Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice
- State University of New York Downstate Medical Center, United States
- New York University, United States
- James A Haley Veterans Hospital, University of South Florida, United States
- University of Texas Medical School at Houston, United States
Figures
Figure 1 with 2 supplements

ZIP reverses LTP maintenance in both wild-type mice and PKMζ-null mice and blocks synaptic potentiation mediated by PKMζ and PKCι/λ.
Bath applications of ZIP (5 µM) reverse (A) wild-type-LTP maintenance and (B) PKMζ-null-LTP maintenance (filled circles). Above insets, numbered representative fEPSP traces correspond to time points noted below. Below, mean ± SEM. For (A), wild-type, n = 5, average response 5 min before ZIP compared to 3.5 hr post-ZIP, t4 = 4.83, p = 0.0084, d = 1.85; for (B), PKMζ-null, n = 4, t3 = 3.34, p = 0.045, d = 2.88. Non-tetanized pathways are stable in the presence of ZIP (open circles). For (A), wild-type non-tetanized pathway: 5 min pre-ZIP vs. 3.5 hr post-ZIP; n = 5, t4 = 1.73, p = 0.16; d = 0.49; for (B), PKMζ-null non-tetanized pathway: n = 4, t3 = 0.82, p = 0.47, d = 0.058. (C) ZIP inhibits both PKMζ and, at higher doses, the autonomous activity of PKCι/λ. The main effects and interactions are all significant (kinase: F1,30 = 85.4, p = 2.77 X 10–10, η2 = 0.036; ZIP concentration: F4,30 = 200.56, p = 3.48 X 10–21, η2 = 0.34; interaction: F4,30 = 26.59, p = 1.98 X 10–9, η2 = 0.045). Post-hoc tests show the kinases respond differently at 1 µM and 2 µM ZIP. (D, E), ZIP blocks EPSC potentiation produced by postsynaptic dialysis of PKMζ or PKCι/λ in CA1 pyramidal cells in hippocampal slices. ZIP (5 µM) is applied to the bath prior to obtaining whole-cell patch. Above insets, numbered representative EPSC traces correspond to time points noted below. Statistical comparisons are at 15 min after whole-cell patch. (D) PKMζ: n’s = 4, F2,11 = 18.07, p = 0.0007, d = 1.78; post-hoc tests: PKMζ vs. baseline, p = 0.0012; PKMζ vs. PKMζ + ZIP, p = 0.0018; PKMζ + ZIP vs. baseline, p = 0.94. (E) PKCι/λ: n’s = 4, F2,11 = 35.2, p = 1.66 X 10–5, d = 1.79; post-hoc tests: PKCι/λ vs. baseline, p<0.0001; PKCι/λ vs. PKCι/λ + ZIP, p<0.0001; PKCι/λ + ZIP vs. baseline, p = 0.86.
