Multi-species, multi-transcription factor binding highlights conserved control of tissue-specific biological pathways
- European Bioinformatics Institute, Wellcome Trust Genome Campus, United Kingdom
- Aix-Marseille Université, UMR1090 TAGC, France
- INSERM, UMR1090 TAGC, France
- SickKids Research Institute, Canada
- University of Cambridge, United Kingdom
- University of Cincinnati, United States
- University of New South Wales, Australia
- Cincinnati Children's Hospital Medical Center, United States
- Wellcome Trust Genome Campus, United Kingdom
- University of Toronto, Canada
Figures
Figure 1 with 3 supplements

Overview of ChIP-seq, CRM construction, and multiple-species comparisons.
ChIP-seq peaks were determined for four liver TFs in five mammals. (A) CRMs were constructed by merging ChIP-seq peaks whose summits occurred within 300 bp and consisted of at least two distinct TFs. Remaining peaks were designated as singletons. (B) Whole genome 9-way EPO multiple sequence alignments (MSA) were used to project CRMs/Singletons across the five species. A CRM was considered shared if its position in the EPO MSA overlapped a CRM in a second species by a minimum of 10 bp. Neither the content nor order of TFs within the CRM was required to be classified as a ‘Shared’ CRM. A singleton in one species was considered ‘Shared’ if it overlapped the same TF in a second species. (C) Relative to human, the average % of shared CRMs is shown. Human CRMs (comprised of any two TFs) that overlap a CRM from a second species are shown with empty circles. Human CRMs containing at least one of each TF (all 4 TFs) were compared to all identified CRMs in a second species (purple circles). (D) The percentage of human CRMs and singletons in different phylogenetic categories that can be found aligned within the EPO MSAs for each of the five species is shown.
-
Figure 1—source data 1
Quality control for ChIP-seq, CRM construction, and multi-species comparisons.
- https://doi.org/10.7554/eLife.02626.004
Figure 1—figure supplement 1
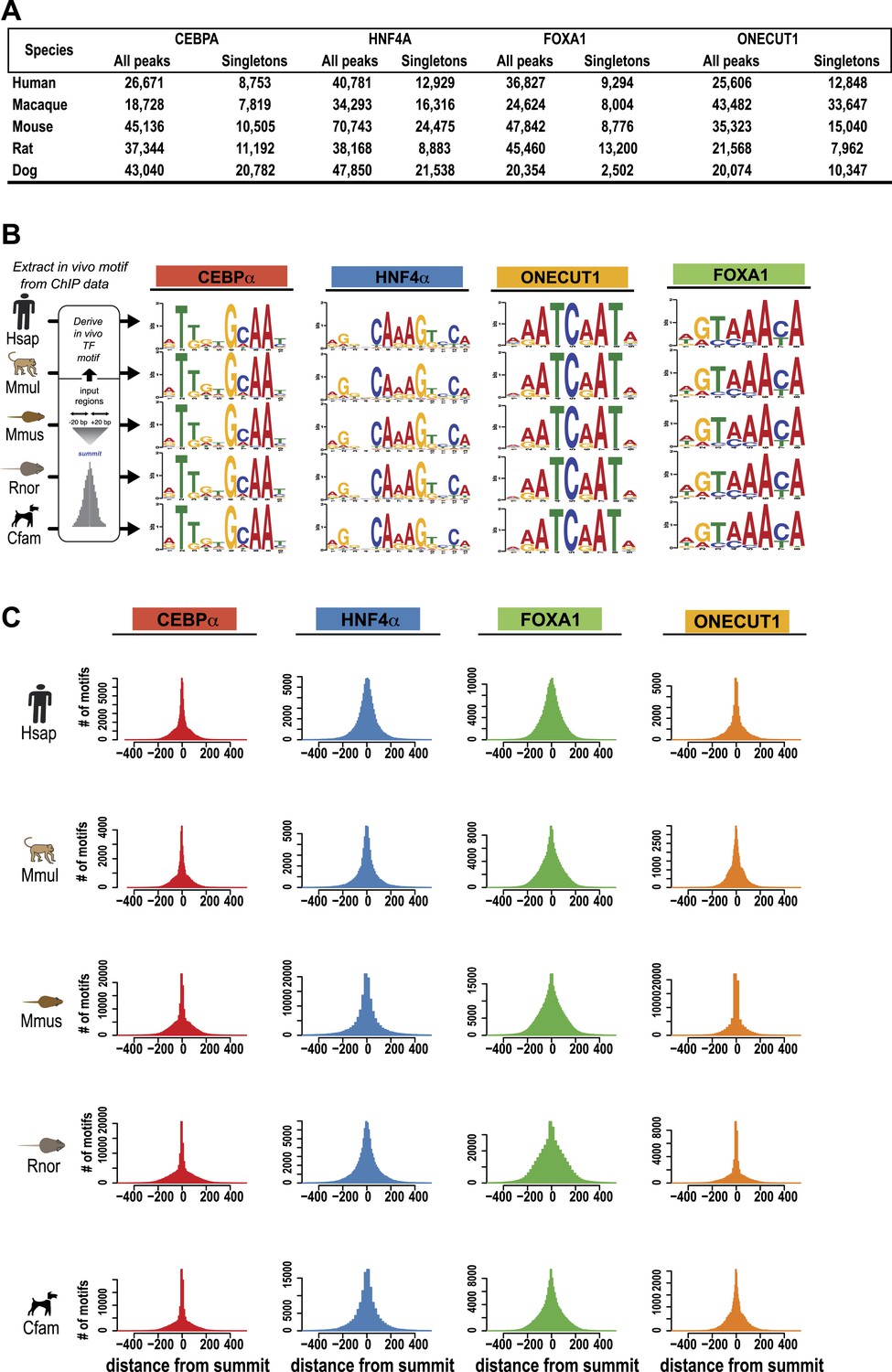
Summary of ChIP-seq peak number and TF motif enrichment.
(A) Number of Peaks (B) Conservation of TF binding motifs. DNA binding specificities of CEBPA, HNF4A, FOXA1, and ONECUT1 are highly conserved. The known sequence motifs were identified de novo in each species interrogated. (C) Central position of motifs under TF binding summits were observed for each factor. Number of motifs identified using the PWM from B (y-axis) vs distance from TF binding summit obtained using the SWEMBL peak caller (x-axis).
Figure 1—figure supplement 2
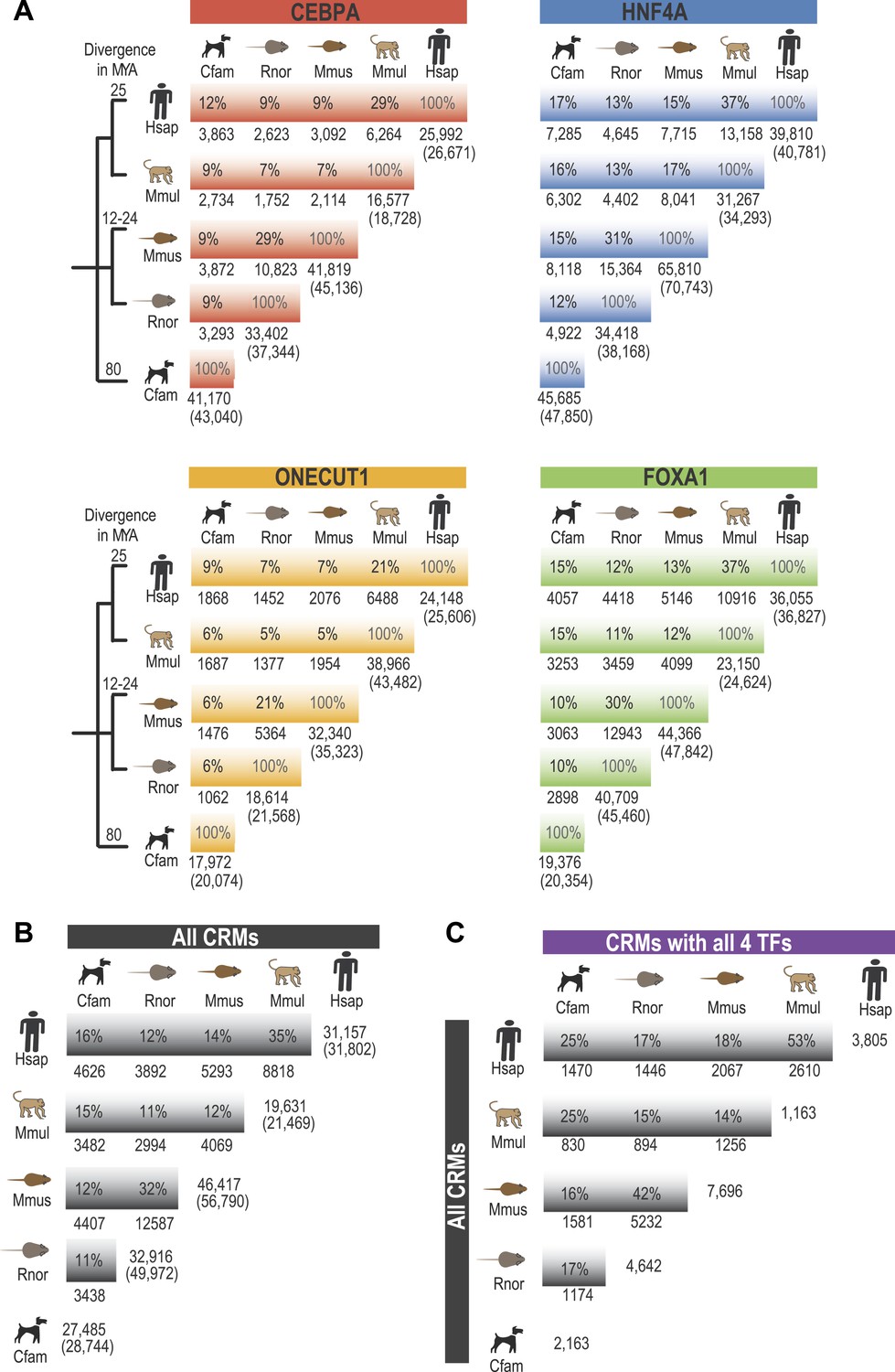
Pairwise analysis of individual TFs using EPO multiple sequence alignment.
