Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin
- National Institutes of Health, United States
- Gwangju Institute of Science and Technology, Republic of Korea
- Indian Institute of Science Education and Research, Pune, India
- Chonnam National University, Republic of Korea
- Korea Basic Science Institute, Republic of Korea
Figures
Figure 1 with 4 supplements
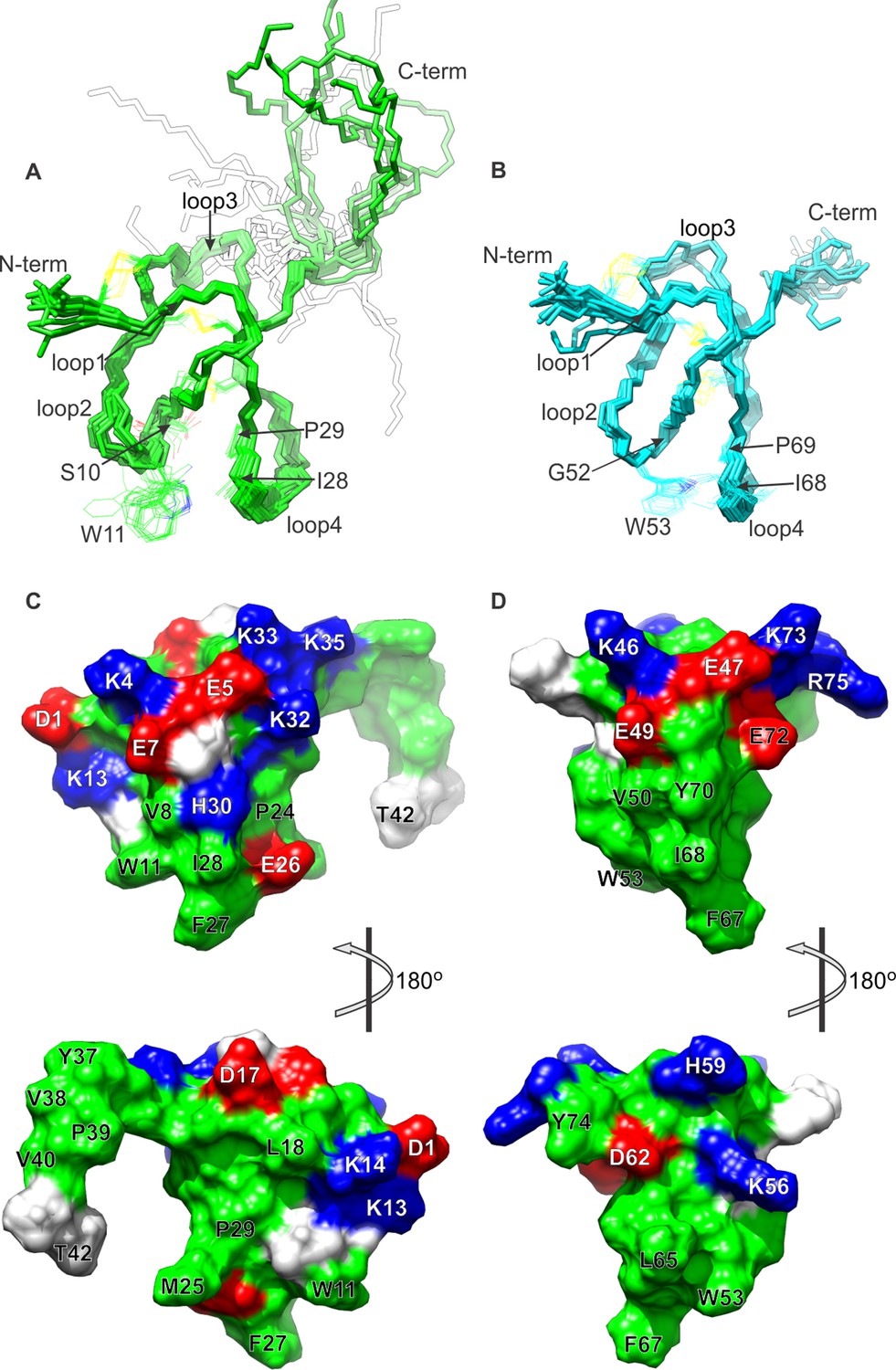
NMR solution structures of K1 and K2.
(A,B) Ensemble of backbone structures of K1 (A), in green, and K2 (B), in cyan. The 20 lowest-energy structures out of 100 energy-minimized structures are shown (PDB entry 2N9Z for K1 and 2NAJ for K2). The distinct orientations of W11 and W53, as well as their interactions with residues in loop 4, are also depicted. The backbone root mean square deviation (RMSD) of the structures in these ensembles relative to the average is 0.4 Å for both K1 and K2 (for the most ordered regions of each protein, which include Cys2-Cys31 in K1 and Cys44-Cys71 in K2). (C,D) Surface representations of energy-minimized structures of K1 (C) and K2 (D). Hydrophobic, basic and acidic residues are colored in green, blue and red, respectively.
Figure 1—figure supplement 1
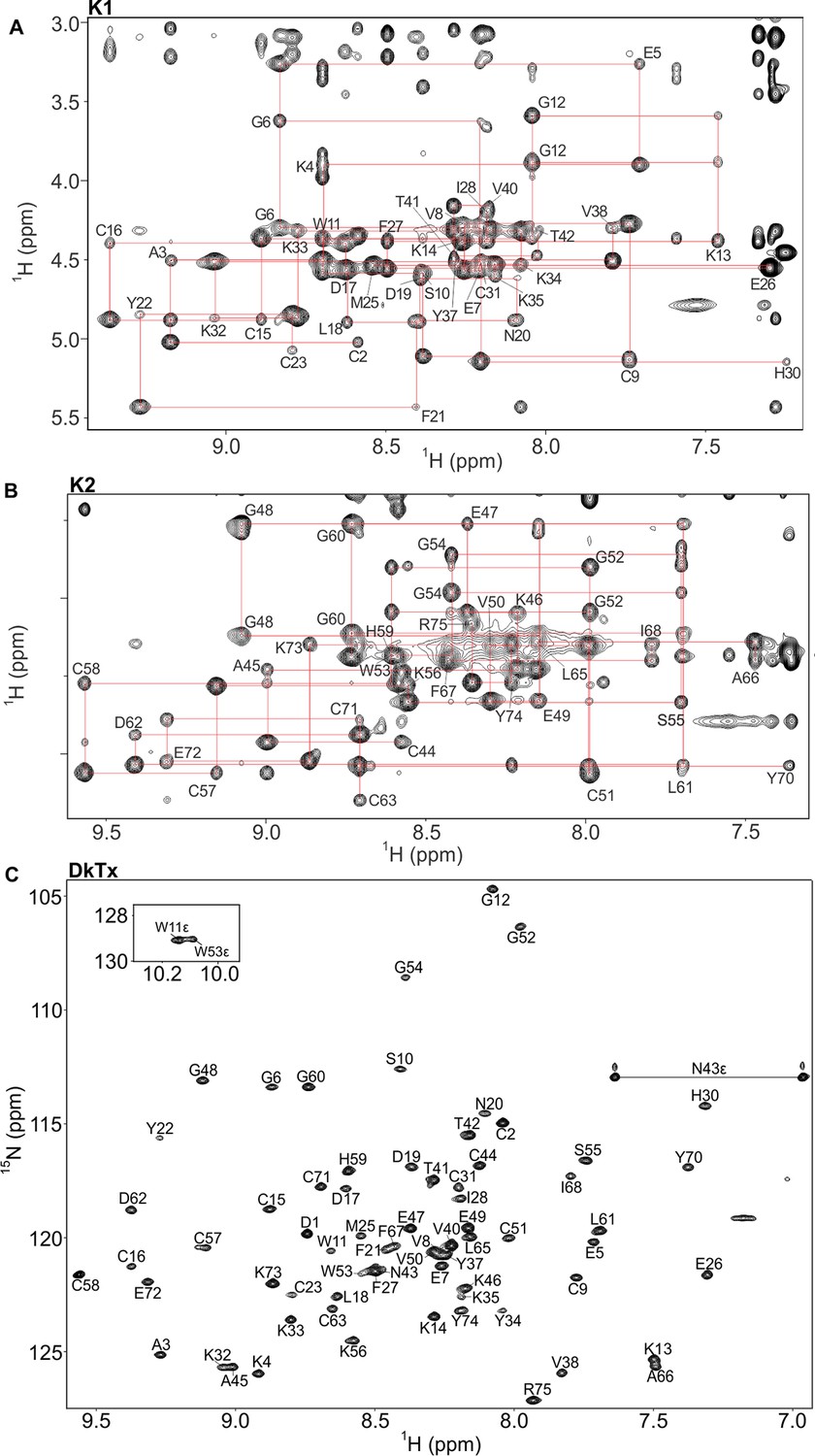
NMR spectra of K1, K2 and DkTx in solution.
(A,B) Fingerprinting regions of 1H-1H NOESY (298 K) spectra of K1 (A) and K2 (B). Sequential dαN(i,i+1) NOE connectivities are shown in red lines. All intra-residue HN-Hα cross peaks are identified (except prolines and N-terminal residue of K1/K2) and labeled as single-letter amino acid with the residue numbering. (C) 1H-15N hetronuclear single quantum coherence (HSQC) spectrum of DkTx. All backbone amide groups in the DkTx were labeled.
Figure 1—figure supplement 2

Backbone assignments of DkTx in solution.
Strips are derived from 1H-15N NOESY-HSQC spectrum of DkTx, and each strip represents single backbone nitrogen resonance. Residue number and 1HN/15N resonances are labeled above and below the stripes, respectively. Sequential NOE connectivity of DkTx is shown in red-dashed lines.