Figure 1—figure supplement 1
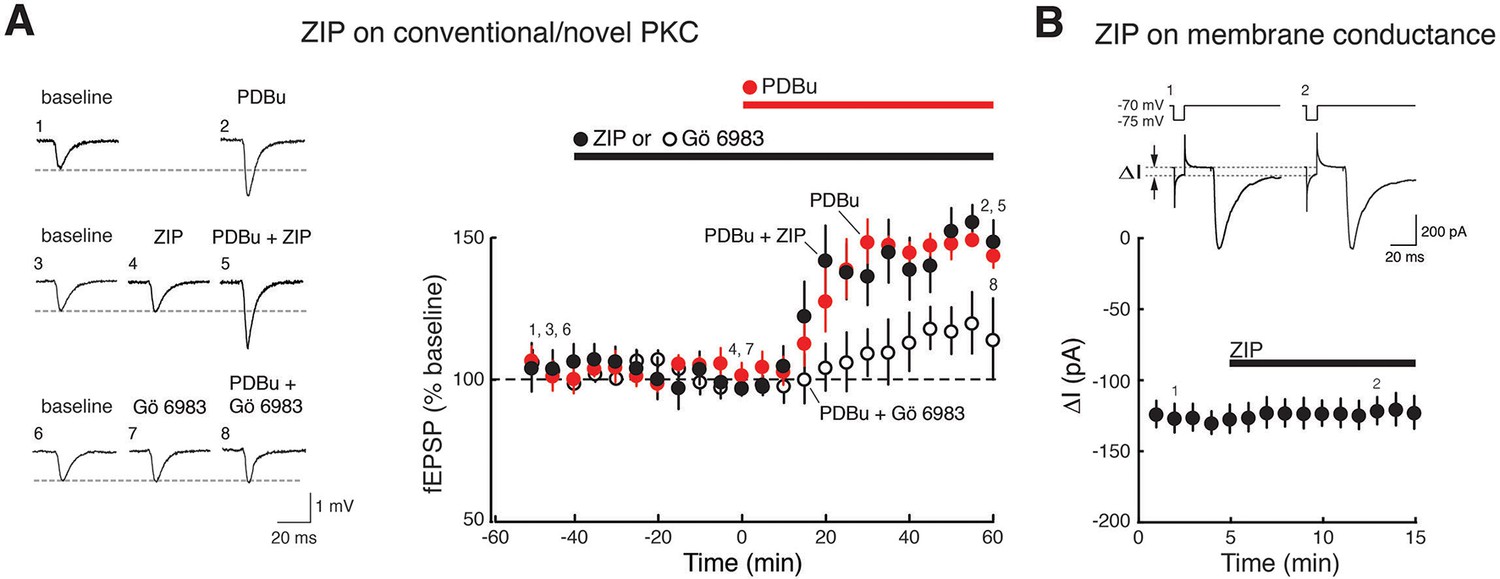
ZIP has no effect on synaptic potentiation induced by activation of conventional/novel PKCs and produces no perturbation of neuronal membrane conductance.
(A) ZIP has no effect on fEPSP potentiation produced by phorbol esters (phorbol 12,13-dibutyrate, PDBu, 1 µM), activators of conventional/novel PKCs. Synaptic potentiation induced by phorbol esters is blocked by the conventional/novel PKC inhibitor Gö 6983 (100 nmol), but not by ZIP (5 µM) (phorbol, n = 5, phorbol + ZIP, n = 5, phorbol + Gö 6983, n = 6, F2,13 = 10.9, p = 0.0017, d = 1.84; phorbol vs. phorbol + ZIP, p = 0.85; phorbol vs. phorbol + Gö 6983, p = 0.0073; phorbol + ZIP vs. phorbol + Gö 6983, p = 0.026). (B) ZIP (5 µM) produces no perturbation of membrane conductance of CA1 pyramidal cells, as expected for recordings of stable baseline fEPSP responses (panel A, Figure 1A,B), as well as previous studies demonstrating membrane stability at this concentration of ZIP that reverses LTP (Wang et al., 2012). Above, representative measurements of membrane conductance (between arrows) in response to current pulse, followed by evoked EPSC. Below, mean ± SEM (n = 4, t3 = 0.33, p = 0.76, d = 0.10). We note that Volk et al. had reported ZIP induces decreases in both tetanized and untetanized pathways in slices (Volk et al., 2013). These results conflict with evidence from a large number of studies showing exclusive actions of ZIP in tetanized or facilitated pathways and not in baseline pathways, including seven studies in brain slices of fEPSPs (Ling et al., 2002; Serrano et al., 2005; Sajikumar et al., 2005; Navakkode et al., 2010; Lin et al., 2012; Panaccione et al., 2013; Chen et al., 2015), four studies of EPSCs (Li et al., 2010; Yao et al., 2013; Velez-Hernandez et al., 2013; Li et al., 2014), two studies in model systems (Cai et al., 2011; Balaban et al., 2015), and four studies in vivo of fEPSPs (Pastalkova et al., 2006; Madronal et al., 2010; Dong et al., 2015) and evoked responses (Cooke and Bear, 2010), as well as evidence of stable basal properties of hippocampal neurons following ZIP injections recorded in vivo (Barry et al., 2012).
