Single molecule compression reveals intra-protein forces drive cytotoxin pore formation
- Shanghai Jiao Tong University, China
Figures
Figure 1

Structure of PFO and the known structural changes during pore formation.
(A) The water-soluble monomer consists of four domains, D1 to D4. (B) AFM images showing the large height difference between prepore and pore complexes. Scale bar: 50 nm.
Figure 2 with 5 supplements
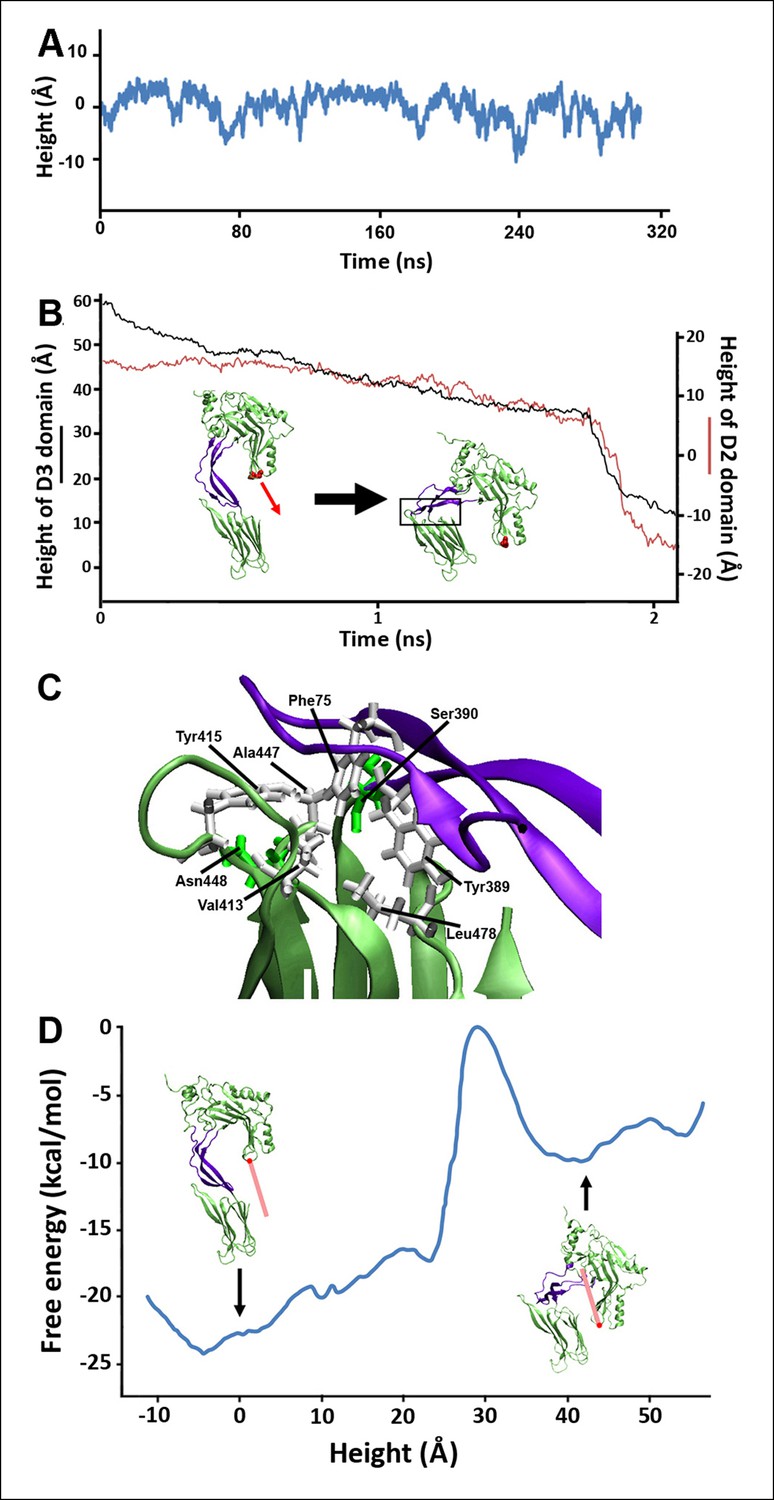
Steered MD simulations and energy landscape associated with the membrane-directed descent of the D3 domain.
(A) The change in height of PFO during extended equilibrium simulations following the removal of the TMHs. (B) The D2 domain restructures as a result of a downward force (here 250 pN) applied to the D3 domain. Inset: the initial and final structures of the steered MD simulations. (C) Detailed view of the D2/D4 interface (the boxed region in [B]). (D) Potential of mean force profile calculated from ABF simulations using the height of the D3 region (specifically the red sphere) as the reaction coordinate.