Shared binding events were initially identified in a pair-wise fashion using the 9-way EPO-MSA. Average % overlap was calculated and the total number of peaks that could be aligned to a second species is shown. The total peak number for each species shown in brackets. A region was identified as shared if its DNA sequence overlapped in a second species by 10 or more base pairs. (A) Average % overlap for the four individual TFs in this study. (B) For each pair of species the average % of CRMs (comprised of any two TFs) that overlap another CRM, regardless of its composition from a second species is shown. The number of CRMs present within the EPO-MSA is given on the right with the total number of CRMs shown in brackets. (C) For each species, CRMs containing at least one of each TF type (all 4 TFs) were compared to all identified CRMs in a second species (Figure 1—source data 1F).
Figure 1—figure supplement 3
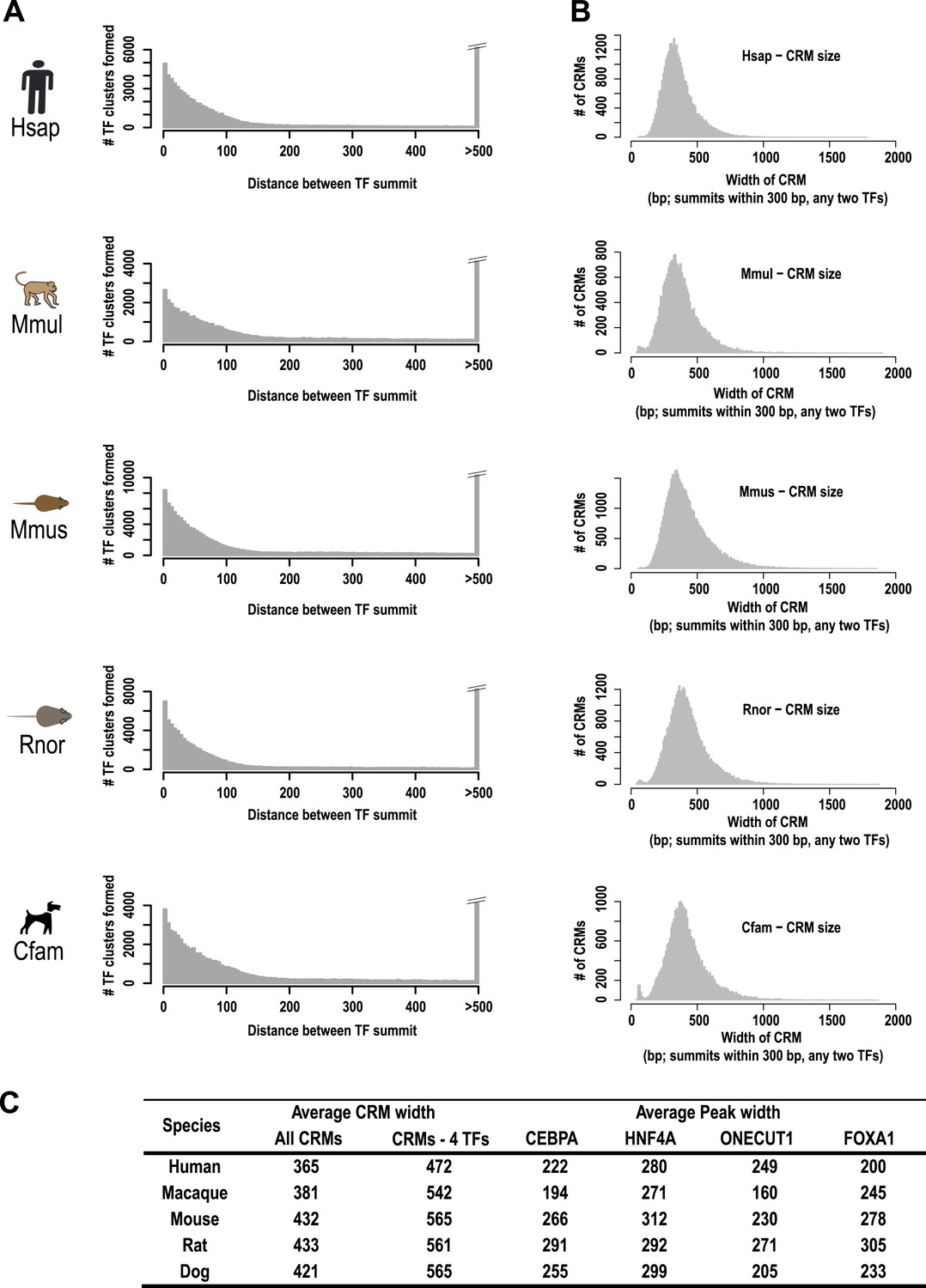
A majority of the four liver-enriched TFs cluster into CRMs.
(A) The number of TF binding event clusters (y-axis) vs the distance between the summits of TF binding events used to generate the clusters (x-axis) is plotted. (B) Number of CRMs (y-axis) vs CRM width (x-axis) is shown for our chosen 300 bp distance-between-summit criteria we used to build CRMs. (C) Average width in base pairs for: all CRMS; CRMs containing all four TFs; and individual peaks are shown.
Figure 2

Annotation of human regulatory regions using interspecies combinatorial transcription factor binding.
(A) Human liver ChIP-seq data from ONECUT1, HNF4A, FOXA1, and CEBPA were assembled into CRMs consisting of at least 2 of the 4 TFs. The CRMs or single TFs were then broken down into categories based on their overlap with ChIP-seq data in macaque, dog, mouse, and rat. Singletons and CRMs were considered shared if they overlapped at least 10 bp with another TF bound region in the EPO multiple sequence alignment (MSA). (B) Experimentally determined combinatorial binding at the blood coagulation F7 locus. Raw sequencing reads from ChIP-seq experiments: CEBPA (red), HNF4a (green), ONECUT1 (yellow), and FOXA1 (green) are overlaid and called peaks are displayed for each species. ChIP-seq determined TF binding events were assembled into CRMs (black bars) underneath the enriched regions (peaks). Grey lines are drawn to illustrate shared CRMs using the EPO-MSA.
-
Figure 2—source data 1
Table of CRMs and singletons along with the phylogenetic categories they were assigned.
File coordinates are for the hg18 assembly.
- https://doi.org/10.7554/eLife.02626.009
Figure 3
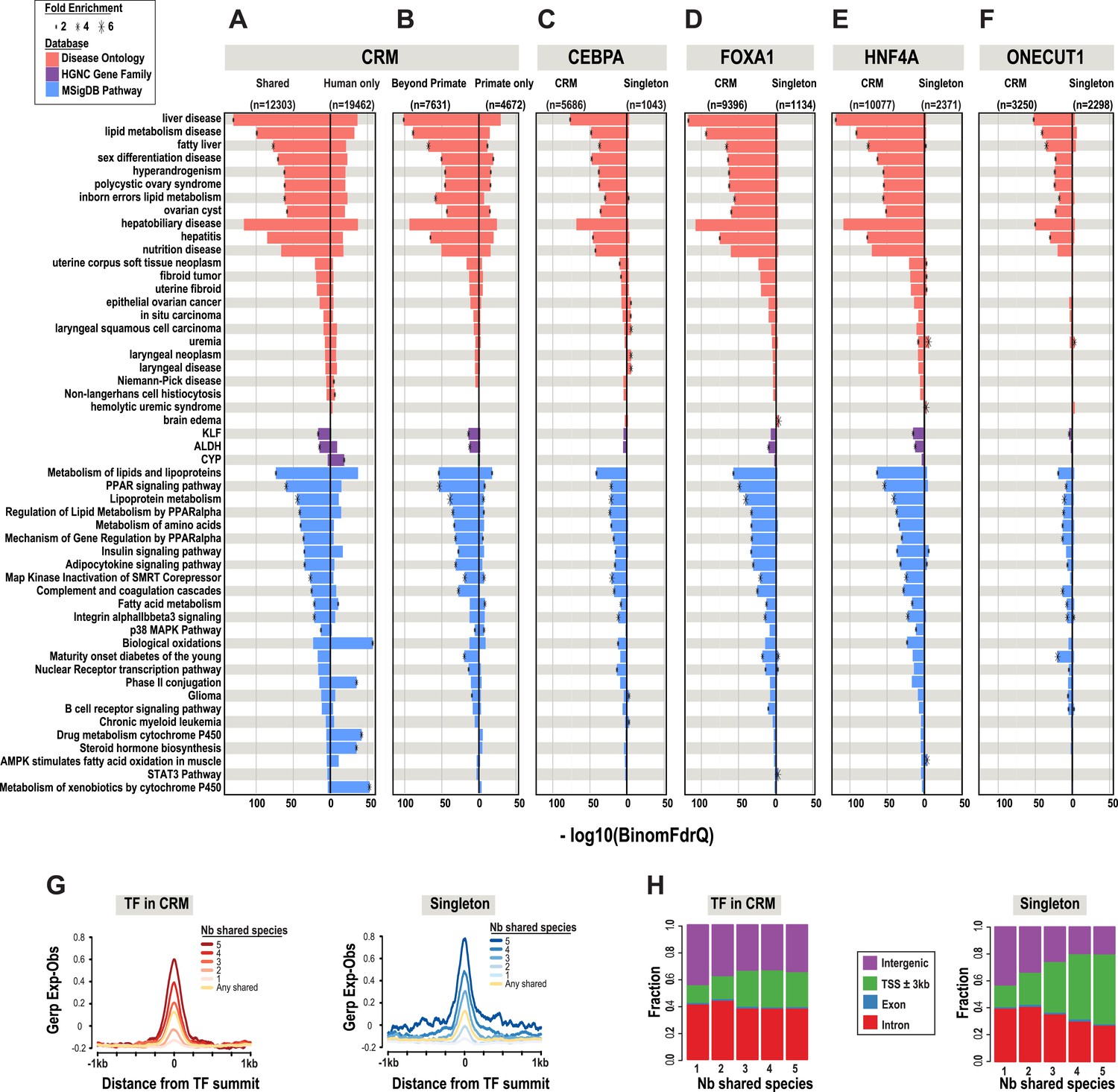
Phylogenetic filtering of experimentally determined liver TF binding events yield distinct functional enrichments.