Figure 1—figure supplement 2
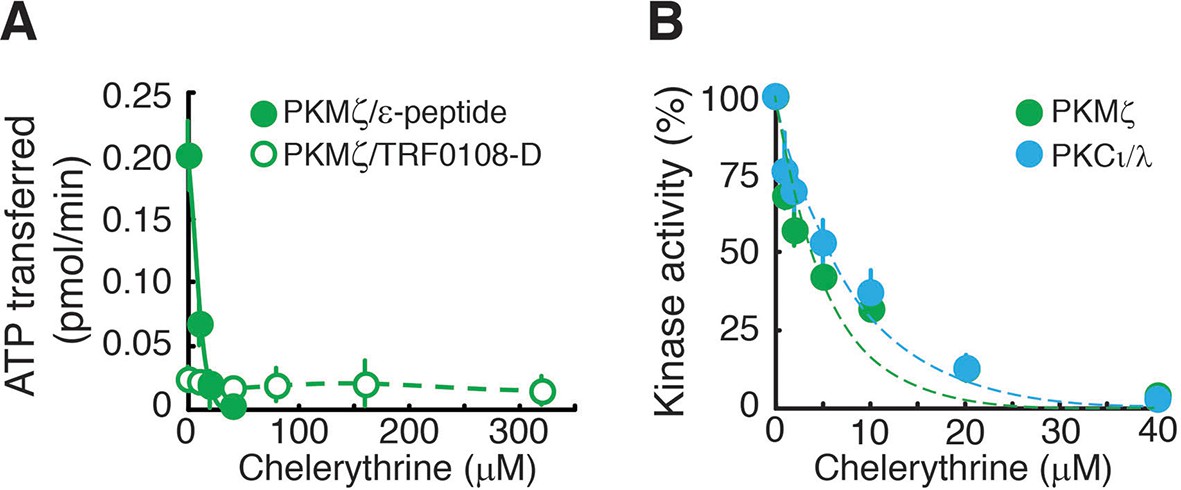
Chelerythrine inhibits both PKMζ and PKCι/λ.
(A) Chelerythrine inhibits PKMζ phosphorylation of the standard atypical PKC substrate, ε-peptide (25 µM, filled circles). In contrast, Lee et al., who had reported no effect of chelerythrine on PKMζ activity (Lee et al., 2013), had performed kinase assays using a substrate based upon the pseudosubstrate sequence of the conventional isoform PKCα (TRF0108-D, PerkinElmer), which we find is a relatively poor substrate for PKMζ. We confirm that the inhibition of PKMζ by chelerythrine cannot be detected using this substrate at the concentration employed in Lee et al. (50 nM, open circles). We note that chelerythrine applied to the bath of hippocampal slices suppresses the synaptic potentiation produced by postsynaptic perfusion of PKMζ in CA1 pyramidal cells, indicating the drug inhibits PKMζ’s phosphorylation of physiologically relevant substrates in neurons (Ling et al., 2002; 2006). (B) Under the appropriate assay conditions for observing inhibition using the standard ε-peptide substrate as described in (A), chelerythrine inhibits both PKMζ and PKCι/λ.