Figure 2—figure supplement 1
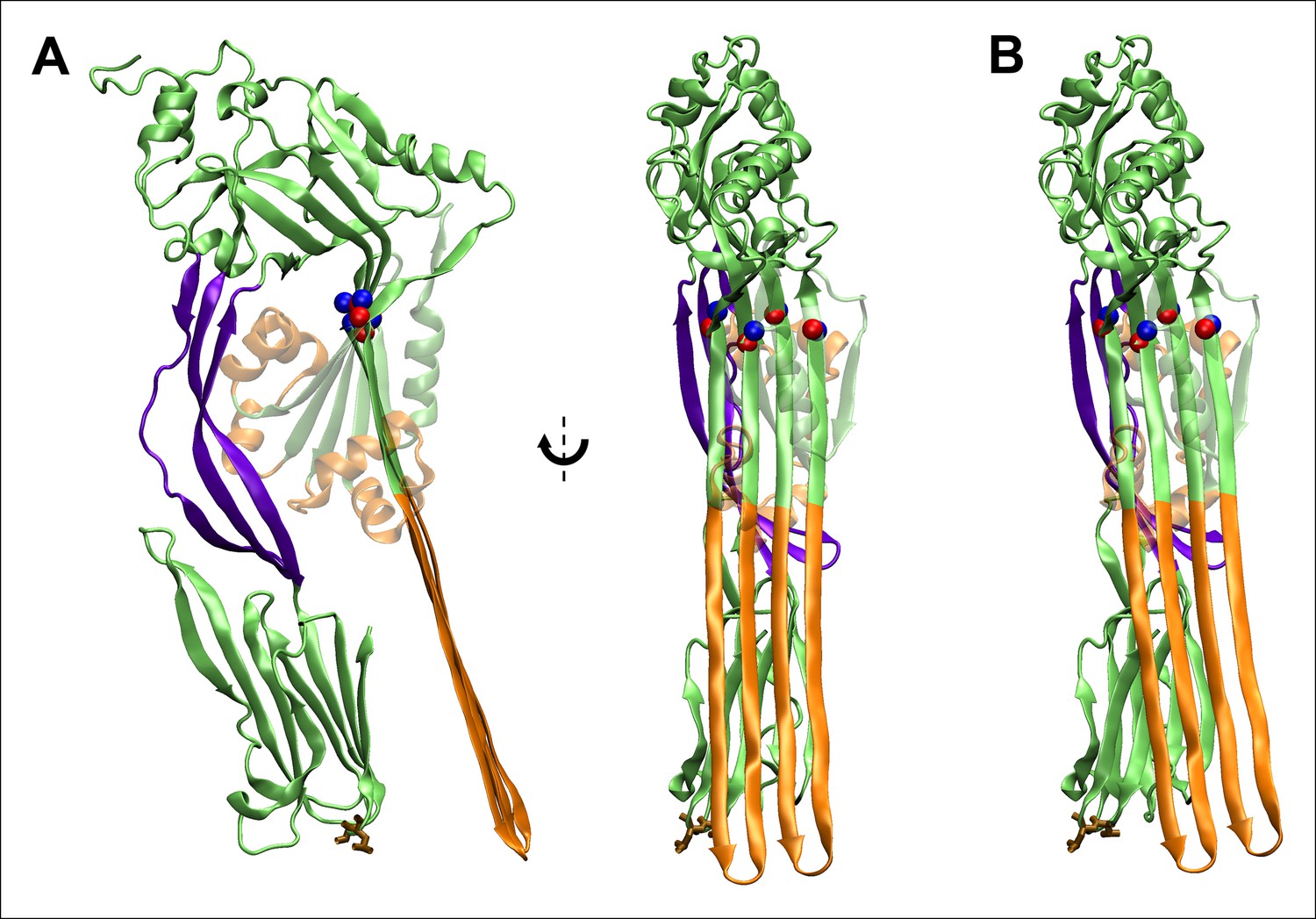
Fully extended TMHs are sufficiently long to touch the membrane surface from their position in the prepore.
(A) The TMHs were modeled as β-strands and positioned in an essentially vertical orientation, overlapping the Cα backbone atoms of residues Gln180, Tyr226, Thr276, and Val322 (red and blue spheres are the native structure and modeled β-strand atoms). Also shown in brown stick representation are the residues, Thr490 and Leu491, in D4 that are believed to directly contact cholesterol (Farrand et al., 2010), and thus indicate the position of the membrane surface. This orientation reflects the approximate path of D3 descent studied in most steered MD simulations and ABF calculations. (B) The TMHs would contact the membrane surface even if tilted by ~∼10° from the orientation depicted in (A).
Figure 2—figure supplement 2
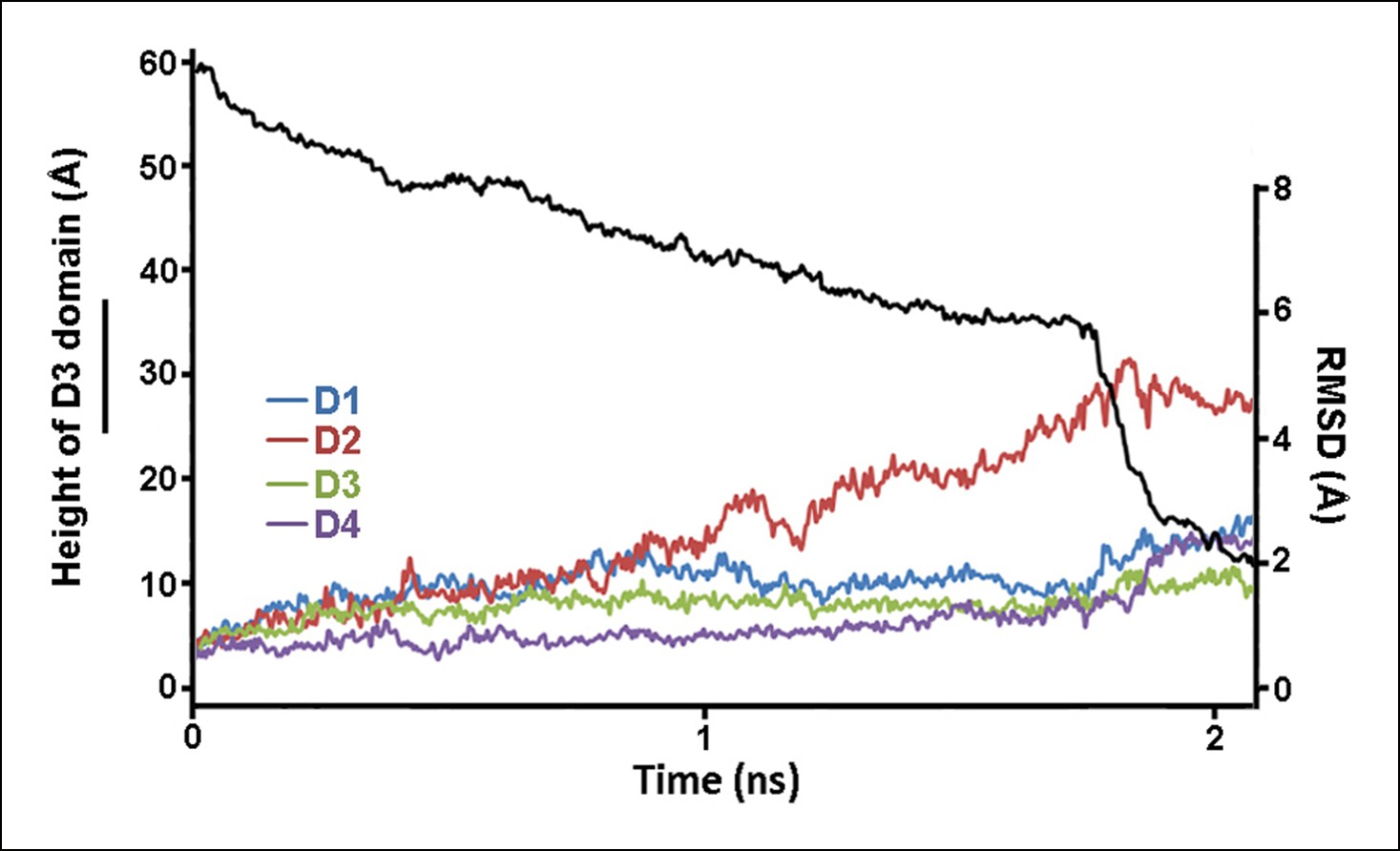
Structural changes in each domain during the steered MD simulations described in Figure 2.