Results were obtained using the programming interface for the online enrichment tool GREAT version 2.02 (McLean et al., 2010) and plotted with custom R scripts. Up to five of the most significant enrichments obtained for each of the six analyses are listed on the left. The −log10 of binomial Q values for Disease ontology, HGNC gene family, and MSigDB are shown along the x-axis. Bars with a black asterisk indicate significant enrichments using GREAT default parameters (binomial and hypergeometric FDR Q-value significance at P ≤ 0.05 with at least twofold region enrichment). The size of the asterisk is proportional to the fold enrichment obtained for the given database. See Figure 3—source data 1 for complete list of Q-values, fold enrichments, genes giving the enrichments along with results from additional databases. (A) Enrichment analysis of any CRM shared in human plus at least one additional species is shown on the left and human only CRMs are shown on the right (Figure 3—source data 1A). (B) Human CRMs (left panel) shared in human and at least one non-primate (Beyond Primates) is shown vs Human CRMs (right panel) shared in human and macaque but no other species (Primate only) (Figure 3—source data 1B). (C) Enrichment analysis of shared CEBPA CRMs and singletons (Figure 3—source data 1C). (D) Enrichment analysis of shared HNF4A CRMs and singletons (Figure 3—source data 1D). (E) Enrichment analysis of shared FOXA1 CRMs and singletons (Figure 3—source data 1E). (F). Enrichment analysis of shared ONECUT1 CRMs and singletons (Figure 3—source data 1F). (G) Human TFs in CRMs and Singletons were categorized by the number of species in which they are shared with. Profiles of constrained elements (sequence conservation) in a 1-kb window around CRMs or singletons were calculated using GERP scores from the 29-way multiple sequence alignments. (H) Genomic location of CRMs and Singletons. Proportion of single TFs located near transcription start sites (TSS) increases to >50%, but remains stable for CRMs at ∼20%.
-
Figure 3—source data 1
Functional enrichment results obtained for CRMs and singletons using GREAT.
- https://doi.org/10.7554/eLife.02626.011
Figure 4 with 1 supplement
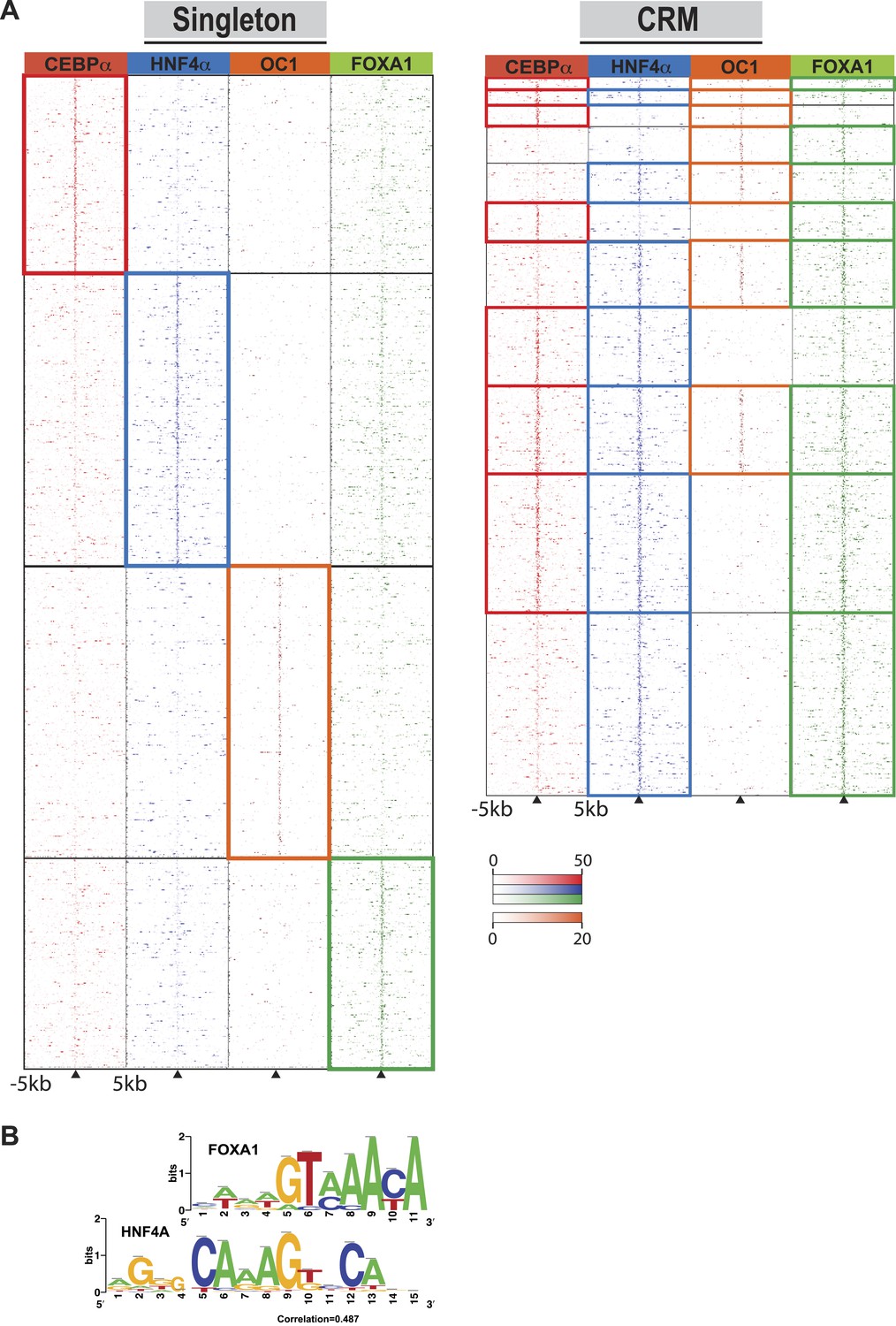
Comparison of TF occupied regions classified as CRMs and singletons.
(A) Regions of ±5 kb are represented around the center of CRMs or singletons. Reads centered on the summit of each TF are counts subtracted by input reads in 100 bp bins plus and minus 5 kb from the summit. Colored boxes indicate CRMs or singletons where a peak was called for a given factor: CEBPA (red), HNF4A (blue), ONECUT1 (orange), and FOXA1 (green). Looking at read counts for all four factors reveal that many of the HNF4A singleton in fact have weak FOXA1 signal. (B) Alignment of FOXA1 de novo ChIP-seq motif to the HNF4A motif. Motif comparison (alignment) was performed using compare-matrices from RSAT. The program calculates the correlation between two matrices shifting positions; the correlation is normalized based on the width of the alignment to avoid high correlation based on few flanking positions.
-
Figure 4—source data 1
Comparison of motif matches between CRMs and singletons. Chi-square test for differences between the number of peaks associated with CRMs and singletons, for each TF, that contained at least one predicted motif using three different p-value thresholds for motif scanning: stringent (10−4), moderate (10−3) and lenient (10−2). Blue shadows highlight siginficnat p-values.