Figure 1—figure supplement 3
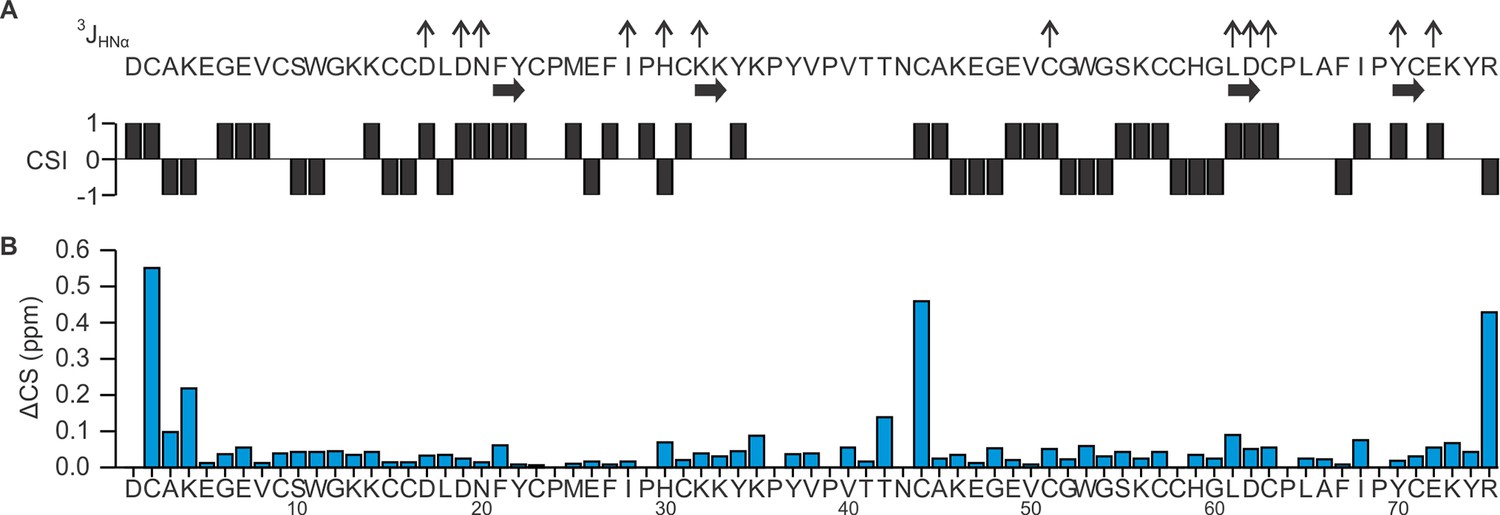
Summary of 3JHNHα coupling constant and proton chemical shifts of DkTx in solution.
(A) Summary of 3JHNHα coupling constants, secondary structure elements, and chemical shift index (CSI) for DkTx. Values of 3JHNHα are represented as ↑ (>8 Hz) or ↓(<5.5Hz). (B) Chemical shift comparison between K1/K2 lobes and DkTx. Chemical shift difference (ΔCS in ppm) between K1/K2 lobe and DkTx was calculated according to ΔCS = [(ΔδHα)2+(ΔδHN)2]1/2, where ΔδHα denotes Hα chemical shift difference between DkTx and K1/K2, and ΔδHN denotes HN chemical shift difference between DkTx and K1/K2.
Figure 1—figure supplement 4
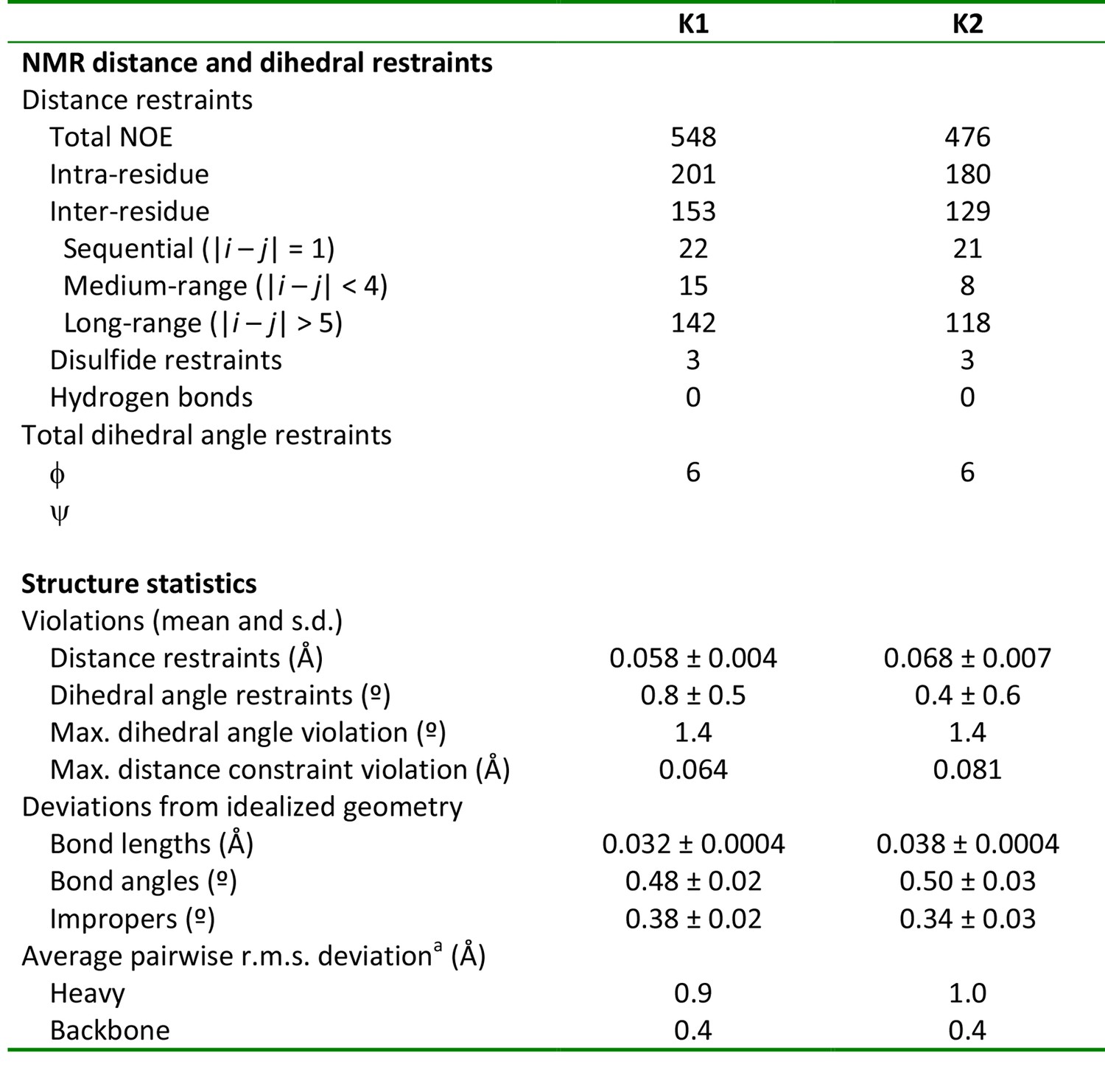
NMR and refinement statistics of K1 and K2
aPairwise r.m.s. deviation to the mean was calculated among 20 refined structures for ordered residues, namely 2–31 in K1 and 71 in K2.
Figure 2 with 3 supplements
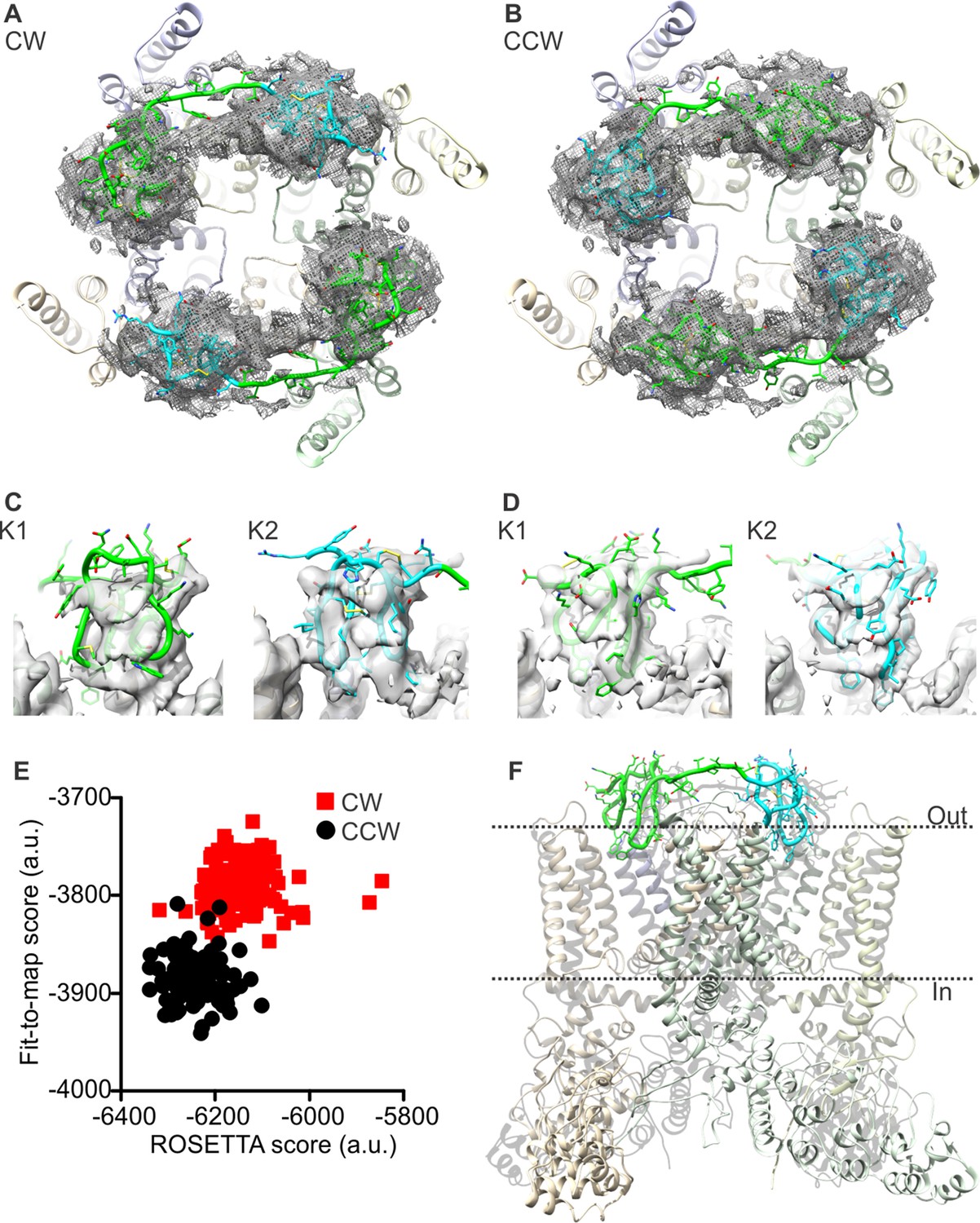
Docking of DkTx into the electron density map of DkTx/RTx-bound TRPV1.