Shown is the root-mean-squared-deviation (RMSD) of the backbone Cα atoms of each domain from their initial conformation throughout the trajectory.
Figure 2—figure supplement 3
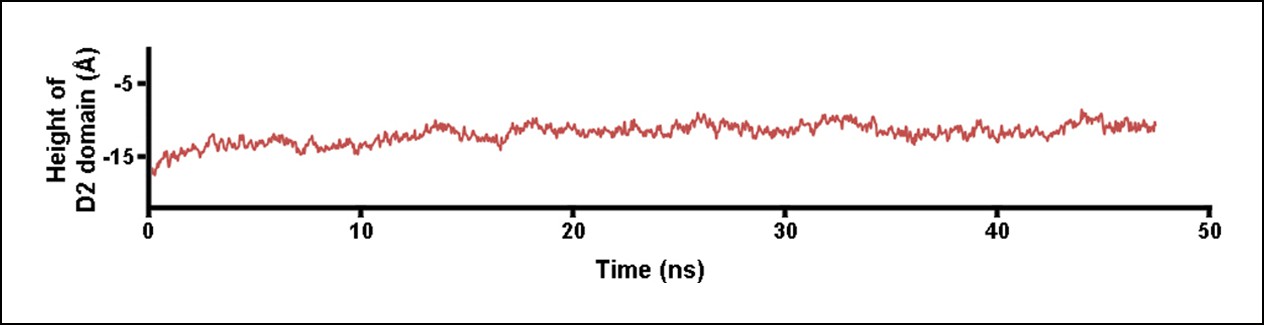
Height of the D2 domain following the release of the constant force on the D3 domain at the end of the steered MD simulations described in Figure 2.
https://doi.org/10.7554/eLife.08421.007
Figure 2—figure supplement 4
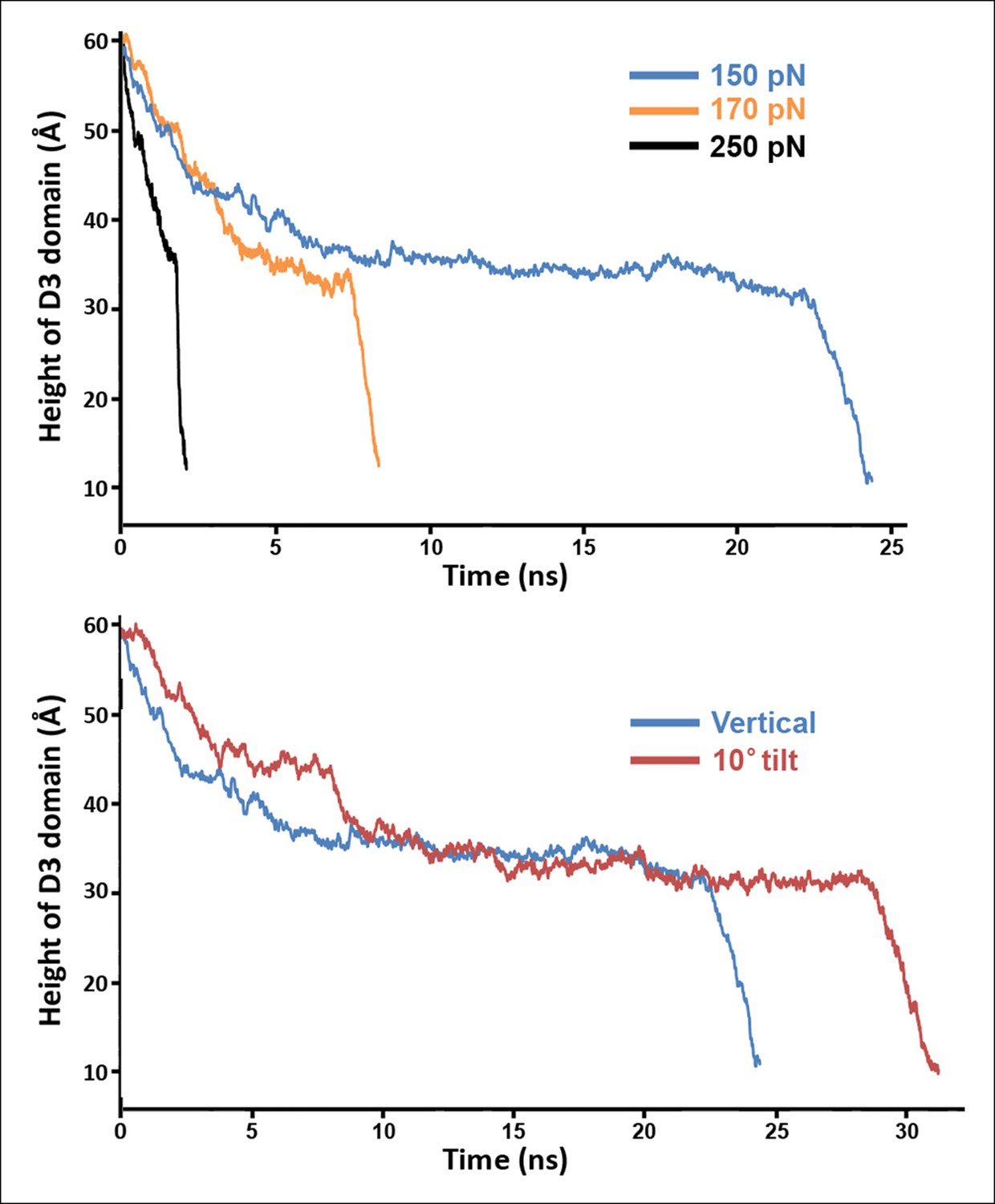
Steered MD simulations with different applied forces and force directions.