Figure 2 with 2 supplements
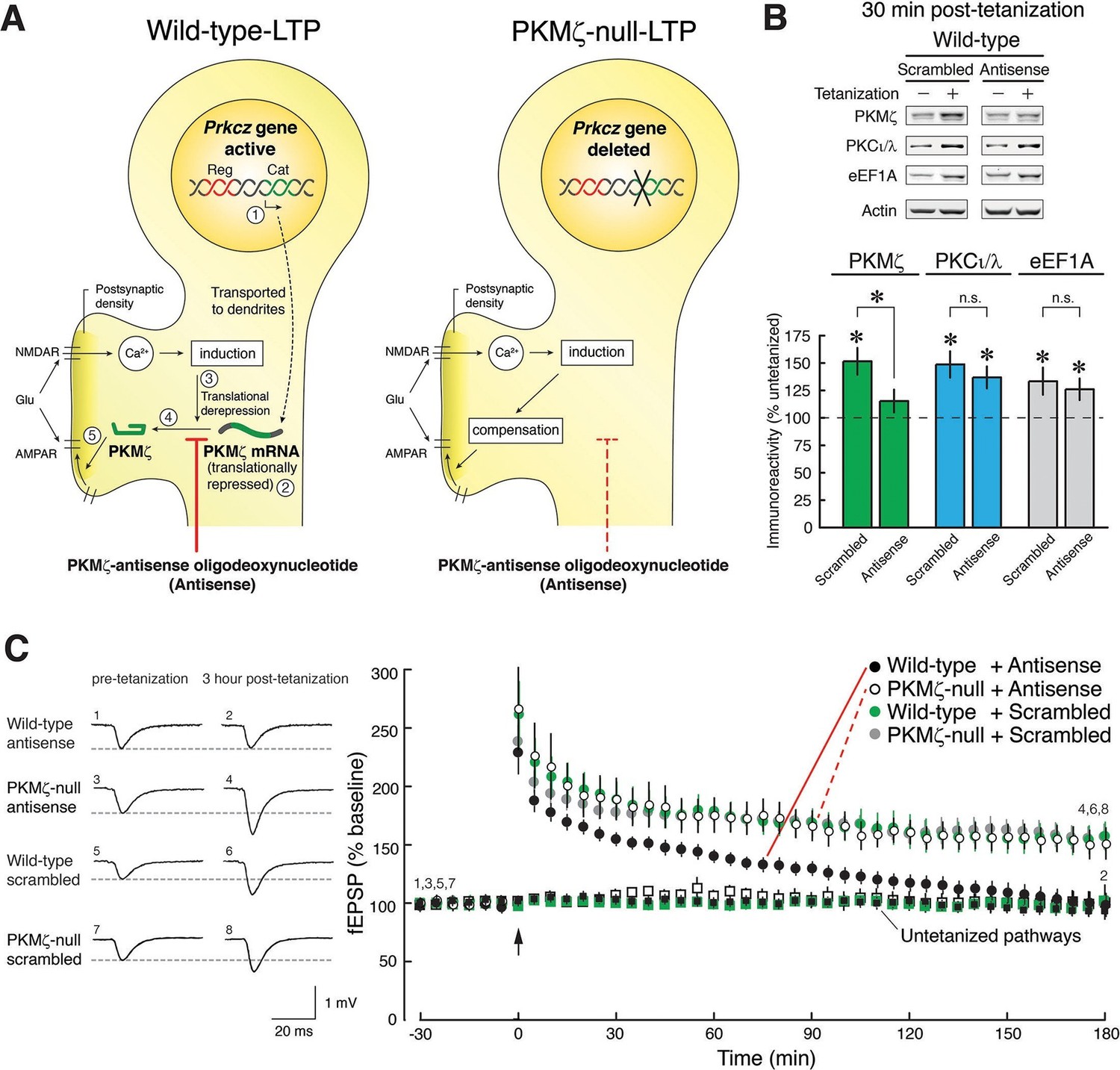
PKMζ is essential for late-LTP in wild-type mice, and compensation accounts for late-LTP in PKMζ-null mice.
