O75122 | CLASP2 | E | 946 | N | |
| (P)HFEPVVPLPDKIEVK | P49792 | RANBP2 | P | 1170 | N | |
| (N)SWFENAEEDLTDPVR | Q13813 | SPTAN1 | N | 2104 | N | |
| (L)AFSEQEEHELPVLSR | O75995 | SASH3 | L | 128 | N | |
| (Q)AIMEMGAVAADKGKK | O95817 | BAG3 | Q | 522 | N | |
| (E)AILEDEQTQR | Q9P2E9 | RRBP1 | E | 1298 | N | |
-
By Fisher′s exact test there is a significant absence of tryptic-like cleavages in the 16 rapidly cleaved peptides (p=0.005).
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Global cellular response to chemotherapy-induced apoptosis
eLife 2:e01236.
https://doi.org/10.7554/eLife.01236




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}