(A) Simulations at different force magnitudes. The direction of the applied force is approximately that of the β-strands depicted in Figure 2—figure supplement 1A. (B) Shown are the trajectories for force applications of 150 pN in the 'vertical' and '10° tilt' directions depicted in Figure 2—figure supplement 1A and B, respectively.
Figure 2—figure supplement 5
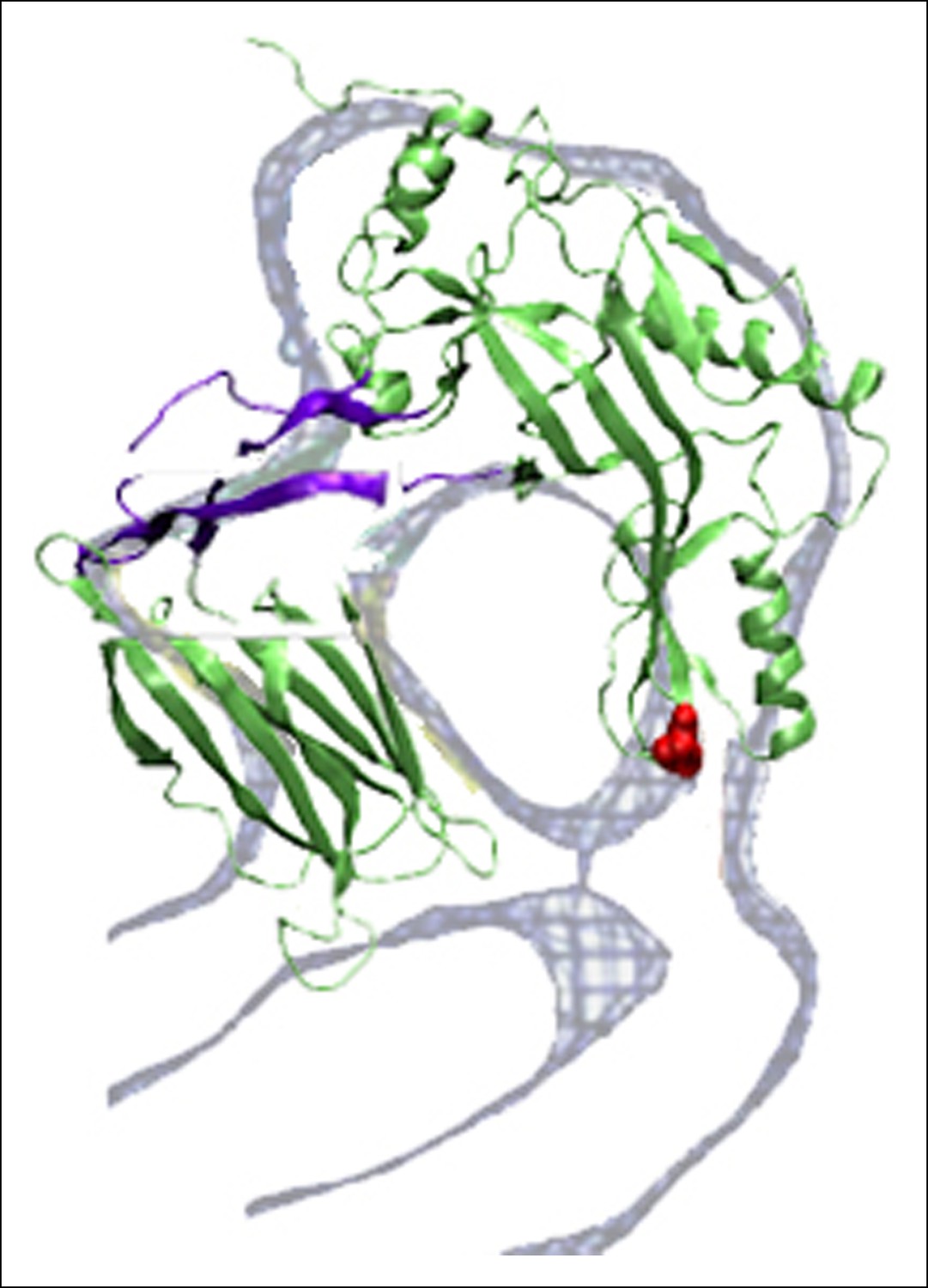
Overlap between the electron density profile from cryo-EM images of the pneumolysin pore (Tilley et al., 2005) and the PFO structure at the final stages of the steered MD simulations.