(A) Diagram illustrating the PKMζ-compensation hypothesis tested by pharmacogenetic analysis of LTP. The Prkcz gene consists of an autoinhibitory PKCζ regulatory domain exon region (Reg, shown in red) and a catalytic domain exon region (Cat, green). In neurons in wild-type mice, PKMζ is produced by an internal promoter within the Prkcz gene, transcribing a PKMζ mRNA that expresses an independent ζ catalytic domain (indicated as step [1] [Hernandez et al., 2003]). The PKMζ mRNA is transported to dendrites (Muslimov et al., 2004) but is translationally repressed ([2] [Hernandez et al., 2003]). During tetanic stimulation, glutamate (Glu) activates the NMDAR to stimulate Ca2+-dependent induction mechanisms that release the translational block ([3] [Hernandez et al., 2003]), resulting in synthesis of PKMζ ([4] [Hernandez et al., 2003]), which potentiates postsynaptic AMPARs ([5] [Ling et al., 2002; Serrano et al., 2005]). If wild-type mice express persistently enhanced AMPAR-mediated synaptic transmission through synthesis of PKMζ and PKMζ-null mice through compensatory mechanisms, then PKMζ-antisense will block LTP in wild-type mice (left) but have no effect in PKMζ-null mice (right). (B) The PKMζ-antisense (20 µM) blocks the new synthesis of PKMζ, but not PKCι/λ or the eukaryotic elongation factor 1A (eEF1A) that are also rapidly synthesized in LTP. In the presence of antisense or scrambled oligodeoxynucleotides, adjacent slices from the same hippocampus are either tetanized or untetanized, and 30-min post-tetanization CA1 regions are assayed by immunoblot. The levels of protein in the untetanized slices are set at 100%. Mean ± SEM; *denotes significance; n.s., no significance. PKMζ: scrambled, tetanized (n = 17) vs. untetanized (n = 19), t34 = 3.81, p = 0.00056, d = 1.27; antisense, tetanized (n = 12) vs. untetanized (n = 18), t28 = 1.35, p = 0.19, d = 0.50; antisense vs. scrambled, t27 = 2.12, p = 0.043, d = 0.80; PKCι/λ: scrambled, tetanized (n = 17) vs. untetanized (n = 18), t33 = 3.72, p = 0.00074, d = 1.26; antisense, tetanized (n = 12) vs. untetanized (n = 17), t27 = 3.59, p = 0.0013, d = 1.35; antisense vs. scrambled, t27 = 0.71, p = 0.49, d = 0.27; eEF1A: scrambled, tetanized (n = 9) vs. untetanized (n = 10), t17 = 2.40, p = 0.028, d = 1.10; antisense, tetanized (n = 12) vs. untetanized (n = 18), t28 = 2.07, p = 0.048, d = 0.77; antisense vs. scrambled, t19 = 0.47, p = 0.64, d = 0.21. (C) PKMζ-antisense blocks late-LTP in wild-type mice but has no effect on LTP in PKMζ-null mice. Left, representative fEPSPs; numbers correspond to time points at right. Right, mean ± SEM. Scrambled version of the oligodeoxynucleotide has no effect on either genotype. Circles show tetanized pathways, and color-coded squares show control untetanized pathways within each slice that receive only test stimulation. Tetanization is at arrow. N’s are: wild-type + antisense: 7; PKMζ-null + antisense: 5; wild-type + scrambled: 8; PKMζ-null + scrambled: 6. The genotype X drug X time ANOVA with repeated measures on time (average of the 5 min ending at 30 min post-tetanization and 180 min post-tetanization) confirmed a significant genotype X drug interaction, F1,22 = 4.7; p = 0.041, η2 = 0.0026. Post-hoc tests confirmed antisense on wild-type at 180 min post-tetanization is significantly less than all other responses.
Figure 2—figure supplement 1

The mutant PKMζ gene expresses neither PKMζ mRNA nor protein.
(A) Strategy for the excision of the exon encoding the PKC/PKMζ catalytic domain ATP-binding site, as previously described (Lee et al., 2013). DNA sequencing of the PKMζ locus in PKMζ-null mice shows upstream and downstream intronic sequences indicating the exon deletion. (B) Agarose gel shows the lowered molecular weight of the DNA PCR amplification product, following excision in PKMζ-null mouse, as compared to wild-type mouse. Primer locations are shown in panel (A). (C) RT-PCR shows no amplification of PKMζ mRNA in the hippocampus of the PKMζ-null. (D) Immunoblot shows no expression of PKMζ protein in the hippocampus of the PKMζ-null. The full-length PKCζ is not expressed in wild-type hippocampus because its promoter is inactive (Hernandez et al., 2003).