(A,B) Top-ranking ROSETTA models of the structure of TRPV1 with DkTx bound in a clockwise (CW, A) or counter-clockwise (CCW, B) configuration. The structures are overlaid on the experimental twofold symmetric cryo-EM map (unpublished data kindly provided by Yifan Cheng and colleagues), and densities close to the toxin molecules are shown at a contour level of 3σ. K1 and K2 are shown in green and cyan, respectively. (C,D) View of K1 and K2 from the membrane plane, either in the CW (C) or CCW (D) configuration. The cryo-EM map is shown as a gray surface. (E) Quantitative evaluation of the CW and CCW configurations of DkTx bound to TRPV1 using ROSETTA. The ROSETTA fit-to-density score and the conventional ROSETTA score (excluding the fit-to-density contribution) are reported for 100 models generated for either the CW or CCW configuration. (F) View of the top-ranking ROSETTA model of the TRPV1-DkTx complex, with the K1 and K2 in a CCW configuration.
Figure 2—figure supplement 1

Evaluation of the CW and CCW orientations for docking of K1 and K2 to TRPV1 using Xplor-NIH.
aCorrelation coefficient between calculated density maps generated with Xplor-NIH for TRPV1-docked K1 and K2 and the twofold cryo-EM map of the TRPV1-DkTx complex. Shown are the mean values over 10 energy-minimized structures ( ± s.d.).
bRMSD of violated NOE distance restraints in the same 10 structures (mean ± s.d.).
cNumber of violated NOE distance restraints in the same 10 structures (mean ± s.d.). The total number of NOE distance restraints is 548 for K1 and 476 for K2.
Figure 2—figure supplement 2

Refinement of cryo-EM structures of apo and DkTx-bound TRPV1 using ROSETTA.
Score distribution for the 12,000 ROSETTA models obtained during the second stage of refinement for the apo (A) and DkTx-bound (B) TRPV1 channel (see Methods). The ROSETTA fit-to-density score (based on the experimental cryo-EM maps) is reported against the total ROSETTA score (excluding the fit-to-density contribution) for all the generated models. Note that the fit-to-density score was not optimized in this second stage. The red circles represent the best ROSETTA model obtained after the first refinement stage; the green circles represents the two final models selected, based on clustering analysis of the two 12,000-model ensembles. These models are the most representative structure of the most populated cluster in each case.
Figure 2—figure supplement 3
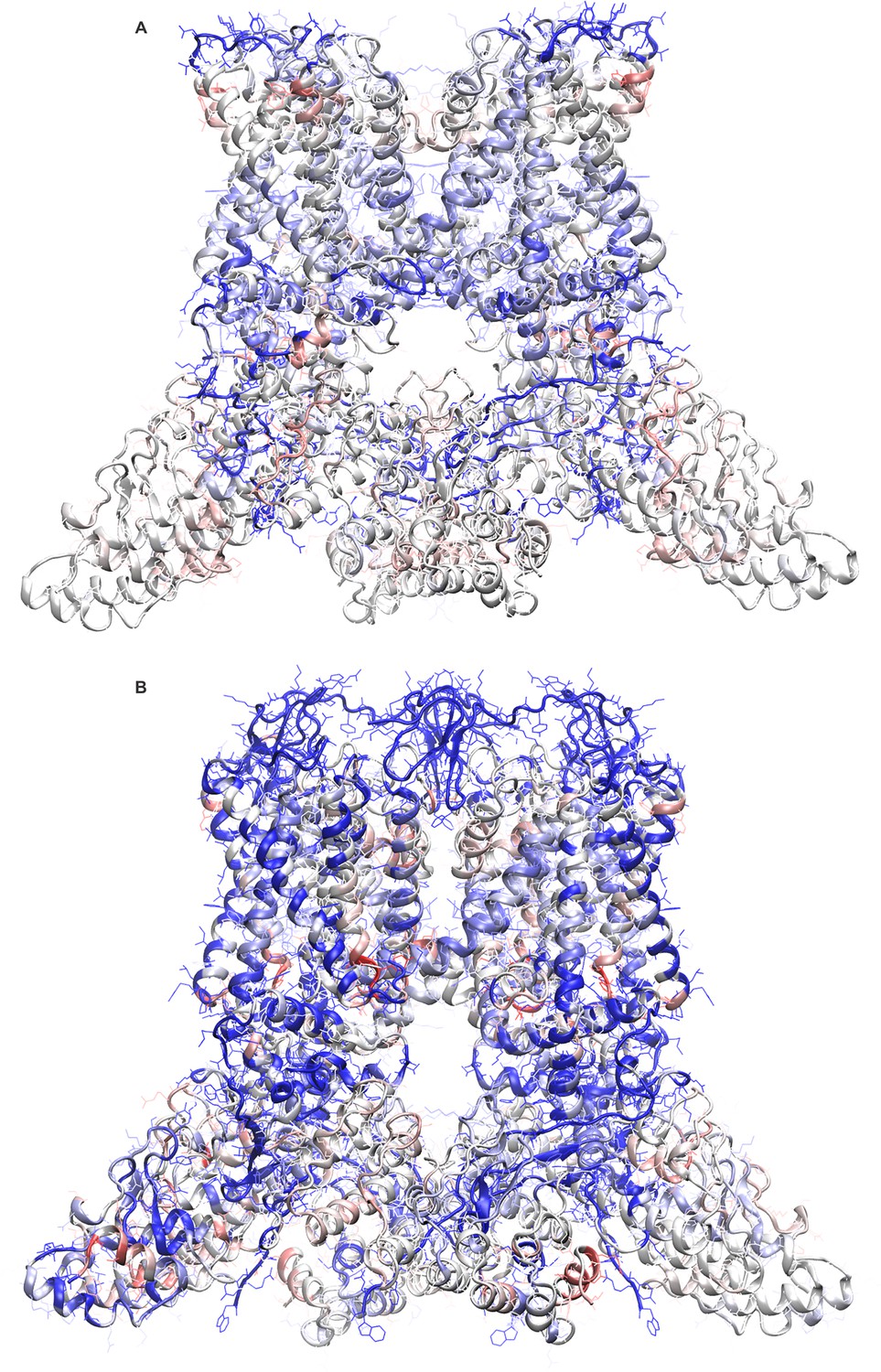
Improvements in the newly refined structural models of apo and DkTx-bound TRPV1.
Final ROSETTA models of apo TRPV1 (A) and of the TRPV1-DkTX complex (B) are shown color-coded according to the change in the calculated ProQM score relative to the initial structures deposited in the PDB (entries 3J5P and 3J5Q, respectively). Blue represents an improved score in the ROSETTA model and/or a region without assigned backbone or side-chain coordinates in the initial structures. Red represents a worse score in the ROSETTA model.
Figure 3
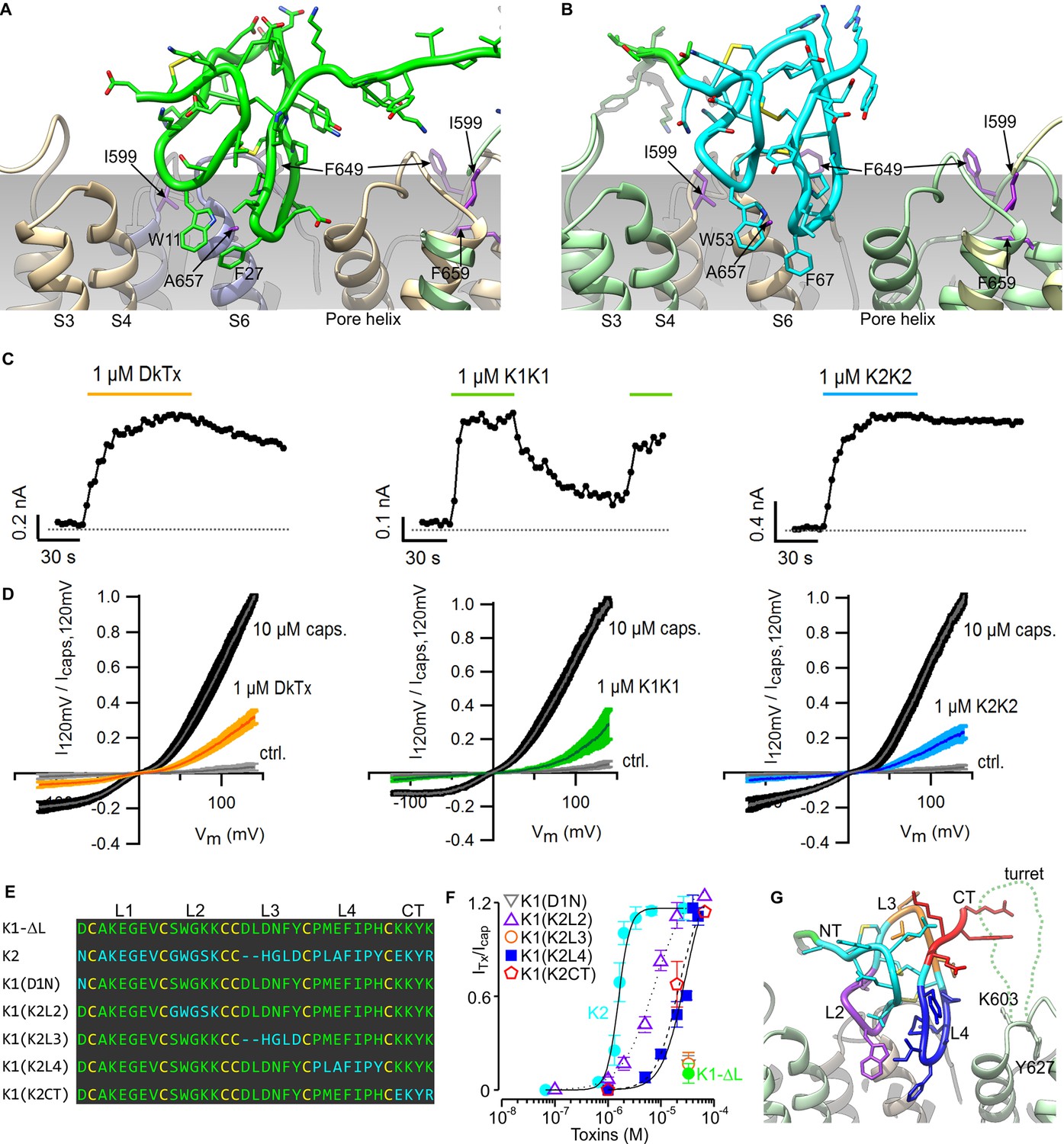
Validation of the proposed structural model of the TRPV1-DkTx complex.