https://doi.org/10.7554/eLife.08421.009
Figure 3 with 4 supplements
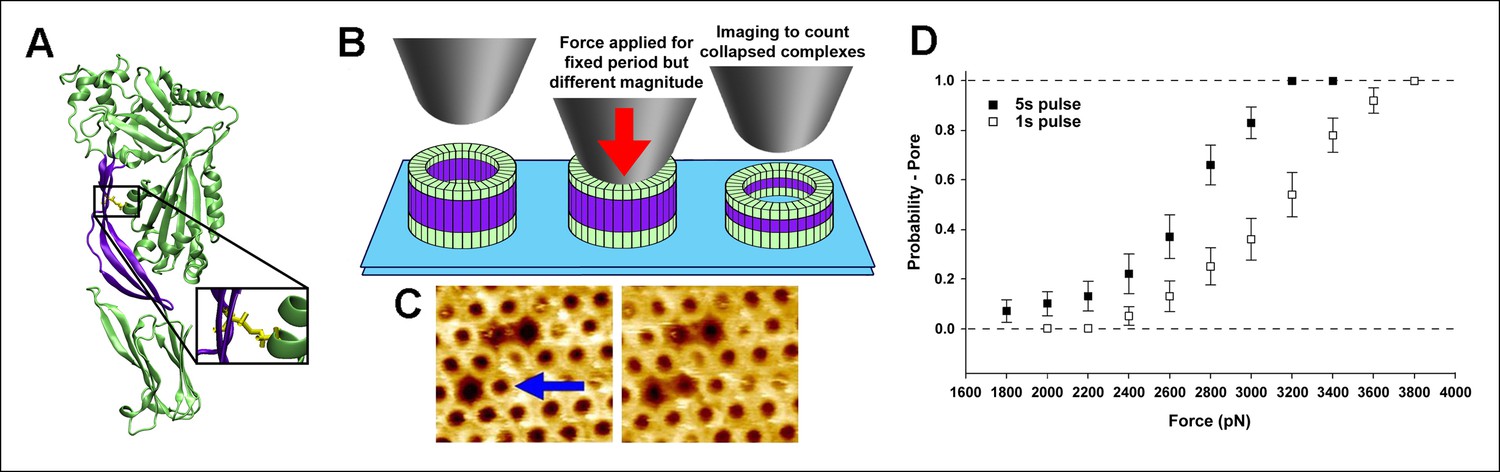
Single molecule compression provides a direct measure of the energy landscape of the D2 collapse.
(A) PFOG57C-S190C prepore-trapped mutant. (B) Schematic of the experiment. (C) A force of 110 pN/monomer (∼4000 pN/complex) applied for 1 s to a single prepore complex (blue arrow) catalyzed the collapse to the pore-like height. Image size: 200 x× 220 nm2. (D) Force and time dependence of the AFM probe-induced D2 collapse.
Figure 3—figure supplement 1

Scanning electron micrograph of the AFM tip used in these experiments shows a symmetric apex with a radius of ∼25 nm, consistent with manufacturer’s specifications.
The compressive force applied with such AFM probes to the center of the PFO complexes (roughly 35 nm in diameter) is thus expected to be essentially equally distributed among all monomers of the complex.
Figure 3—figure supplement 2
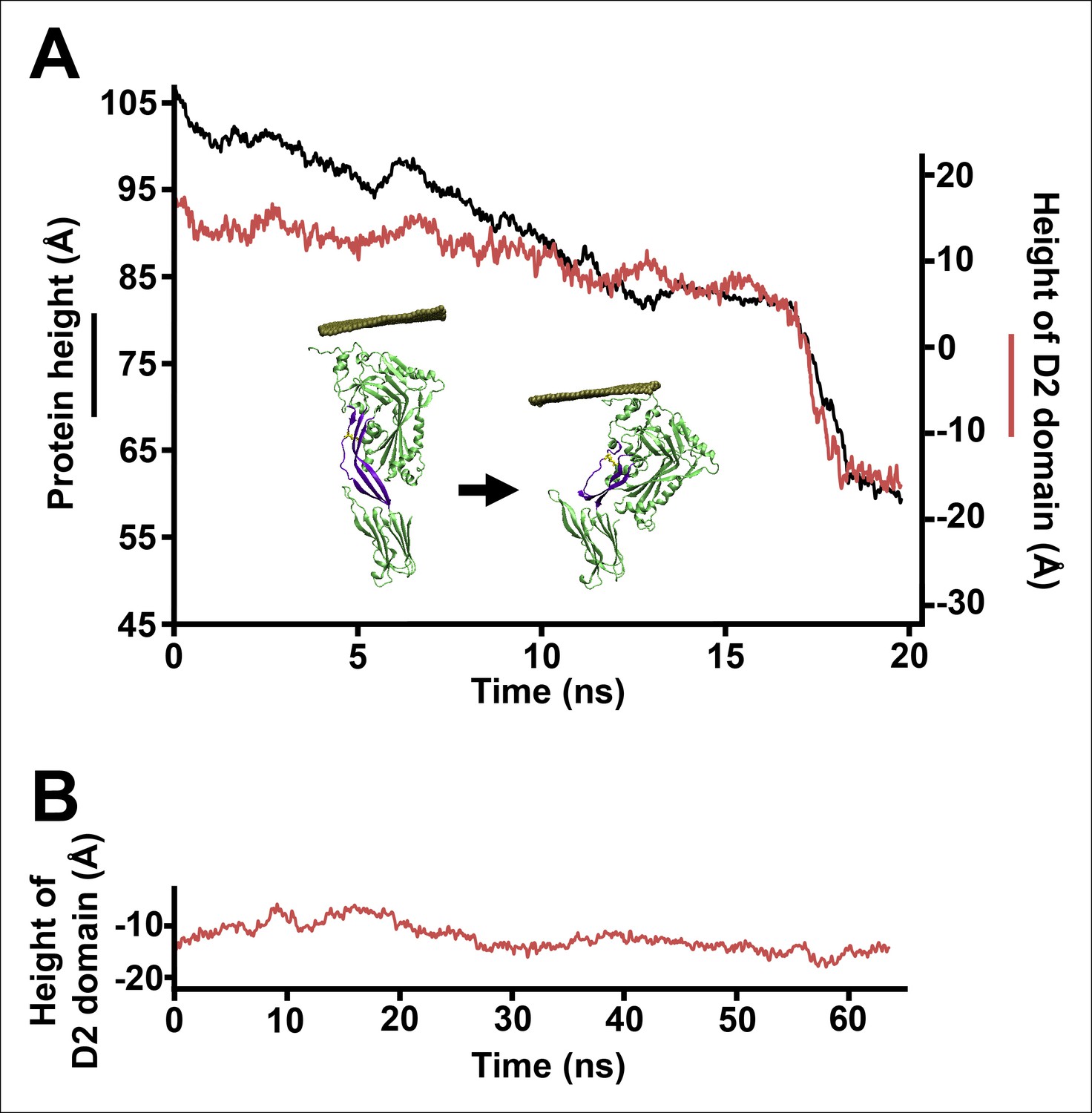
Steered MD simulations reveal the collapse of the D2 domain under the application of compressive force to PFOG57C-S190C.