- https://doi.org/10.7554/eLife.02626.013
Figure 4—figure supplement 1
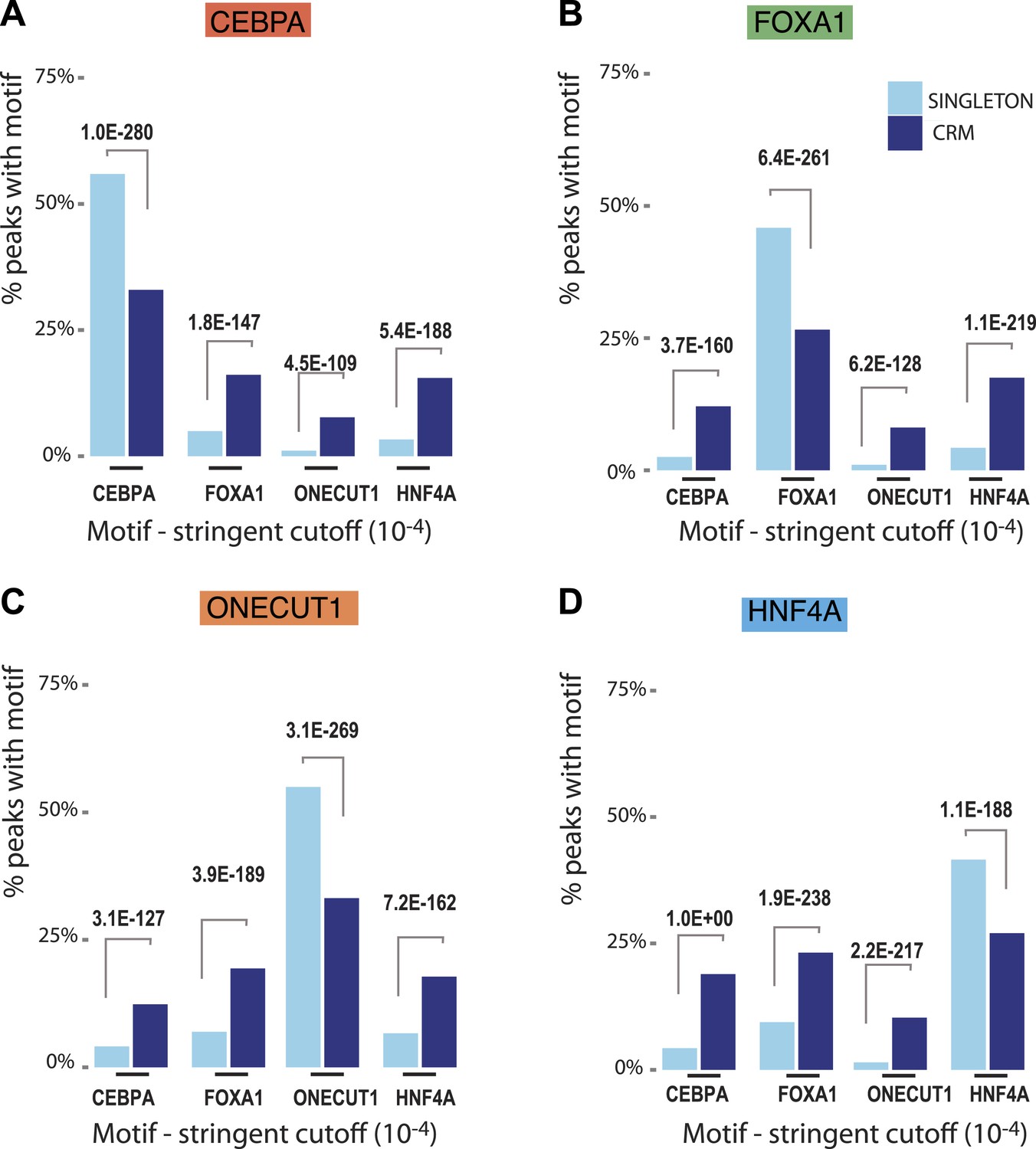
Comparison of stringent motif matches between CRMs and singletons.
Each TF peak set was scanned using the RSAT tool matrix-scan with PWMs for the four TFs. A stringent motif cutoff (10−4) was applied. The figure shows the percentage of peaks with at least one detected site for each TF below the corresponding p-value threshold (y-axis) and the motif PWM scanned (x-axis). (A) Motif scanning in CEBPA peaks; (B) Motif scanning in FOXA1 peaks; (C) Motif scanning in ONECUT1 peaks; and (D) Motif scanning in HNF4A peaks. For the stringent thresholds, significant adjusted p-values comparing singletons and CRMs are given. See all X2 test p-values for lenient (10−2) and moderate cutoffs (10−3) in Figure 4—source data 1.
Figure 5 with 1 supplement
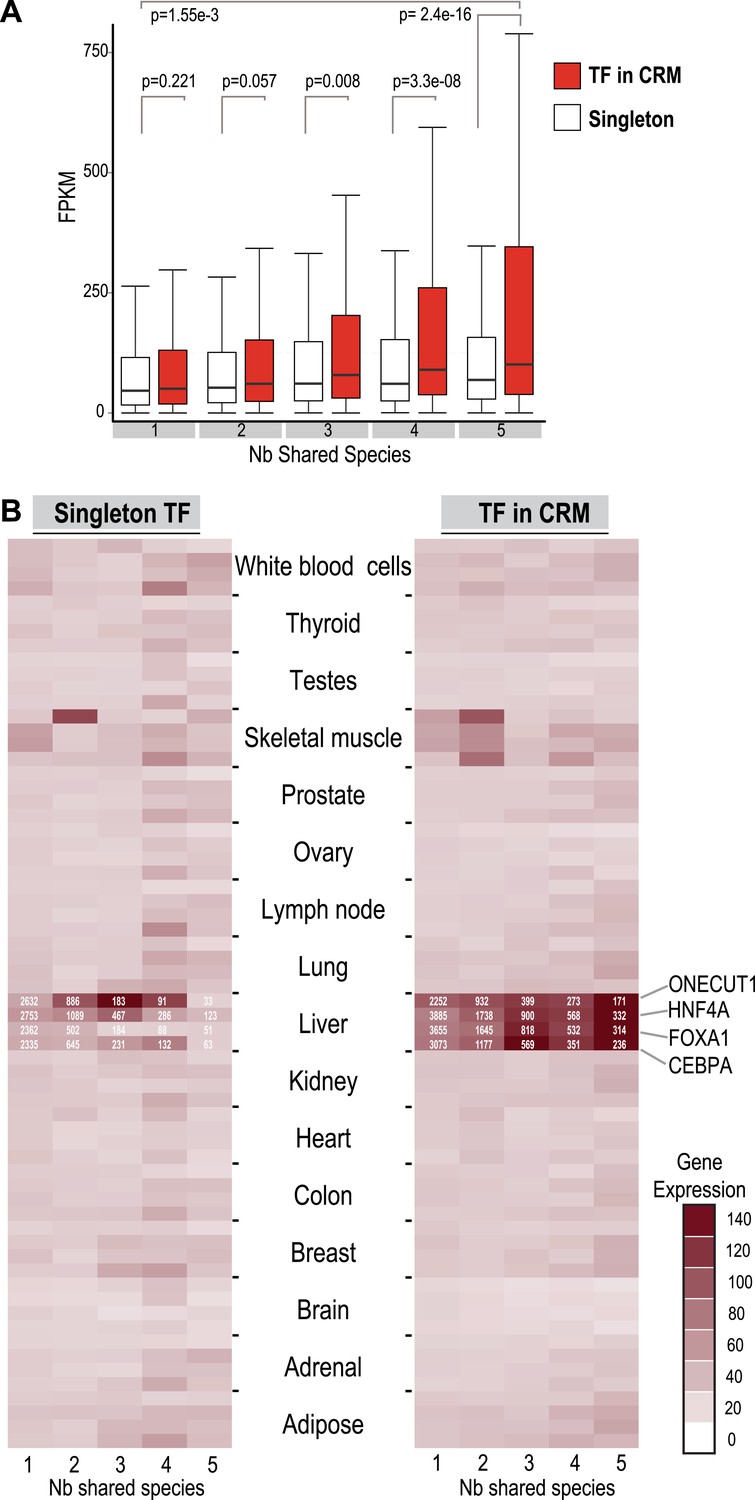
TFs in Deeply shared CRMs are near genes highly expressed in a tissue-specific manner.
(A) Association of shared TFs in CRMs and Singletons with human gene expression obtained by RNA-seq in human liver (Kutter et al., 2011; E-MTAB-424). TFs in CRMs or Singletons were assigned to the nearest gene, and the FPKM (Fragments Per Kilobase of exon per Million reads) was recorded. In contrast to Singletons, TFs in Deeply CRMs are associated with highly expressed genes (adjusted p-values shown). The numbers of target gene associations for the singletons and CRMs in categories 1 to 5 are: 19354(S), 32706(CRM); 6325(S), 14669(CRM); 1935(S), 5755(CRM); 1005(S), 3292(CRM); and 459(S), 2530(CRM). (B) Comparison with CRMs and Singletons to a reference mRNA-seq data from 16 human tissues (E-MTAB-513) further shows that relative to singletons, liver-specific CRMs are highly expressed in liver, and that each TF contributes to this specificity. The number of gene associations for each category in the liver data is shown in white text within the heat map.