Close-up of K1 (A) and K2 (B) in the top-ranking ROSETTA model of DkTx bound to TRPV1. Side chains of TRPV1 residues where mutation disrupts DkTx-induced channel activation (I599, F649, A657 and F659) (Bohlen et al., 2010) are highlighted in purple. (C) Representative time courses of TRPV1 activation at +120 mV by either DkTx (left panel), K1K1 (middle panel) or K2K2 (right panel) measured from a train of voltage-ramps in the whole-cell configuration with the TRPV1 expressed in HEK cells. In the case of K1K1, toxin was reapplied to verify that the observed decrease in current upon removal of the toxin was due to toxin dissociation from the channel rather than desensitization. Voltage ramps were from −120 to +140 mV over 1 s and were applied every 3 s from a holding potential of −90 mV. The colored horizontal lines denote the application of toxins. The dotted gray line denotes the zero-current level. Fractional dissociation 1 min after toxin removal was quantified across cells as the current amplitude 1 min after toxin removal divided by the steady-state current amplitude in the presence of the toxin. Fractional dissociation values (mean ± sem) were 0.8 ± 0.05 (n = 8) for DkTx, 0.3 ± 0.04 (n = 5) for K1K1 and 0.96 ± 0.03 for K2K2 (n = 6). (D) Mean normalized I-V relations obtained from voltage ramps at steady-state for control solution (gray), toxins (yellow, green or blue) and saturating capsaicin (10 µM; black) in the whole-cell configuration using transiently transfected HEK293 cells. The thin colored curves represent the mean and the thicker envelope the standard error (n = 5–6 for each panel). (E) Primary sequence of K1 without linker (K1-ΔL), K2 and gain-of-function chimeras between K1 and K2. Residues of K1-ΔL and K2 are shown in green and cyan, respectively, and cysteines are shown in yellow. Loops between cysteine residues are labeled as L1, L2, L3 or L4. CT denotes C-terminal region. (F) Concentration-dependence for activation of TRPV1 by K1, K2 and chimeras in two-electrode voltage clamp recordings of oocytes expressing TRPV1. Activation of the full-length TRPV1 channel by the toxin at different concentrations was measured in oocytes at a holding voltage of −60 mV. Toxin-induced currents (ITx) were normalized against the current activated by a saturating concentration of capsaicin (Icap at 10 μM), in the same cell. Note that the data for K1(D1N) and K1(K2L3) are obscured by the data for K1-ΔL. Error bars represent SEM (n = 3). (G) Close-up of the structure of K2 bound to TRPV1 with regions of the toxin colored with the same scheme as in (F). Side chains of K603 and Y627 indicate the region where the pore turret (residues 604–626) would likely reside if not deleted in the construct used for cryo-EM and in our model.
Figure 4 with 2 supplements
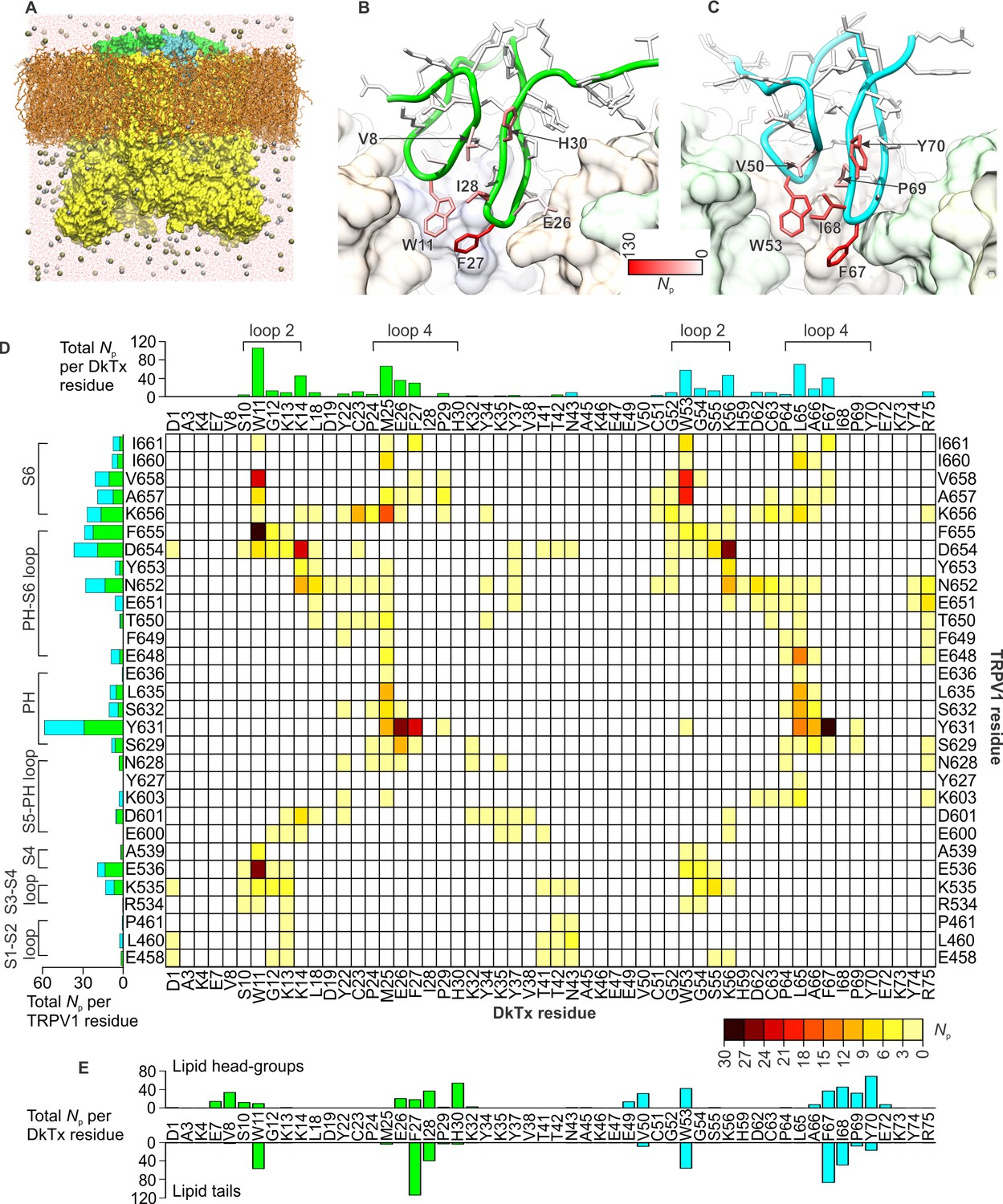
DkTx interactions with TRPV1 and the surrounding lipid bilayer from MD simulations.
(A) Molecular system employed in the simulation of the TRPV1-DkTx complex. The molecular surface of the channel is shown in yellow, while the two bound DkTx molecules are shown in green (K1) and cyan (K2). Electrolyte ions are shown as silver (Na+) and tan (Cl-) spheres, while water molecules are indicated by red dots, and the POPC bilayer in orange. A comparable system was used to simulate the apo channel. (B, C) Residues of K1 (B) and K2 (C) that continue to interact with the membrane after DkTx recognizes TRPV1. The side-chains in the toxin are color-coded according to the number of contacting atom pairs, Np, that were observed for each of these side-chains and any lipid molecule in the membrane (see Methods). The values plotted are averages over the 200 ns accelerated MD simulation of the TRPV1-DkTx complex. (D) Analysis of the side-chain contacts between TRPV1 and DkTx, as observed in the accelerated MD simulation of the complex. The color-coded matrix reports on the contacts between specific side-chain pairs in the toxin and channel. The color-key reflects the number of contacting atom pairs in each case, Np, averaged over the simulated time (see Methods). The bar charts report on the total value of Np for each residue in either the toxin (top) or the channel (left), when any residue in the other protein is considered. The data for K1 and K2 are colored differently (green and cyan, respectively) in the bar graphs. (E) Contacts between DkTx residues and lipid molecules in the surrounding membrane. The bar plot reflects the number of contacting atom pairs between any of the lipid head-groups or tails, and each toxin residue.
Figure 4—figure supplement 1
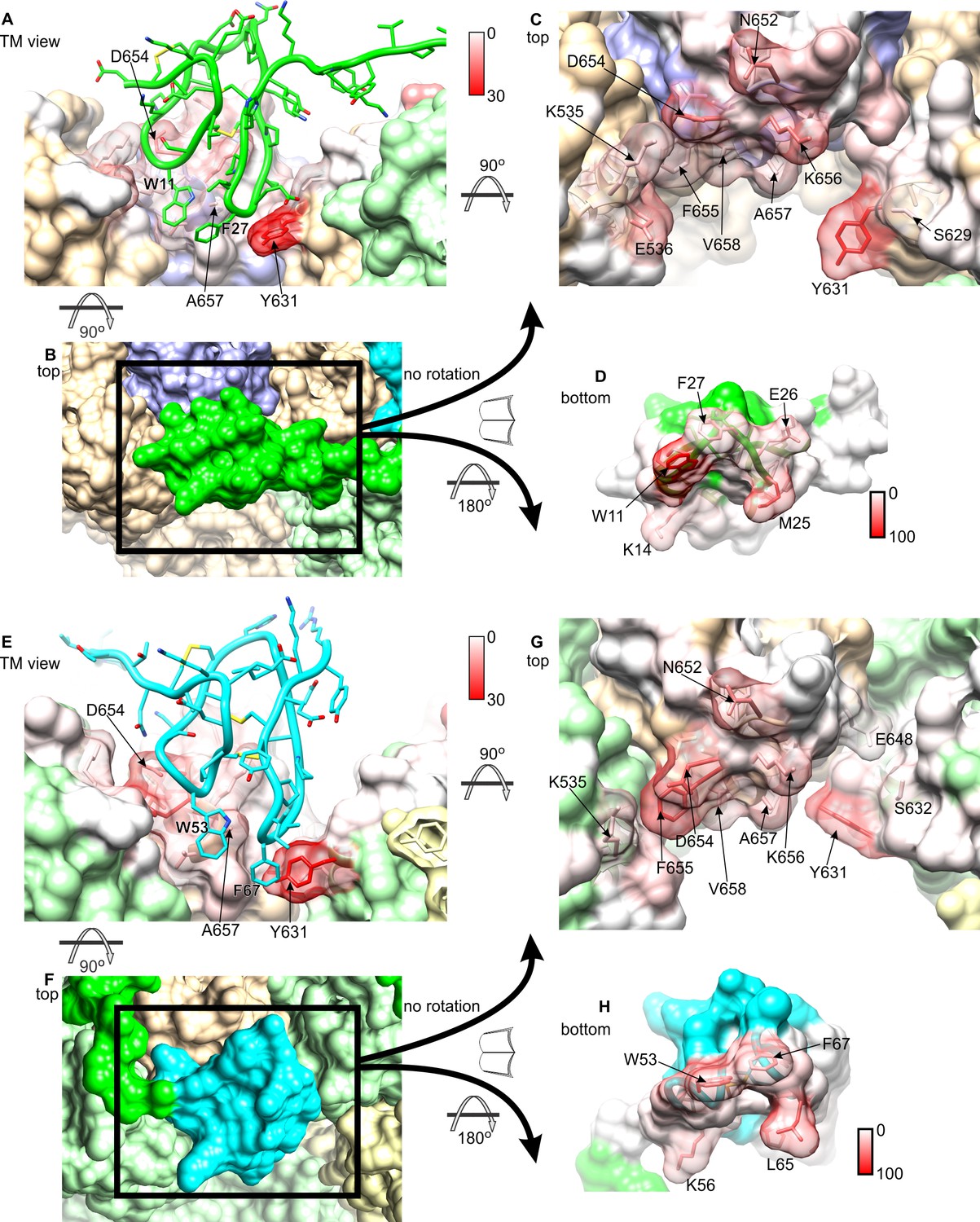
Interactions between DkTx and TRPV1 from MD simulations.