(A) The D2 domain restructures as a result of a downward force (175 pN in total) applied to a section of a carbon nanotube that directly contacts the top of the D1 domain. The disulfide bridge linking G57C and S190C that remains present throughout the simulations is shown in yellow. (B) Height of the D2 domain following the release of the constant force on the nanotube (removing the nanotube atoms from the simulation) at end of the steered MD simulations described (A).
Figure 3—figure supplement 3
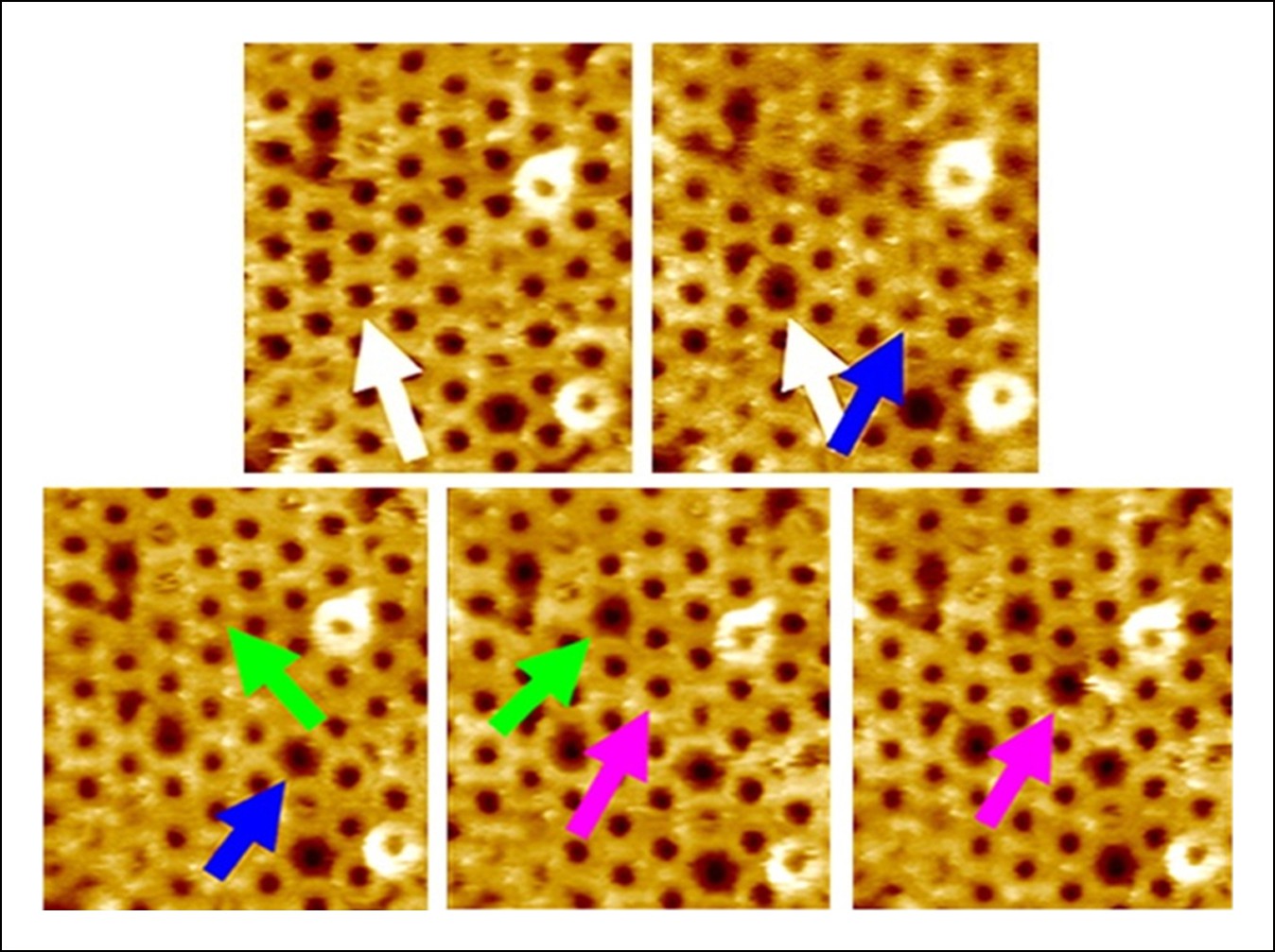
Additional AFM images showing the forced collapse of prepore-locked PFOG57C-S190C complexes.