Figure 2—figure supplement 2
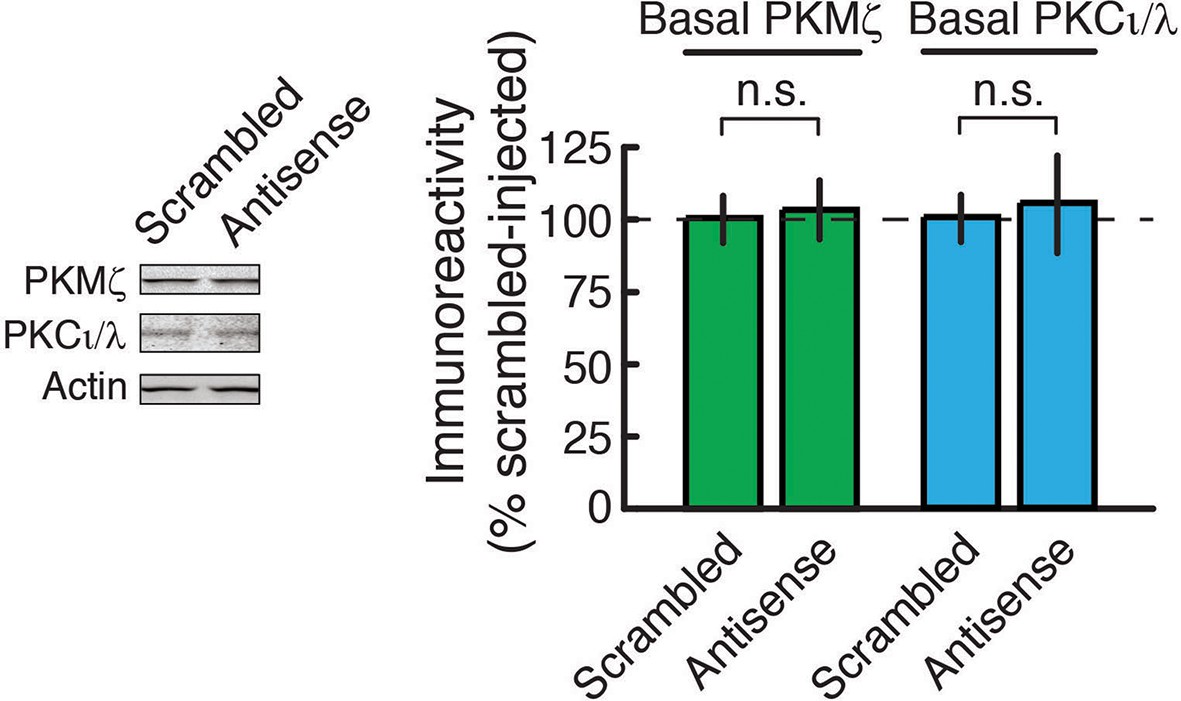
PKMζ-antisense bath-applied to hippocampal slices for 2 hr does not affect the amount of basal PKMζ or PKCι/λ.
Left, representative immunoblots; right, mean ± SEM (PKMζ: n’s = 8; t14 = 0.16, p = 0.88, d = 0.08; PKCι/λ: n’s = 8; t14 = 0.24, p = 0.82, d = 0.12; n.s., no significance).