Figure 5—figure supplement 1
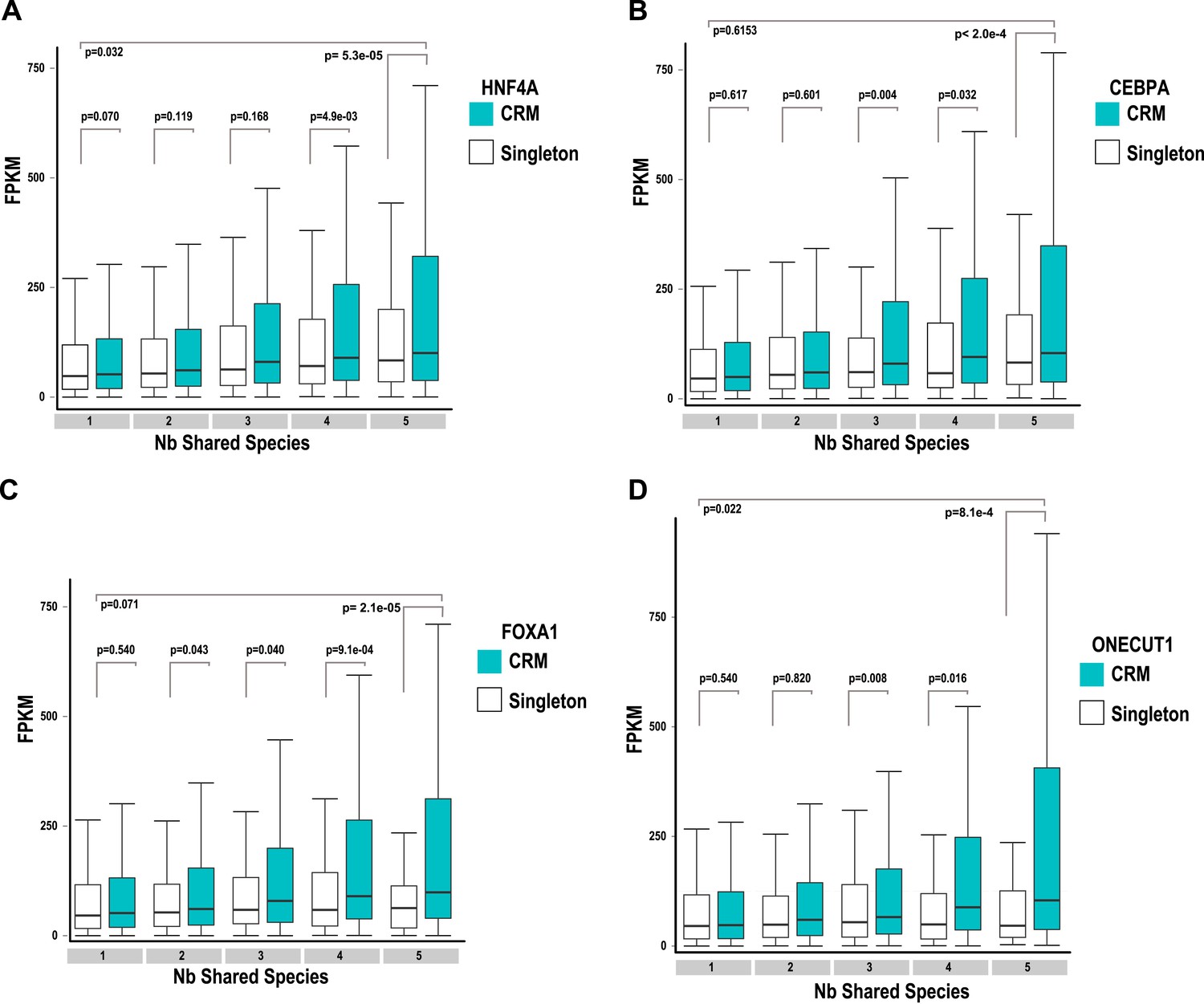
Association of shared TFs in CRMs and Singletons with human gene expression obtained by RNA-seq in human liver (Kutter et al., 2011; E-MTAB-424) broken down by the transcription factor in the CRM: (A) HNFA; (B) CEBPA; (C) FOXA1 and (D) ONECUT1.
TFs in CRMs or singletons were assigned to the nearest gene and the FPKM (Fragments Per Kilobase of exon per Million reads) was recorded for CRMs and TFs broken down by transcription factor. Adjusted p-values are shown and like Figure 5, the CRMs were associated with more highly expressed genes in the 5-way and 4-way–shared categories.
Figure 6
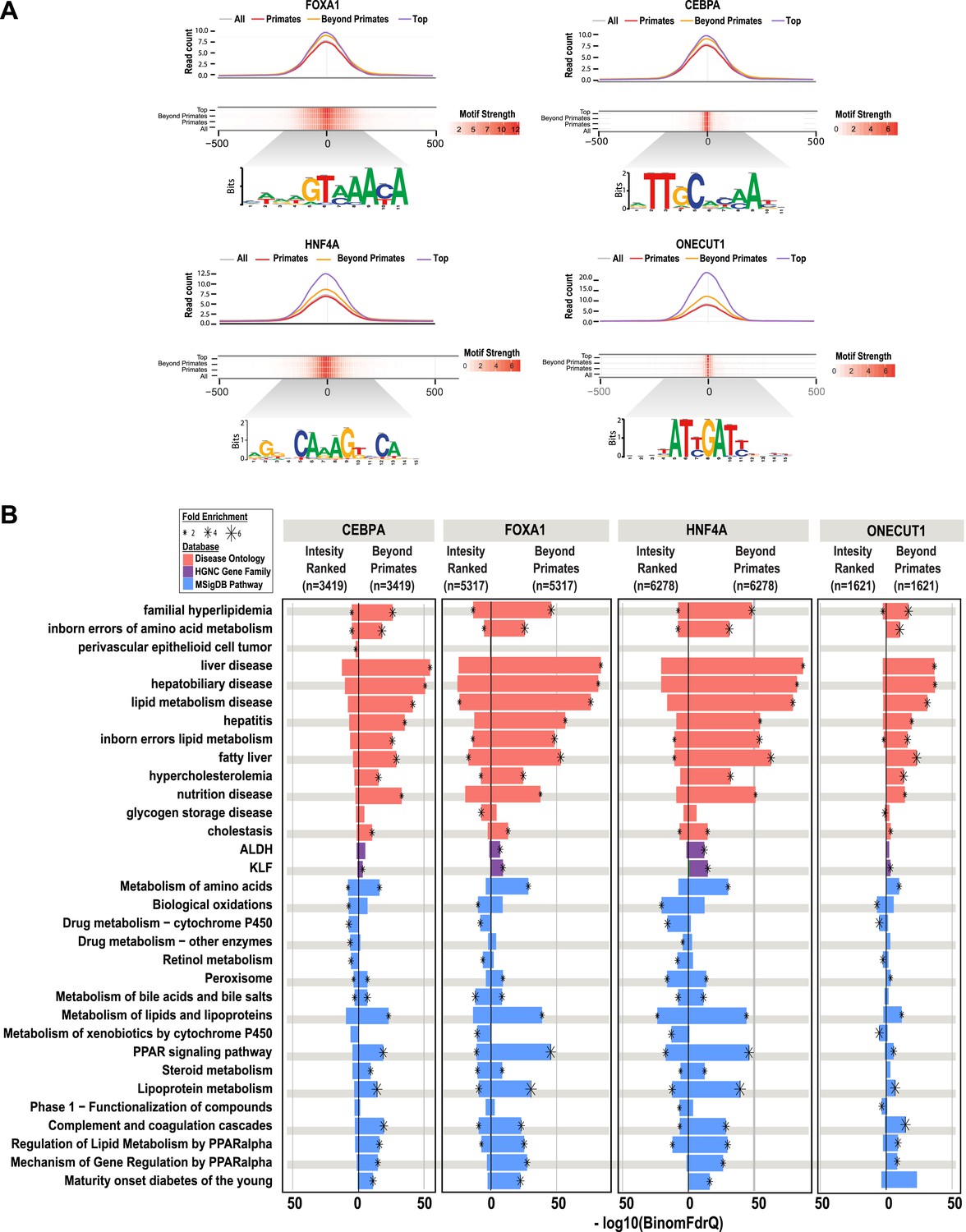
Shared HNF4A CRMs unravel more liver related functional classes than do the equivalent number of CRMs with the best peak enrichment scores.
CRMs containing each TF were analyzed separately. (A) Read count and motif binding weight scores were calculated for: (1) all CRMs (All); (2) CRMs shared in human and at least one additional non-primate (Beyond primates); (2) human CRMs shared in macaque only (Primates); and (3) the equivalent number of CRMs (equal to the number of Beyond primate CRMs) ranked by the SWEMBL peak intensity score for the TF in question (Top). (B) Functional enrichments were performed using GREAT comparing the Beyond primate category to the top ranked category. The top five enrichments for all comparisons performed were collected and the enrichments, if available are plotted. Databases used for GREAT enrichment analyses are indicated by color and are ranked according to the −log10 binomial FDR q-values plotted on the x-axis. Significant enrichments are labelled with an asterisk which is sized according to fold enrichment of the given database category.
Figure 7 with 2 supplements
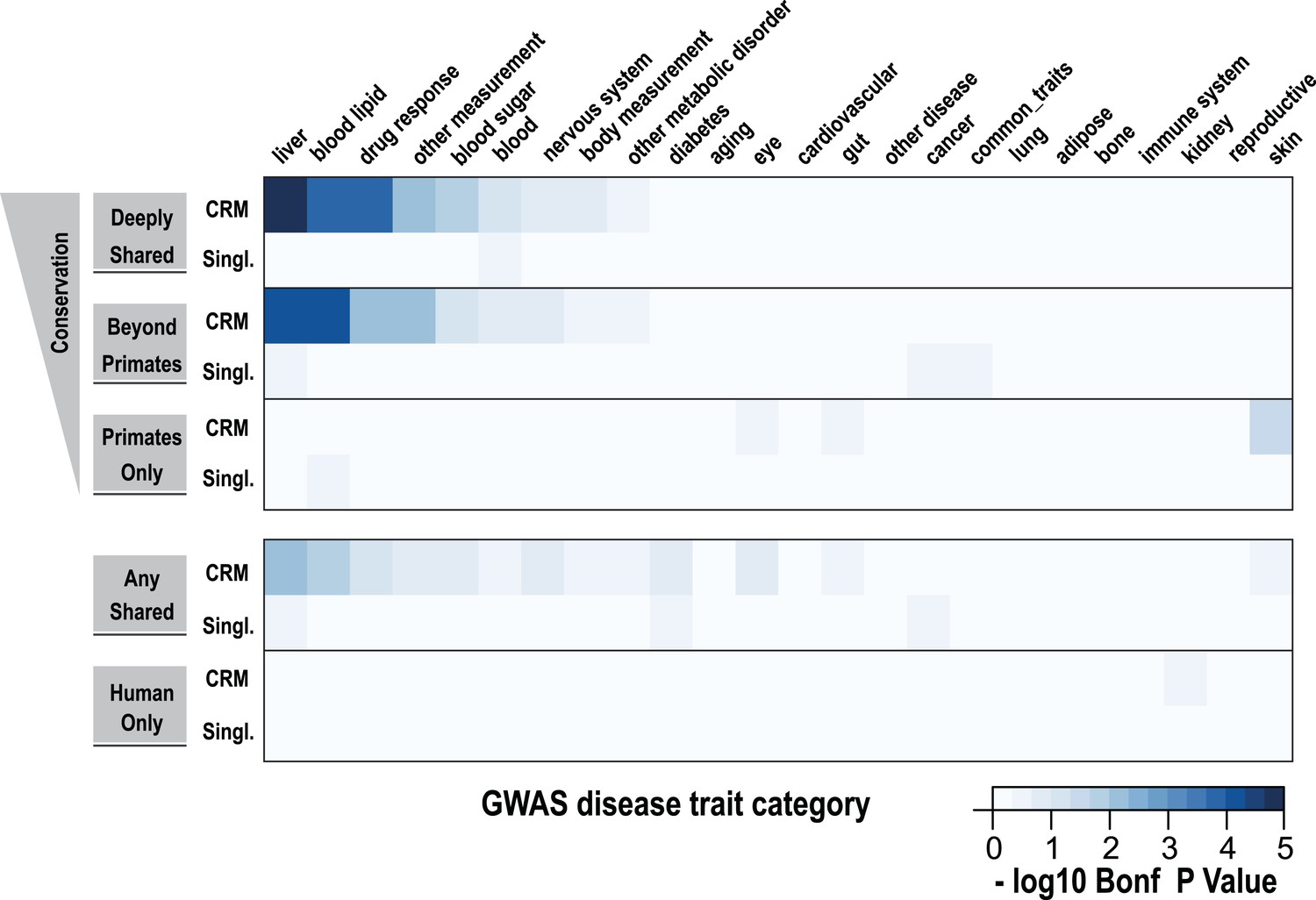
Using a window of ±2.5 kb, lead SNPs obtained by GWAS were enriched for shared CRMs in a tissue/disease specific manner.