The interaction analysis described in Figure 4 is represented graphically for K1 and TRPV1 (A–D), and K2 and TRPV1 (E–H). See also Video 2. Residues of TRPV1 (A, C, E, G) are color-coded according to the number of contacting atom pairs with one or more toxin residues (color keys are shown between panels A/C and E/G). Conversely, residues of K1 (D) and K2 (H) are color-coded based on the number of contacting atom pairs with one or more channel residues. (A, E) TRPV1-bound K1 (green) and K2 (cyan) are viewed sideways, from the plane of the membrane. (B, F) TRPV1-bound K1 (green) and K2 (cyan) are viewed from the extracellular side. Subunits of TRPV1 are colored in light brown, purple, light yellow and light green. (C, G) TRPV1 residues in contact with K1 (C) or K2 (G), viewed from the extracellular side. Some of the residues in the channel that form persistent interactions with the toxin are shown as transparent surfaces and labeled. (D, E) Surfaces of K1 and K2 that interact with TRPV1 are shown with the color key.
Figure 4—figure supplement 2
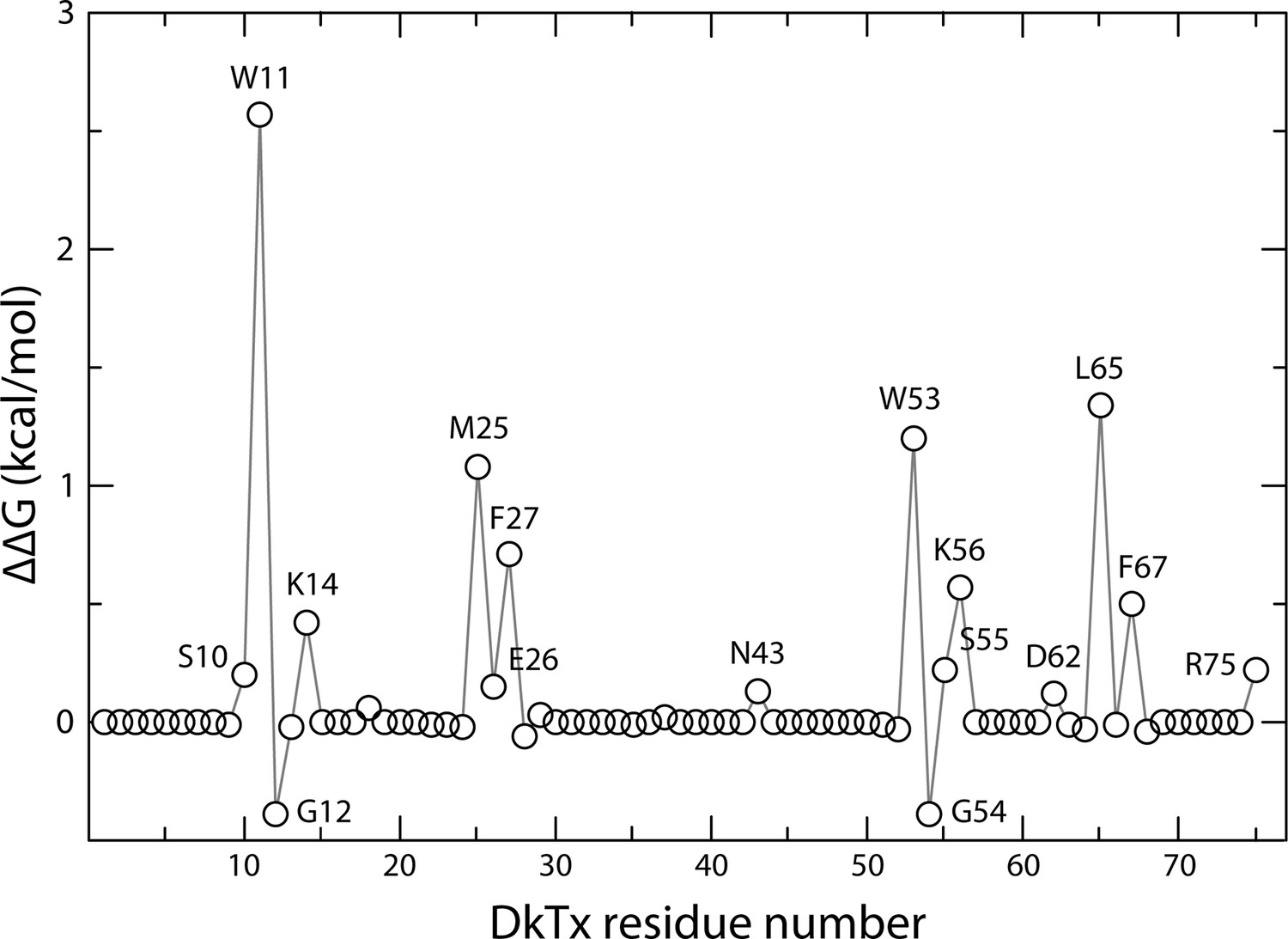
Computational alanine-scan of the DkTx-TRPV1 interface.
The plot reports the estimated change in the association free energy of the toxin-channel complex upon alanine substitution of each of the residues in the toxin, calculated with ROSETTA (see Methods). Positive values indicate a destabilizing effect. The estimates are averages over 200,000 snapshots of the complex extracted from a MD simulation trajectory in which the experimental cryo-EM map is used as a three-dimensional restraint.
Figure 5 with 1 supplement
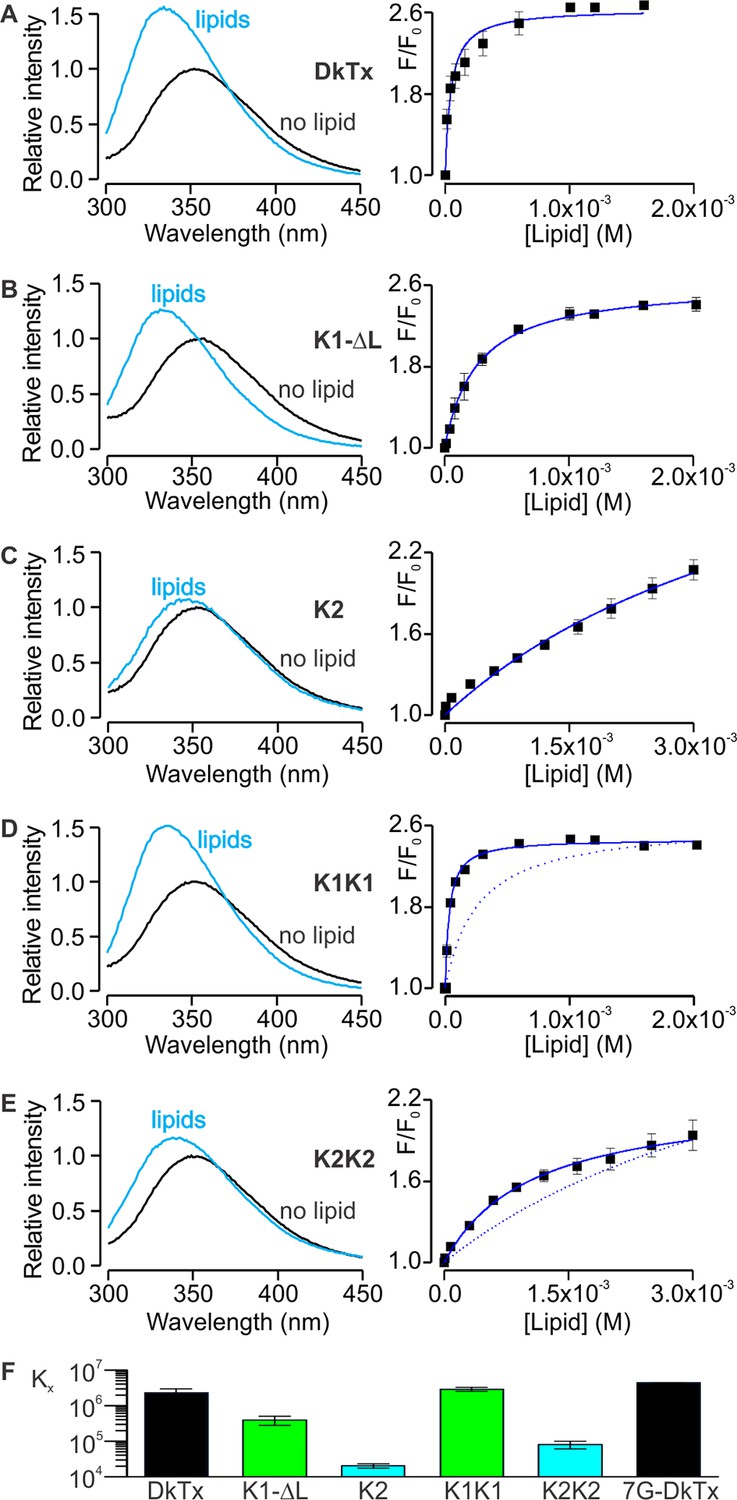
Interaction of DkTx and toxin constructs with lipid membranes measured using Trp fluorescence.