Figure 3 with 2 supplements
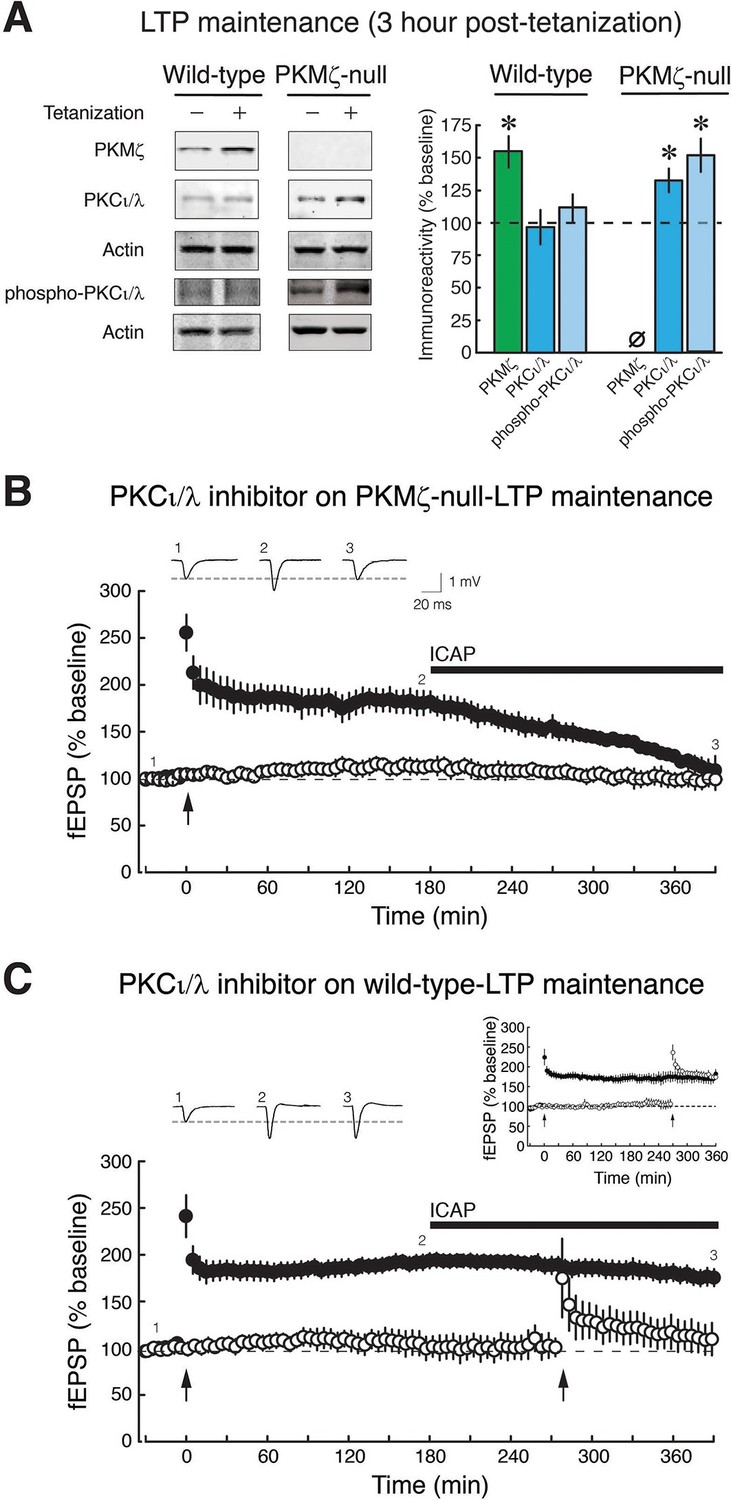
PKCι/λ inhibitor reverses the maintenance of late-LTP in PKMζ-null mice, but not in wild-type mice.