Heatmap representation of the −log10 of Bonferroni corrected p-values from hypergeometric testing for enrichment of CRMs or single TFs (broken down into categories related to their degree of conservation) by lead GWAS SNPs obtained from the NHGRI catalog (Hindorff et al., 2009). The NHGRI catalog disease traits were summarized into 25 categories prior to enrichment. Each GWAS lead SNP was given a ±2.5-kb window prior to identifying overlapping CRMs/singletons.
-
Figure 7—source data 1
Full tables of 2.5 kb and LD GWAS enrichments performed in Figure 7 and Table 1.
The file also includes the categories used to annotate the NHGRI catalog.
- https://doi.org/10.7554/eLife.02626.019
Figure 7—figure supplement 1
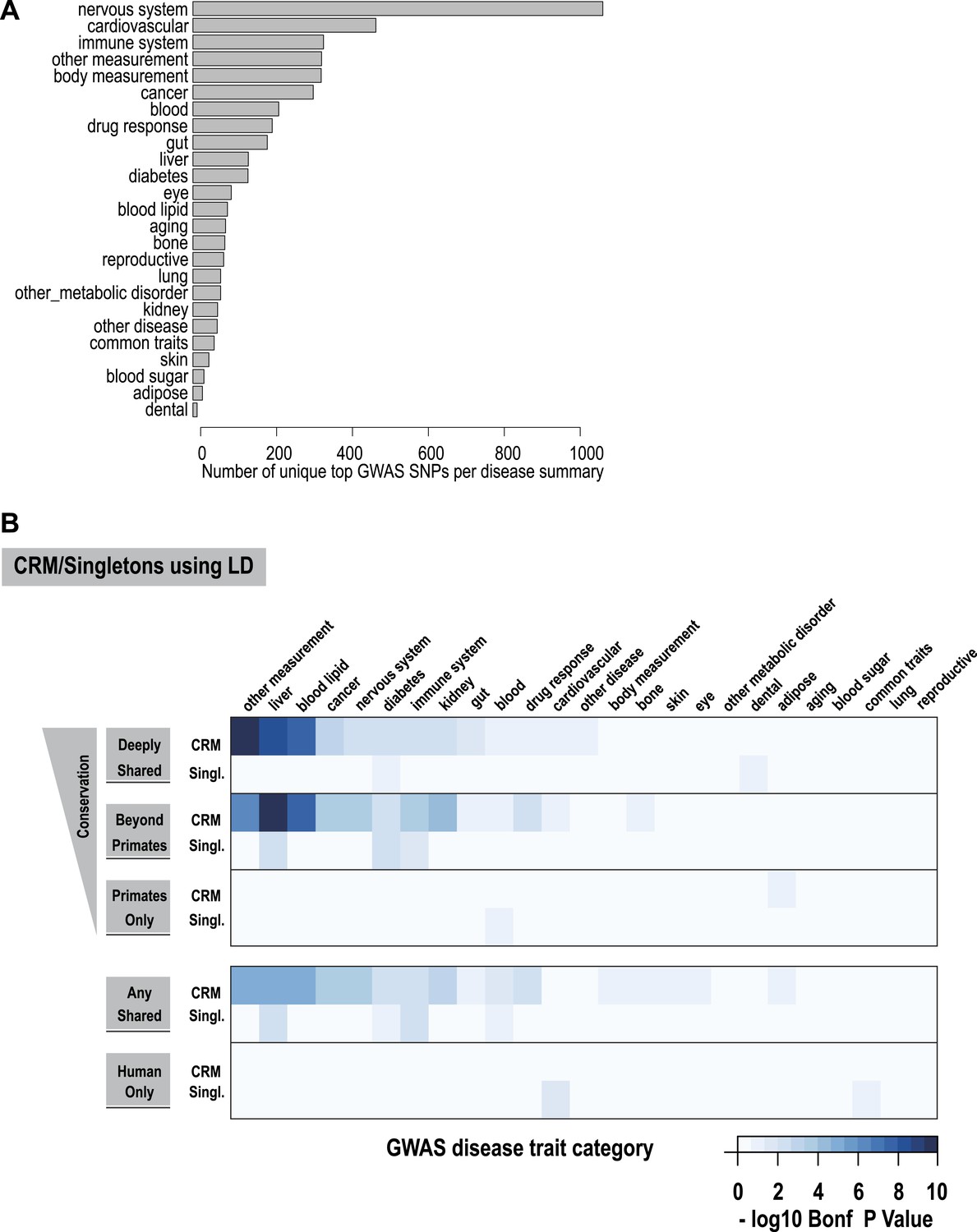
Using linkage disequilibrium (r2 ≥ 0.8), GWAS lead SNPs were enriched for shared CRMs in a tissue/disease specific manner.
(A) Number of lead SNPs reported in the NHGRI catalog for each of the summarized GWAS categories. (B) CRM regions were classified depending on their conservation pattern. These categories were then intersected with summarized disease traits obtained from the NHGRI catalogue (Hindorff et al., 2009). Linkage disequilibrium measures were calculated for lead SNPs from the NHGRI GWAS catalog up to a maximum of 100 kb flanking the lead SNP. Windows around each SNP were created based on the largest genomic interval between two SNPs in linkage disequilibrium (r2 ≥ 0.8). The heatmap shows the −log10 of Bonferroni corrected p-values for the enrichments of CRMs and singletons broken down into categories related to their degree of conservation determined by our multi-species ChIP-seq comparisons.
Figure 7—figure supplement 2
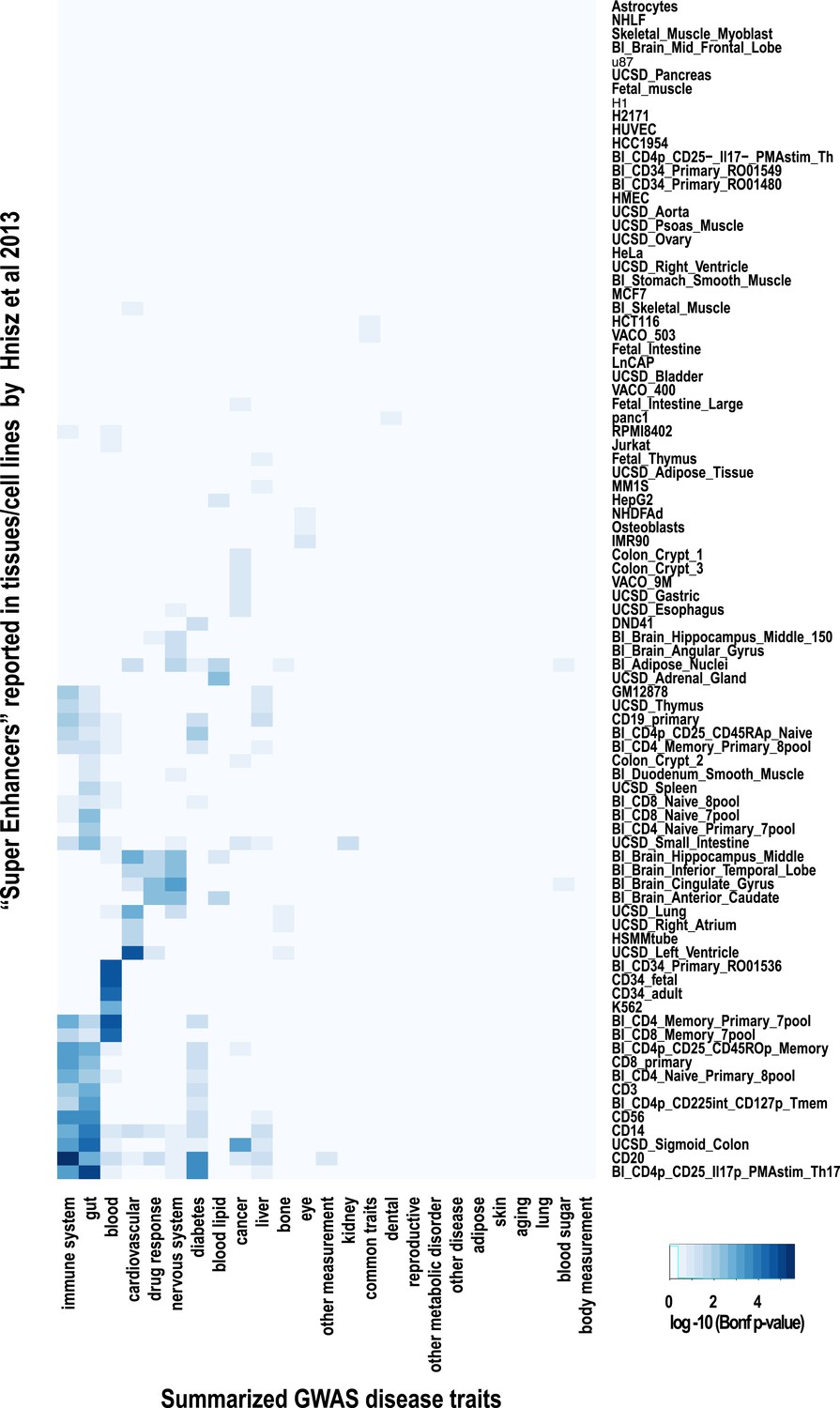
Super-enhancer enrichments obtained by GWAS lead SNPs.