(A–E, left panel) Trp emission spectra of DkTx (A), K1 without linker (B), K2 (C), bivalent K1 (K1K1) (D) and bivalent K2 (K2K2) (E) in the absence (black) and presence (blue, 1.6 mM for DkTx, 2 mM for K1-ΔL and K1K1, and 3 mM for K2 and K2K2) of lipid vesicles (1:1 mix of POPC:POPG). (A–E, right panel) Relative fluorescence intensity at 320 nm (F/F0) as a function of available lipid concentrations (60% of total lipid concentration). Smooth curves correspond to partition functions with Kx = (2.3 ± 0.7) × 106 and F/F0max = 2.65 ± 0.08 for DkTx, Kx = (3.9 ± 1.1) × 105 and F/F0max = 2.63 ± 0.07 for K1-ΔL, Kx = (2.0 ± 0.3) × 104 and F/F0max = 3.71 ± 0.40 for K2, Kx = (2.9 ± 0.3) × 106 and F/F0max = 2.47 ± 0.03 for K1K1 and Kx = (8.1 ± 2) × 105 and F/F0max = 2.77 ± 0.55 for K2K2. Error bars correspond to SEM (n = 3 or 4). Partition functions for K1-ΔL and K2 are shown as dotted lines for comparison in D and E, respectively. (F) Comparisons of mole-fraction partition coefficient (Kx) of toxins. 7G-DkTx denotes a construct of DkTx whose linker (PYVPVTT) is replaced with 7 Gly residues. Error bars correspond to SEM (n = 3 or 4).
Figure 5—figure supplement 1
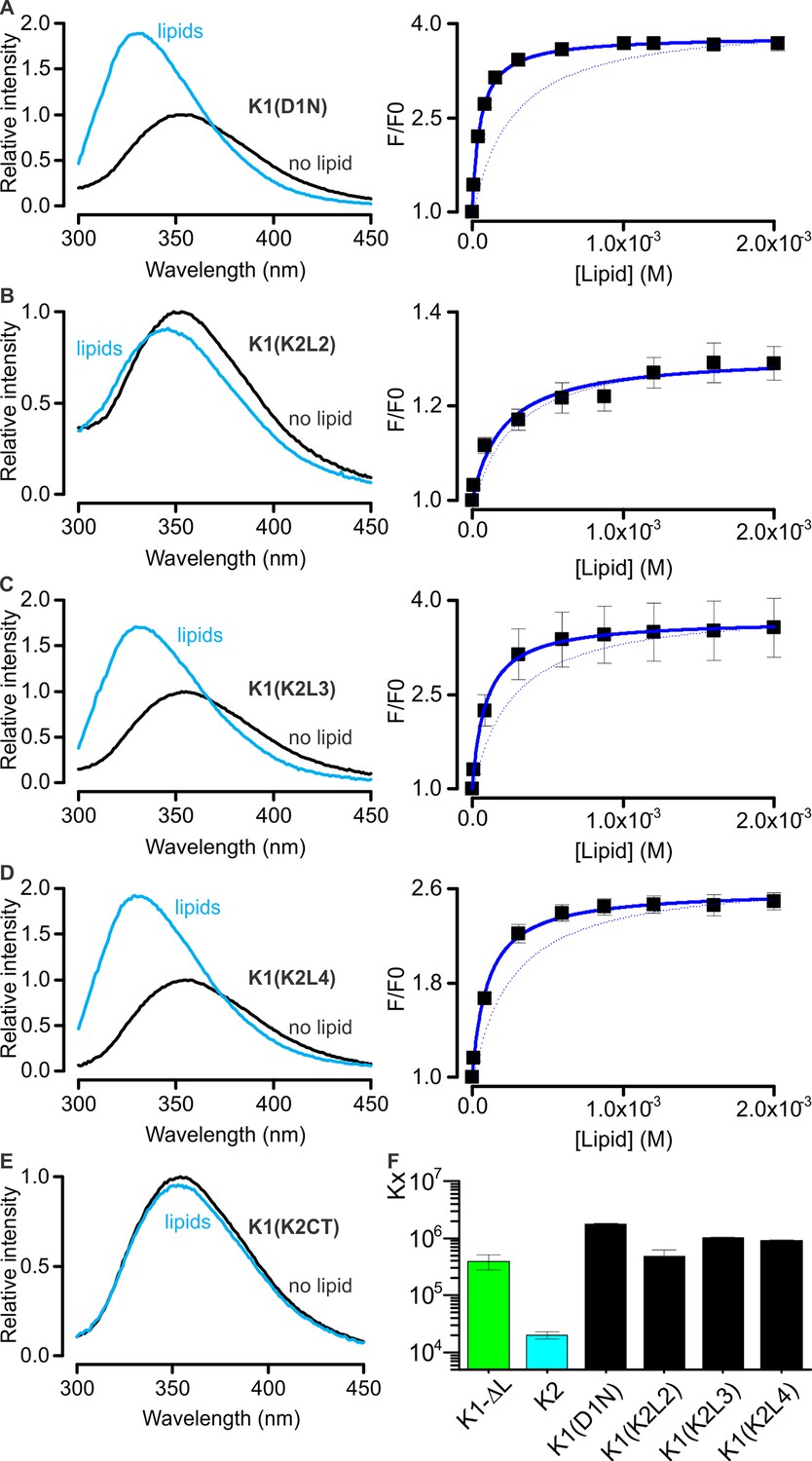
Interaction of K1-K2 chimeras with lipid membranes measured using Trp fluorescence.
(A–E, left panel) Trp emission spectra of K1(D1N) (A), K1(K2L2) (B), K1(K2L3) (C), K1(K2L4) (D) and K1(K2CT) (E) in the absence (black) and presence (blue, 2 mM) of lipid vesicles (1:1 mix of POPC:POPG). (A–D, right panel) Relative fluorescence intensity at 320 nm (F/F0) as a function of available lipid concentrations (60% of total lipid concentration). Smooth curves correspond to partition functions with Kx = (1.76 ± 0.06) × 106 and F/F0max = 3.79 ± 0.02 for K1(D1N), Kx = (4.77 ± 1.39) × 105 and F/F0max = 1.31 ± 0.02 for K1(K2L2), Kx = (1.02 ± 0.04) × 104 and F/F0max = 3.69 ± 0.02 for K1(K2L3), and Kx = (0.90 ± 0.04) × 106 and F/F0max = 2.59 ± 0.02 for K1(K2L4). Error bar corresponds to SEM (n = 3 or 4). Smooth curve of K1-ΔL was shown as a dotted line for comparison throughout the figures. (F) Comparisons of mole-fraction partition coefficient (Kx) of toxins. Error bars correspond to SEM (n = 3 or 4).
Figure 6
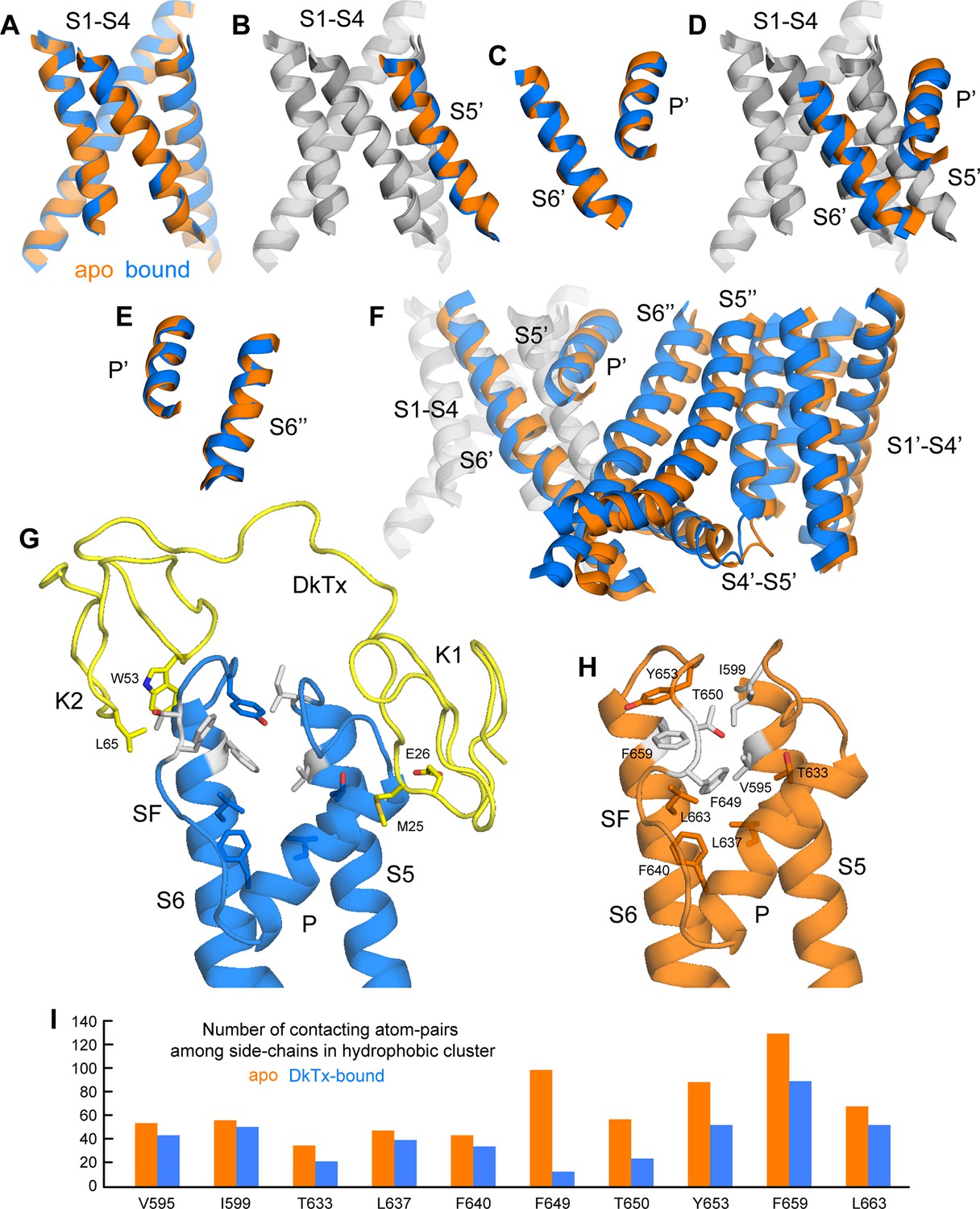
Conformational mechanism of activation of TRPV1 inferred from the cryo-EM structures of apo and DkTx-bound channel.