From the top-left to bottom-right panels, compressive forces (∼110 pN/monomer or ∼4000 pN/complex, 1s) were applied sequentially to the complexes marked by the white, blue, green, and pink arrows. In each case, the complex collapsed to the smaller pore-like height. Image size: 300 x× 325 nm2.
Figure 3—figure supplement 4
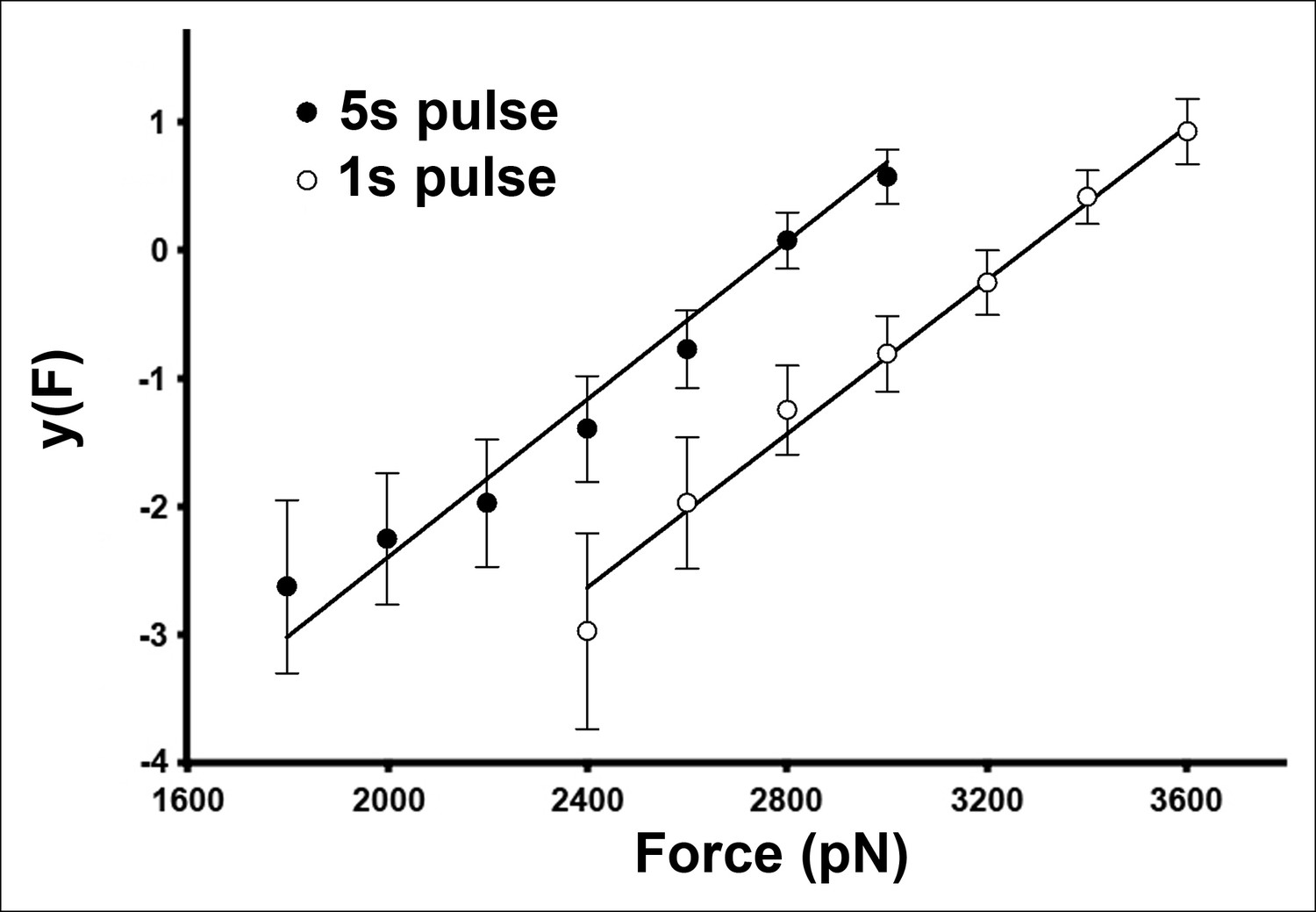
The AFM compressive data is well described by a two-state, single energy barrier model.
For this plot, the data shown in Figure 3D were transformed according to Equation 3 (see Materials and methods).