‘Super-enhancers’ were directly obtained from Hnisz et al. (2013) and overlapped with lead GWAS SNPs annotated in the NHGRI catalogue (Hindorff et al., 2009). Each lead GWAS SNP was given a 5-kb window prior to identify overlapping ‘Super Enhancers’. The heatmap shows hierarchical clustering of the −log10 of corrected p-values for the enrichments of super-enhancers. A Hypergeometric test with Bonferroni correction was employed to assess the significance of enrichments in all categories. The heatmap shows significant enrichment of lead SNPs by immune cell super-enhancers. Super-enhancers from the liver cancer cell line HepG2 were not enriched for liver-related GWAS lead SNPs but were enriched for blood lipid related SNPs.
Figure 8 with 1 supplement
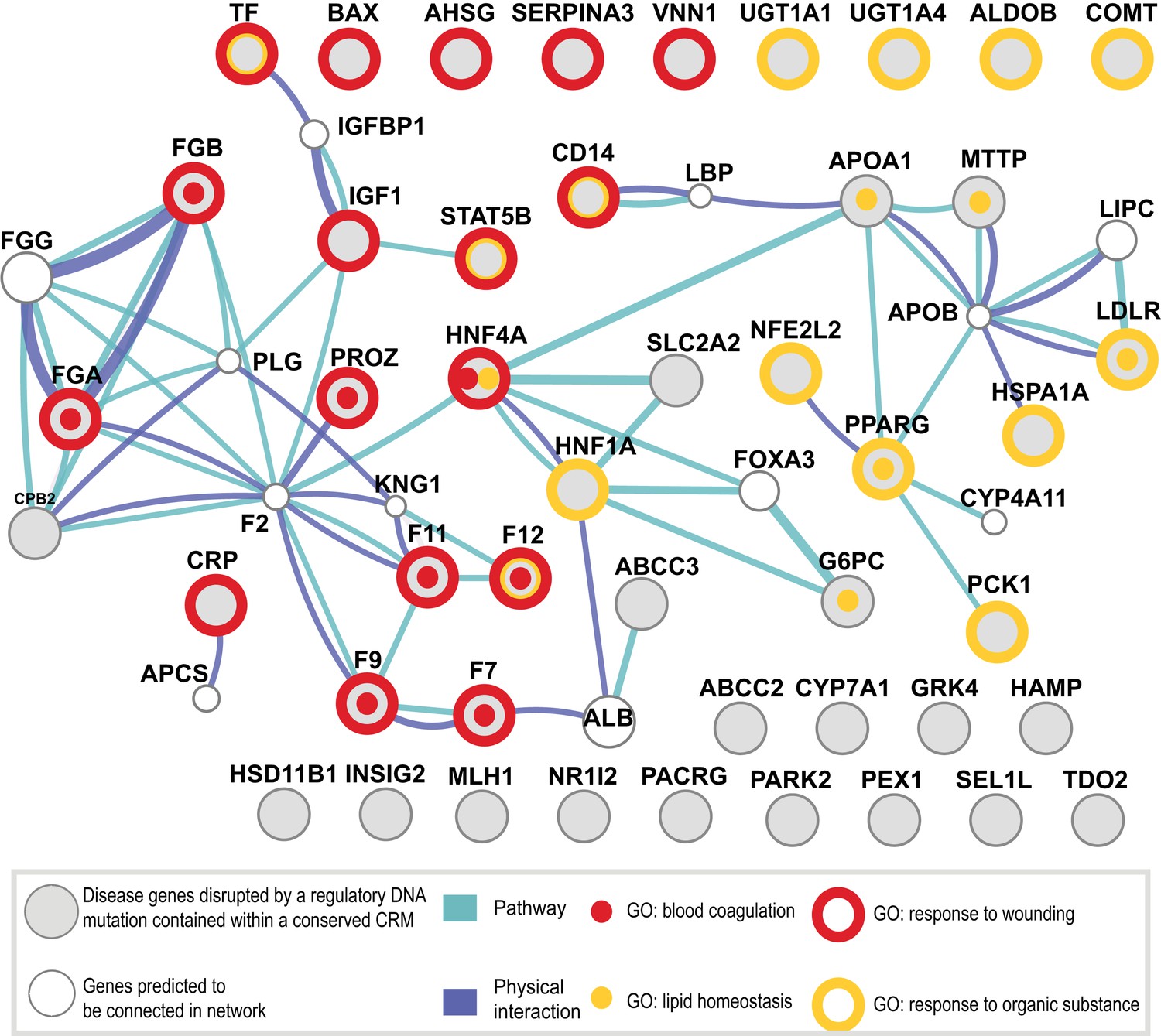
Shared CRMs link TF binding with disease-causing variants in coagulation and lipid regulation in the liver.
Human CRMs that had TF binding in syntenic regions in at least two additional species (n = 5046) were intersected with the HGMD database. All protein coding genes associated with a regulatory mutation were analysed. Relationships among these genes were investigated and a representative analysis obtained using GeneMANIA (‘Materials and methods’). Genes (large grey circles) are connected by pathways and protein–protein interactions are shown. The smaller white circles are genes predicted by GeneMANIA to be in the network. The 47 unique genes were associated into 35 clusters using DAVID (Huang da et al., 2009). Eight Gene Ontology terms from the 35 clusters had an adjusted p-value of less than 0.005 (Figure 8—source data 1A; Supplementary file 3). 4 of the 8 significant GO categories containing the most genes are illustrated: response to wounding (open red circle, p = 3.16 × 10−9; 9.9-fold enriched); blood coagulation (red dot, p = 9.34 × 10−6; 22.0-fold enriched); response to organic substance (open yellow circle, p = 1.05 × 10−5; 6.4-fold enriched); and lipid homeostasis (yellow dot, p = 6.93 × 10−5; 36.2-fold enriched).
-
Figure 8—source data 1
Table of DAVID enrichments used to annotate Figure 8 and the HGMD genes that overlapped our CRMs and singletons in the different phylogenetic categories.
- https://doi.org/10.7554/eLife.02626.025
Figure 8—figure supplement 1
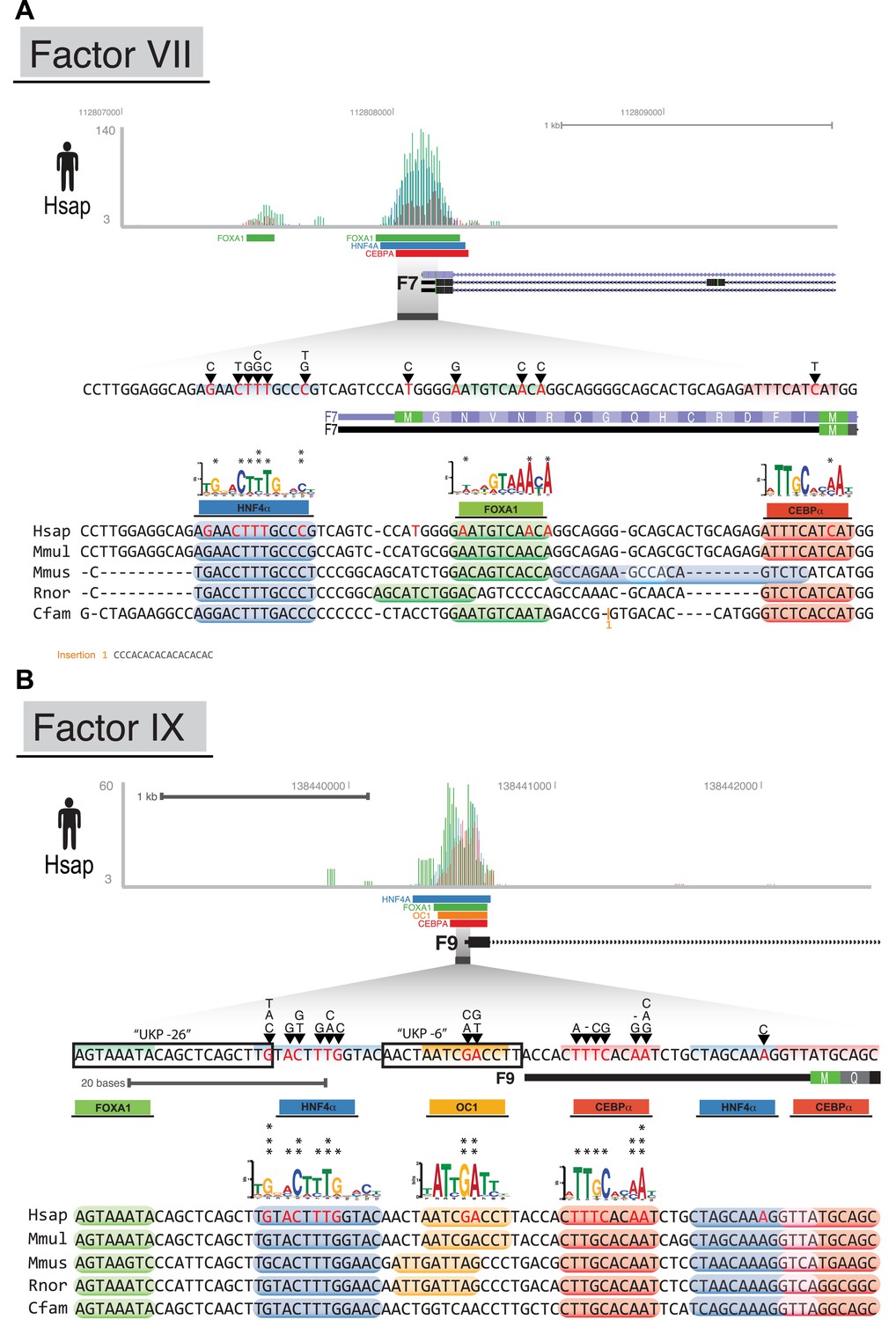
CRMs link TF binding with disease-causing variants in blood coagulation.