(A) Overlay of the S1-S4 unit in the apo (orange) and toxin-bound (marine) structure; the root-mean-square difference (RMSD) of the C-α trace is only ~0.7 Å, indicating the internal structure of this unit is not altered upon toxin binding. (B) Overlay of the S1-S4 unit, plus the transmembrane portion of the neighboring S5, which belongs to the adjacent protomer (hence referred to as S5’); the RMSD is again ~0.7 Å. (C) Overlay of the pore helix (P') and the transmembrane portion of S6'; the RMSD is also ~0.7 Å. (D) Overlay of the S1-S4 unit, plus S5', the pore helix P' and S6’. Only the S1-S4 unit and S5' (in gray) are used for fitting, to highlight the relative displacement of S6' and the P' helix, which, as shown in panel (C), move as a largely rigid unit. (E) Overlay of the pore helix P’ and the transmembrane portion of S6 across the protomer interface, i.e. S6"; the RSMD is ~ 0.6 Å. (F) Overlay of two adjacent channel subunits, using the same fit as in panel (D). Owing to the quasi-rigid structure of the P-S6 interface across subunits (see panel E), the displacements induced upon toxin binding on the pore and S6 helices result in a change in the relative orientation of the S1-S4 units, which we propose leads to a change in the crossing-angle of S5 and S6 on the intracellular side, and thus the opening of the lower gate. (G,H) Close-up view of the changes induced upon toxin (yellow) binding on a hydrophobic cluster formed by side-chains from S5, the pore helix, the selectivity filter (SF) and S6. Mutation of the side-chains colored in gray has been shown to diminish toxin-induced TRPV1 activation (see text). (I) Evaluation of the compactness of the hydrophobic cluster shown in (G,H) during MD simulations of apo and toxin-bound TRPV1, in terms of the number of contacting atom pairs of a given side-chain with all others. Fewer contacts imply a less compact arrangement with greater exposure to the solvent, and potentially, an increased heat-capacity of the channel.
Figure 7
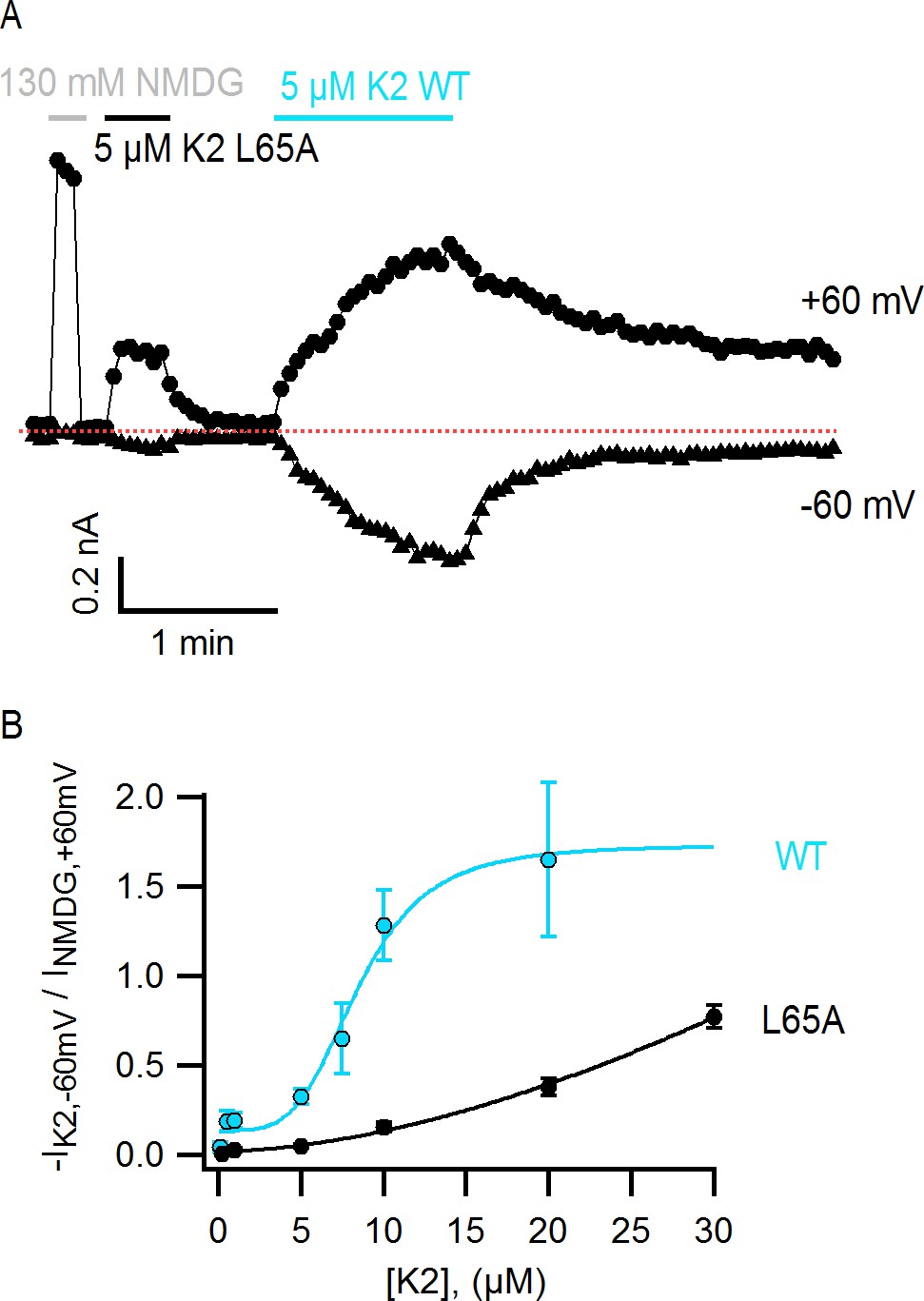
The L65A mutation in K2 perturbs activation of the TRPV1 channel.
(A) Representative TRPV1 current time-course in HEK293 cells depicting activation of the channel by L65A and WT K2 toxins in the whole-cell configuration at +60 and −60 mV. Currents were measured every 3 s by applying voltage-ramps of 1 s duration from −120 to +140 mV starting at a holding potential of −90 mV. Currents were recorded under constant perfusion with control solution or with the test solutions containing either NMDG+ instead of Na+ as permeant ion (gray), or the K2 toxins together with 130 mM NaCl (black and blue), as indicated by the continuous horizontal lines. The dotted red line denotes the zero-current level. Removing external Na+ leads to constitutive activation of TRPV1 to a similar extent as a saturating concentration of DkTx or K2 (Jara-Oseguera, 2016), and therefore provides a convenient means of normalizing response to the toxins. (B) Normalized concentration-response relation for TRPV1 activation at −60 mV by WT K2 toxin (blue) or the L65A mutant (black) obtained in the whole-cell configuration using HEK293 cells. Currents at −60 mV were measured from voltage-ramps as in (A), and normalized to the current value at +60 mV measured before application of the toxin, in the presence of an external solution containing 130 mM NMDGCl instead of NaCl (see (A), gray). Initial currents measured in control solution at +60 and −60 mV were subtracted from the data. A single toxin variant (WT or L65A) was evaluated per experiment to construct dose-response curves, and one or more toxin concentrations were tested per cell. Data are shown as mean ± sem, with n = 3–14 for each data point. The continuous curves are fits to the Hill equation with parameters: WT, KD = 8.5 µM, n = 4.0; L65A, KD > 0.2 mM, n = 1.7.
Figure 8
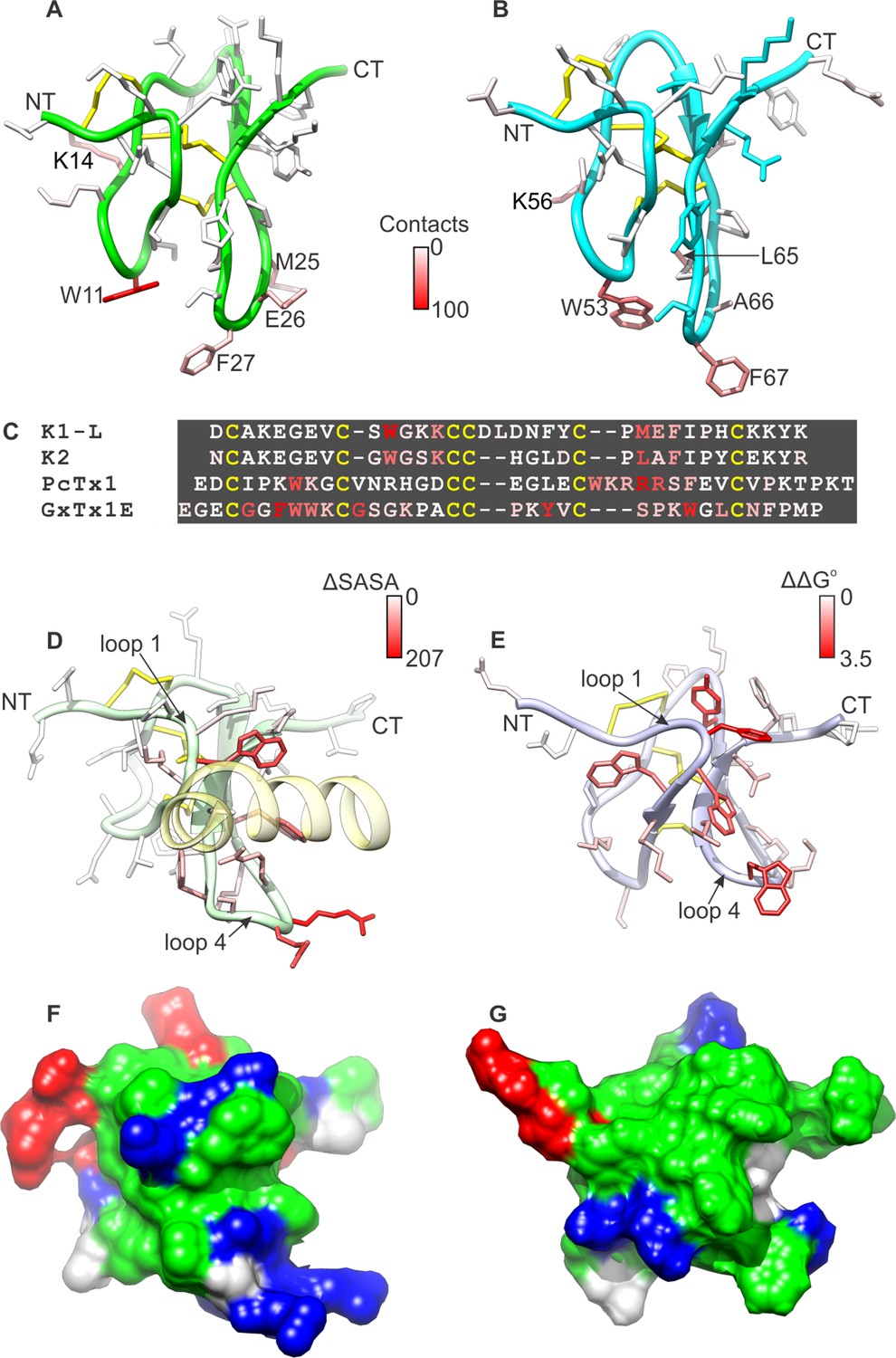
Comparison of active surfaces of different classes of tarantula toxins.