Figure 4
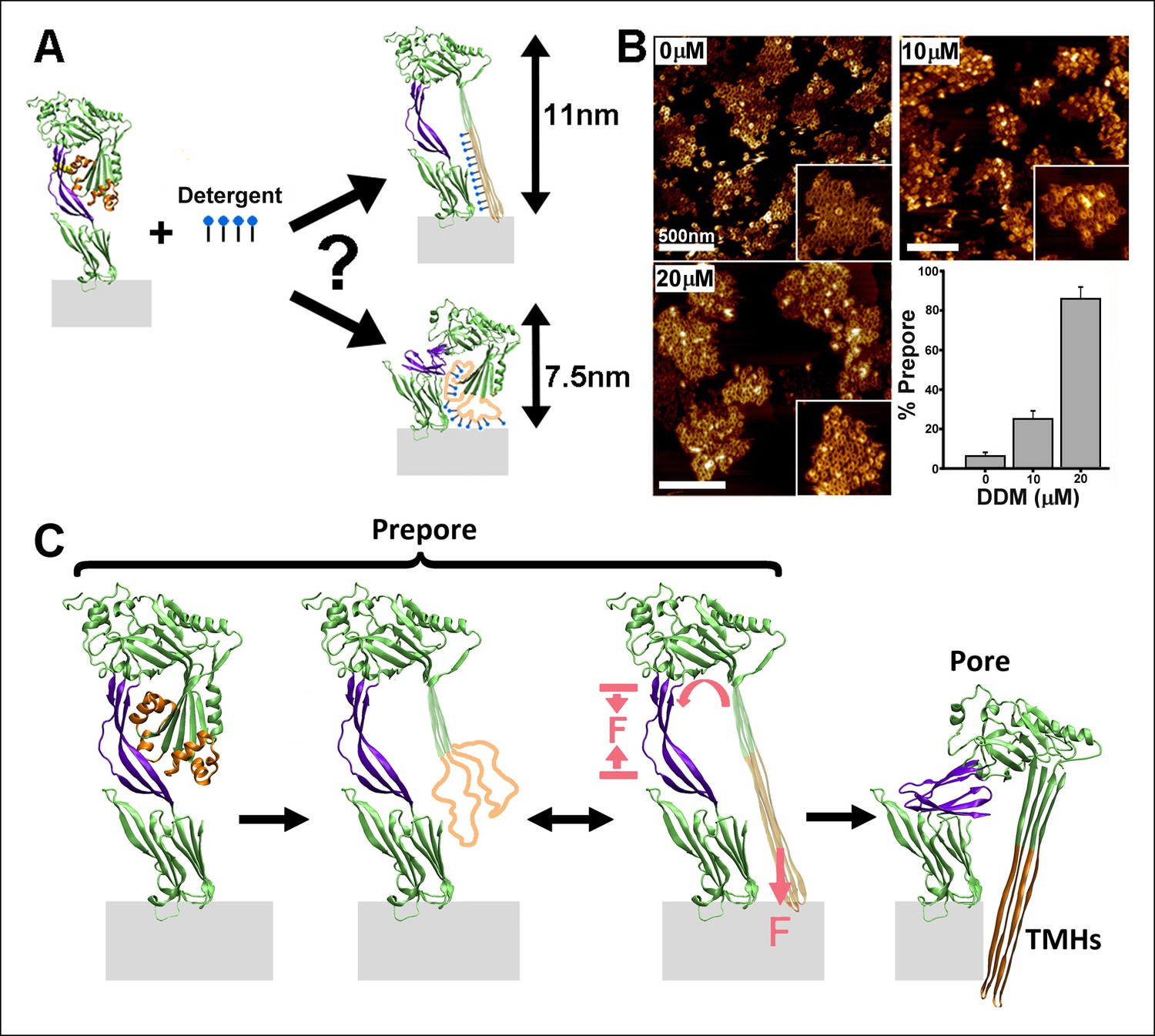
Effect of detergent on the prepore-to-pore transition and final model.
(A) Potential outcomes of binding of detergent to hydrophobic residues within the TMHs. (B) AFM images showing the inhibition of the pore-like height with increasing concentration of the detergent, DDM. Inset image size: 550 x× 550 nm2. (C) Schematic model of the mechanism by which conformational changes in the D2 and TMHs are coordinated. Compressive intra-protein stresses generated by the bilayer-directed forces on the unfolded TMHs catalyze the restructuring of the D2 domain to its collapsed conformation in the pore.
Author response image 1
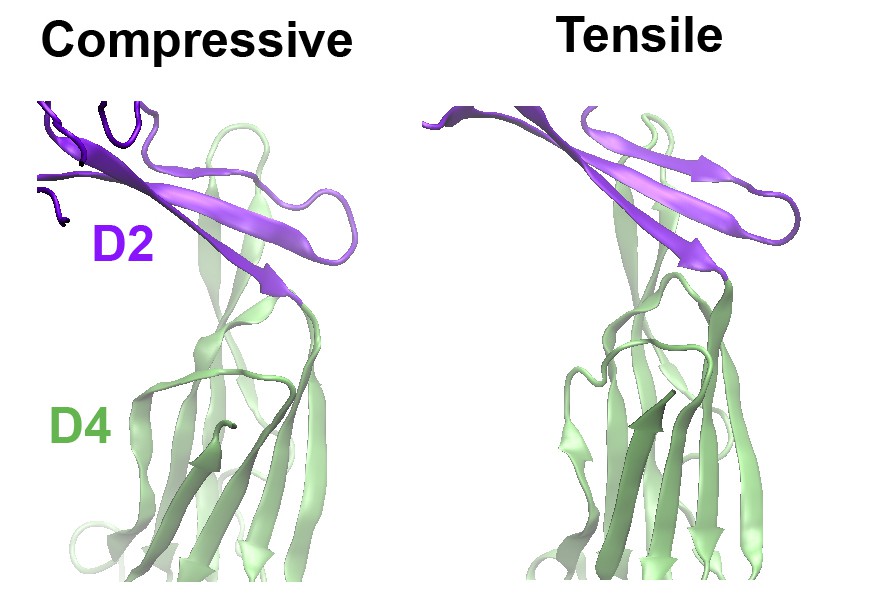
Comparison of the structures of the D2 domain and the D2/D4 interface, following the application of compressive or tensile forces.
Shown are snapshots of the structures 40 ns after the release of the applied force.
Additional files
-
Supplementary file 1
Sequence similarity of the hydrophobic residues in PFO at the D2/D4 interface in the collapsed D2 conformation among members of the CDC family.
The proteins were aligned using BLAST. Shown in parentheses are the residue numbers for each protein.
- https://doi.org/10.7554/eLife.08421.016
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Single molecule compression reveals intra-protein forces drive cytotoxin pore formation
eLife 4:e08421.
https://doi.org/10.7554/eLife.08421




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}