Raw human ChIP-seq signals (bed graph format) are shown at the proximal promoters of three blood coagulation genes: (A) F7; and (B) F9. TF reads and significantly bound peak regions are shown as colored rectangles according to the TF (CEBPA = red; HNF4A = blue; FOXA1 = green; and ONECUT1 = yellow). All unique regulatory mutations in the HGMD overlapping human CRMs are shown with black arrows. Asterisks are shown above the position weight matrices for each TF at the position homologous to a unique human regulatory mutation (i.e., an A → T is only counted once). For each species, in silico predicted motif positions for each TF are shown above the multiple sequence alignment at each promoter when they overlap regions that have evidence of ChIP signal.
Tables
Table 1
Table of GWAS Disease Traits that significantly enriched for Deeply shared CRMs
| Disease trait (NHGRI) | Category (this study) | Number of Deeply shared CRMs | Deeply shared CRM enrichment (adjusted p-value) | Fold enrichment | Closest genes |
|---|---|---|---|---|---|
| Other metabolic traits | other measurement | 11 | 4.42E-05 | 6.42 | CRP, HNF1A, PANK1 |
| LDL cholesterol | blood lipid | 20 | 4.84E-05 | 3.71 | TRIB1, ABCG8 PSRC1, DOCK7, APOB, HNF1A, LDLR, TOMM40, HNF1A, APOA5 |
| C-reactive protein | liver | 17 | 1.17E-04 | 3.97 | MLXIPL, RORA, CRP, HNF1A, TOMM40, BAZ1B, IRF1 |
| D-dimer levels | liver | 11 | 6.87E-04 | 4.99 | NME7, FGG, EDEM2, FGA |
| Fibrinogen | liver | 15 | 3.40E-03 | 3.40 | FGB, FGA, FGG |
| Lung cancer | cancer | 14 | 4.39E-03 | 3.46 | C2, CRP, HSPA1A, TP63 |
| Mean corpuscular hemoglobin | blood | 8 | 8.57E-03 | 5.02 | GCDH, USP49, RCL1, SLC17A1, TFRC, MPST |
| Protein quantitative trait loci | other measurement | 14 | 2.49E-02 | 2.93 | CRP, IFT81, BCO2 |
| Serum markers of iron status | other measurement | 13 | 2.67E-02 | 3.03 | TCP1, MRPL18 , TF, SLC17A1, HIST1H4C, MPST, GHR |
| Triglycerides | blood lipid | 12 | 2.84E-02 | 3.16 | TRIB1, MLXIPL, DOCK7, BAZ1B, GALNT2, TRIB1, APOA5 |
| Select biomarker traits | other measurement | 8 | 3.89E-02 | 4.08 | CRP, OR10J5 |
-
Lead GWAS SNPs and their associated Disease Traits were obtained directly from the NHGRI catalog (Hindorff et al., 2009). An LD window (r2 ≥ 0.8) around each SNP was obtained and regions with identical Disease Traits were collapsed into a single interval. These Disease-Trait–associated intervals were then intersected with all CRMs and Singleton categories as in Figure 7. This table shows Disease Traits that were significantly enriched for Deeply shared CRMs. The summarized disease category used in Figure 7, the number of Deeply shared associated CRMs, the Bonferroni corrected p-values from the hypergeometric test, fold enrichment of Deeply shared CRMs, and the nearest gene to the Deeply shared CRM, if it is protein coding, are shown. Figure 7—source data 1G contains detailed enrichment information including SNP ID and primary GWAS publication (PMID).
Table 2
HGMD disease variants falling within motifs of shared CRMs and singletons
| Coordinates | Gene name | HGMD (regulatory, disease mutations) | Disease mutations | |
|---|---|---|---|---|
| Shared CRM | 3:30622781-30623121 | TGFBR2 | Marfan syndrome II | 1 |
| 3:37009728-37010105 | MLH1 | Colorectal cancer, non-polyposis | 8 | |
| 3:95175129-95175686 | PROS1 | Protein S deficiency | 2 | |
| 3:172227012-172227583 | SLC2A2 | Diabetes | 1 | |
| 4:187423808-187424193 | F11 | Factor XI deficiency | 1 | |
| 5:176769019-176769427 | F12 | Factor XII deficiency | 2 | |
| 7:75769704-75769860 | HSPB1 | Amyotrophic lateral sclerosis | 1 | |
| 9:35647685-35648108 | RMRP | Cartilage-Hair hypoplasia* | 60 | |
| 9:103237817-103238177 | ALDOB | Fructose intolerance | 1 | |
| 11:57121333-57121845 | SERPING1 | Angioneurotic oedema | 3 | |
| 11:116213420-116213833 | APOA1 | Apolipoprotein A1 deficiency; Atherosclerosis with coronary artery disease | 2 | |
| 12:119900361-119900931 | HNF1A | Diabetes | 9 | |
| 13:112807935-112808278 | F7 | Factor VII deficiency | 15 | |
| 17:39777939-39778164 | GRN | Amyotrophic lateral sclerosis; Frontotemporal dementia | 2 | |
| 19:11060834-11061300 | LDLR | Hypercholesterolaemia | 23 | |
| 19:40465042-40465277 | HAMP | Haemochromatosis | 2 | |
| 19:50140788-50141367 | APOC2 | Apolipoprotein C2 deficiency | 1 | |
| 20:42417527-42417906 | HNF4A | Diabetes | 13 | |
| X:138440311-138440689 | F9 | Haemophilia B | 22 | |
| Shared singleton | 1:55277551-55277787 | PCSK9 | Hypercholesterolaemia, autosomal dominant | 1 |
| 2:47483486-47483635 | MSH2 | Colorectal cancer, non-polyposis | 1 | |
| 5:147191404-147191657 | SPINK1 | Pancreatitis | 5 | |
| 11:107598900-107599060 | ATM | Ataxia telangiectasia | 1 | |
| 17:3486175-3486485 | CTNS | Cystinosis | 2 | |
| 17:27840752-27841062 | CDK5R1 | Mental retardation | 1 | |
| 19:54160105-54160315 | FTL | Cataract, bilateral | 1 | |
| X:66680386-66680665 | AR | Prostate cancer | 1 | |
| Human only CRM | 1:113300254-113300535 | SLC16A1 | Exercise-induced hyperinsulinism | 1 |
| 1:224075148-224075491 | EPHX1 | Hypercholanaemia | 1 | |
| 3:170965492-170965762 | TERC | Aplastic anaemia; Dyskeratosis congenita; Myelodysplastic syndrome | 3 | |
| 8:64161127-64161334 | TTPA | Ataxia, isolated vitamin E deficiency | 1 | |
| 10:27429312-27429620 | ANKRD26 | Thrombocytopaenia | 12 | |
| 13:59636050-59636246 | DIAPH3 | Auditory neuropathy | 1 | |
| X:146800975-146801150 | FMR1 | Fragile X mental retardation syndrome | 3 | |
| X:153643911-153644318 | DKC1 | Dyskeratosis congenita, X-linked | 1 |
-
HGMD disease variants falling within shared CRMs, shared singletons and human only CRMs. The number of unique regulatory mutations designated as ‘disease-mutations’ in the HGMD database recorded within each CRM or singleton is shown.
-
*
RMRP is a non-coding RNA that is found in HGMD associated to several related diseases.
Additional files
-
Supplementary file 1
Motif enrichments of CRMs and singletons as well as overlaps of CRMs and Singletons with ENCODE data.
- https://doi.org/10.7554/eLife.02626.027
-
Supplementary file 2
Deeply conserved CRMs within 2.5 kb of a lead GWAS SNP were overlapped with RegulomeDB annotated SNPs. SNPs with a high confidence evidence of regulatory potenital are shown (evidence levels 1 and 2 from Regulome DB).
- https://doi.org/10.7554/eLife.02626.028
-
Supplementary file 3
Tally of genes with HGMD regulatory mutations that overlap liver CRMs or singletons.
- https://doi.org/10.7554/eLife.02626.029
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Multi-species, multi-transcription factor binding highlights conserved control of tissue-specific biological pathways
eLife 3:e02626.
https://doi.org/10.7554/eLife.02626




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}