(A,B) Active surfaces of K1 (A) and K2 (B). Side-chains in DkTx are colored based on the probability of finding TRPV1 channel residues within 6 Å in a simulation of the complex (except cysteine residues colored in yellow). The value specified in the scale bar reflects the number residues of the channel with one or more atoms within 6 Å of the toxin residue, on average. Orientation of K1 and K2 are the same as in Figure 1A–D, with loops 2 and 4 pointing down. (C) Sequences of tarantula toxins. Conserved cysteine residues are shown in yellow and residues are colored according to schemes in A, B, D or E as appropriate. (D) Structure of PcTx1 bound to helix-5 of the ASIC1a channel (pastel yellow) oriented the same as K1 and K2 in A and B by superimposing backbones (PDB entry 4FZ0). Backbone of the toxin is colored in pastel green, and side-chains are colored by changes in solvent-accessible surface area between the structure of PcTx1 when the toxin is bound to ASIC1a and the structure of toxin without channel (Gupta et al., 2015) (ΔSASA in Å2). (E) Solution structure of GxTx1E oriented the same as K1 and K2 in A and B by superimposing backbones (PDB entry 2WH9). Side-chains are colored according to perturbation in free energy of binding (ΔΔG° in kcal mol−1) (Gupta et al., 2015). (F) Surface representation of the PcTx1 structure where hydrophobic residues are colored green, basic residues blue and acidic residues red. Orientation is the same as in D. (G) Surface representation of the GxTx-1E structure where hydrophobic residues are colored green, basic residues blue and acidic residues red. Orientation is the same as in E.
Figure 9

Proposed mechanism of DkTx partitioning into membranes and binding to TRPV1.
Partitioning of DkTx into the lipid membrane (gray) before recognition of TRPV1 is preferentially driven by the K1 lobe (green), whereas K2 (cyan) has a higher affinity for binding to the channel. After binding to TRPV1, both K1 and K2 retain a significant number of interactions with the lipid membrane, presumably so as to stabilize the complex, which features a relatively small protein-protein interface.
Videos
Video 1
The top-ranking ROSETTA model of the TRPV1-DkTx complex, with DkTx in a CCW configuration.
The K1 knot and linker is colored green and the K2 knot is colored blue.
Video 2
Close up of the interface between TRPV1 and DkTx, color-coded according to the persistence of protein-protein contacts from MD simulations.
See also Figure 4—Supplement 1.
Video 3
Structural changes in TRPV1 resulting from DkTx binding, and close-up view of a cluster of buried hydrophobic residues that become exposed upon channel opening.
https://doi.org/10.7554/eLife.11273.023Tables
Table 1
Evaluation of structural models of apo TRPV1.
| PDB entry 3J5P | ||||
| Number of atoms: 18,628 | ||||
| Correlation coefficient with cryo-EM map EMD-5778 (3.4 Å): 0.71 | ||||
| All-atom contacts | Clashscore, all atomsa | 76.31 | 16th percentileb | |
| Protein geometry | Poor rotamers | 524 | 26.57% | Ideal: <1% |
| Ramachandran outliers | 0 | 0.00% | Ideal: <0.05% | |
| Ramachandran favored | 2204 | 94.35% | Ideal: >98% | |
| MolProbity scorec | 3.82 | 36th percentileb | ||
| Cβ deviations >0.25 Å | 0 | 0.00% | Ideal: 0 | |
| Bad backbone bonds | 166/19032 | 0.87% | Ideal: 0% | |
| Bad backbone angles | 72/25760 | 0.28% | Ideal: < 0.1% | |
| Partial ROSETTA modeld | ||||
| Number of atoms: 18,628 | ||||
| Correlation coefficient with cryo-EM map EMD-5778 (3.4 Å): 0.71 | ||||
| All-atom contacts | Clashscore, all atomsa | 3.45 | 97th percentileb | |
| Protein geometry | Poor rotamers | 4 | 0.20% | Ideal: <1% |
| Ramachandran outliers | 12 | 0.51% | Ideal: <0.05% | |
| Ramachandran favored | 2200 | 94.18% | Ideal: >98% | |
| MolProbity scorec | 1.53 | 94th percentileb | ||
| Cβ deviations >0.25 Å | 0 | 0.00% | Ideal: 0 | |
| Bad backbone bonds | 44/19024 | 0.23% | Ideal: 0% | |
| Bad backbone angles | 56/25744 | 0.22% | Ideal: < 0.1% | |
| Complete ROSETTA model | ||||
| Number of atoms: 19,632 | ||||
| Correlation coefficient with cryo-EM map EMD-5778 (3.4 Å): 0.71 | ||||
| All-atom contacts | Clashscore, all atomsa | 3.77 | 96th percentileb | |
| Protein geometry | Poor rotamers | 8 | 0.37% | Ideal: <1% |
| Ramachandran outliers | 32 | 1.33% | Ideal: <0.05% | |
| Ramachandran favored | 2236 | 92.70% | Ideal: >98% | |
| MolProbity scorec | 1.62 | 92nd percentileb | ||
| Cβ deviations >0.25 Å | 0 | 0.00% | Ideal: 0 | |
| Bad backbone bonds | 52/20068 | 0.26% | Ideal: 0% | |
| Bad backbone angles | 148/27144 | 0.55% | Ideal: < 0.1% | |
-
aClashscore is the number of serious steric overlaps (>0.4 Å) per 1,000 atoms.
-
b100th percentile is the best score among structures of comparable resolution; 0th percentile is the worst score. For clashscore, the comparative set of structures was selected in 2004, for MolProbity score in 2006.
-
cThe MolProbity score combines the clashscore, rotamer and Ramachandran evaluations into a single score, normalized to be in the same scale as X-ray resolution.
-
dThe partial ROSETTA model is identical to the complete ROSETTA model, except that it includes only the set of atoms resolved in PDB entry 3J5P.
Table 2
Evaluation of structural models of the TRPV1-DkTx complex.
| PDB entry 3J5Q | ||||
| Number of atoms: 18,244 | ||||
| Correlation coefficient with cryo-EM map EMD-5776 (fourfold symmetric, 3.8 Å): 0.78 | ||||
| All-atom contacts | Clashscore, all atomsa | 101.85 | 10th percentileb | |
| Protein geometry | Poor rotamers | 457 | 27.33% | Ideal: <1% |
| Ramachandran outliers | 20 | 0.82% | Ideal: <0.05% | |
| Ramachandran favored | 2264 | 92.33% | Ideal: >98% | |
| MolProbity scorec | 4.04 | 29th percentileb | ||
| Cβ deviations >0.25 Å | 0 | 0.00% | Ideal: 0 | |
| Bad backbone bonds | 106/18000 | 0.59% | Ideal: 0% | |
| Bad backbone angles | 50/24444 | 0.20% | Ideal: < 0.1% | |
| Partial ROSETTA modeld | ||||
| Number of atoms: 18,244 | ||||
| Correlation coefficient with cryo-EM map relion_ct16_halfSUM_mr (twofold symmetric, 4.3 Å): 0.77 | ||||
| All-atom contacts | Clashscore, all atomsa | 4.06 | 96th percentileb | |
| Protein geometry | Poor rotamers | 6 | 0.35% | Ideal: <1% |
| Ramachandran outliers | 36 | 1.48% | Ideal: <0.05% | |
| Ramachandran favored | 2234 | 91.56% | Ideal: >98% | |
| MolProbity scorec | 1.69 | 89th percentileb | ||
| Cβ deviations >0.25 Å | 4 | 0.17% | Ideal: 0 | |
| Bad backbone bonds | 43/18610 | 0.23% | Ideal: 0% | |
| Bad backbone angles | 100/25258 | 0.40% | Ideal: < 0.1% | |
| Complete ROSETTA model | ||||
| Number of atoms: 20,814 | ||||
| Correlation coefficient with cryo-EM map relion_ct16_halfSUM_mr (twofold symmetric, 4.3 Å): 0.76 | ||||
| All-atom contacts | Clashscore, all atomsa | 6.39 | 89th percentileb | |
| Protein geometry | Poor rotamers | 12 | 0.53% | Ideal: <1% |
| Ramachandran outliers | 72 | 2.81% | Ideal: <0.05% | |
| Ramachandran favored | 2284 | 89.29% | Ideal: >98% | |
| MolProbity scorec | 1.92 | 80th percentileb | ||
| Cβ deviations >0.25 Å | 10 | 0.41% | Ideal: 0 | |
| Bad backbone bonds | 150/21288 | 0.70% | Ideal: 0% | |
| Bad backbone angles | 261/28792 | 0.91% | Ideal: < 0.1% | |
-
aClashscore is the number of serious steric overlaps (>0.4 Å) per 1,000 atoms.
-
b100th percentile is the best score among structures of comparable resolution; 0th percentile is the worst score. For clashscore, the comparative set of structures was selected in 2004, for MolProbity score in 2006.
-
cThe MolProbity score combines the clashscore, rotamer and Ramachandran evaluations into a single score, normalized to be in the same scale as X-ray resolution.
-
dThe partial ROSETTA model is identical to the complete ROSETTA model, except that it includes only the set of atoms resolved in PDB entry 3J5Q.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin
eLife 5:e11273.
https://doi.org/10.7554/eLife.11273




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}