(A) Immunoblots show that in wild-type-LTP maintenance, PKMζ persistently increases for 3 hr, whereas PKCι/λ, as determined by both total and activation loop phosphorylated-PKCι/λ antisera, are at baseline. In PKMζ-null-LTP maintenance, both total and activation loop phosphorylated-PKCι/λ persistently increase for 3 hr. Wild-type: PKMζ, untetanized vs. tetanized (n’s = 9), t16 = 4.51, p = 0.00036, d = 2.13; PKCι/λ, untetanized (n = 6) vs. tetanized (n = 7), t11 = 0.19, p = 0.85, d = 0.11; phospho-PKCι/λ, untetanized vs. tetanized (n’s = 9), t16 = 0.86, p = 0.40, d = 0.41. PKMζ-null: PKCι/λ, untetanized vs. tetanized (n’s = 7), t12 = 2.41, p = 0.033, d = 1.29; phospho-PKCι/λ, untetanized (n = 8) vs. tetanized (n = 9), t15 = 4.35, p = 0.00058, d = 2.11. PKMζ-null: tetanized total PKCι/λ vs. phospho-PKCι/λ, t14 = 1.83, p = 0.09, d = 0.92. Tetanized wild-type vs. PKMζ-null PKCι/λ, t12 = 2.28, p = 0.042, d = 1.22; tetanized wild-type vs. PKMζ-null phospho-PKCι/λ, t16 = 3.27, p = 0.0048, d = 1.54. (B) PKMζ-null late-LTP maintenance (filled circles) is reversed by PKCι/λ-antagonist ICAP (10 µM) applied 3 hr post-tetanization. Insert above, representative fEPSPs; numbers correspond to time points below. Below, mean ± SEM. Comparing average responses of the 5 min before drug and 3.5 hr after drug, n = 4, t3 = 5.4, p = 0.012, d = 3.22. ICAP has no effect on a second, independent synaptic pathway recorded within the slices that received no tetanization (open circles). (C) ICAP has no effect on wild-type LTP maintenance (filled circles; n = 7, t6 = 1.88, p = 0.11, d = 0.67), but blocks the initial potentiation following tetanization (right arrow) in the second synaptic pathway (open circles). The effect of ICAP is different on LTP maintenance in the wild-type and PKMζ-null; t9 = 2.75, p = 0.023, d = 1.129. Right insert above, inhibition of LTP induction is not due to prolonged perfusion in vitro because tetanization of a second pathway recorded for equivalent periods of time induces LTP.
Figure 3—figure supplement 1

Analysis of the complete PKC isoform family shows increases in basal expression of PKCι/λ and PKCβI in the dorsal hippocampus of PKMζ-null mice.
(A) Total PKCι/λ-, activation loop-phosphorylated PKCι/λ-, and ζ-specific antisera show PKMζ-null mice with increased total and phospho-PKCι/λ in dorsal hippocampus, but not in the contralateral hemibrain. In previous studies, the amounts of basal PKCι/λ and phospho-PKCι/λ in the PKMζ-null and wild-type mice were reported to be indistinguishable on immunoblots of total brain homogenates (Lee et al., 2013; Volk et al., 2013). If the increases are regionally selective, however, immunoblots of total brain homogenates may not be sensitive enough to detect the basal increases in dorsal hippocampus (dorsal and ventral hippocampus together constitute ~5% of total mouse brain [Kovacevic et al., 2005]). Using a 'split-brain' preparation, we find the basal increases of PKCι/λ and phospho-PKCι/λ in dorsal hippocampus are below the level of detection if the entire contralateral hemibrain is homogenized and assayed. Dorsal hippocampus: *denotes significance, total PKCι/λ, n’s = 8, t14 = 4.38, p = 0.00063, d = 2.19; phospho-PKCι/λ, PKMζ-null, n = 11, wild-type, n = 7, t16 = 2.77, p = 0.014, d = 1.34; hemibrain: total PKCι/λ, PKMζ-null, n = 9, wild-type, n = 8, t15 = 0.73, p = 0.47, d = 0.36; phospho-PKCι/λ, PKMζ-null, n = 7, wild-type, n = 6, t11 = 0.014, p = 0.89, d = 0.077. (B) The conventional PKCβI increases in dorsal hippocampus of PKMζ-null mice. Initial analysis of all seven conventional/novel PKC isoforms expressed in dorsal hippocampus with n’s of 3–4 revealed an increase in PKCβI (t6 = 2.53, p = 0.045, d = 1.79). To correct for multiple comparisons, the n’s were increased to wild-type, 10, and PKMζ-null, 13, resulting in t21 = 3.40, p = 0.0026, d = 1.43, which reached significance with Bonferroni correction. Earlier studies of the PKMζ-null mice used antisera that do not distinguish between PKCβI and PKCβII isoforms (Lee et al., 2013; Volk et al., 2013). PKCβI is transiently activated by translocation to the cell membrane early in LTP induction (Sacktor et al., 1993). The amount of the novel PKCθ isoform is below the level of detection by immunoblot in dorsal hippocampus (Naik et al., 2000).
Figure 3—figure supplement 2
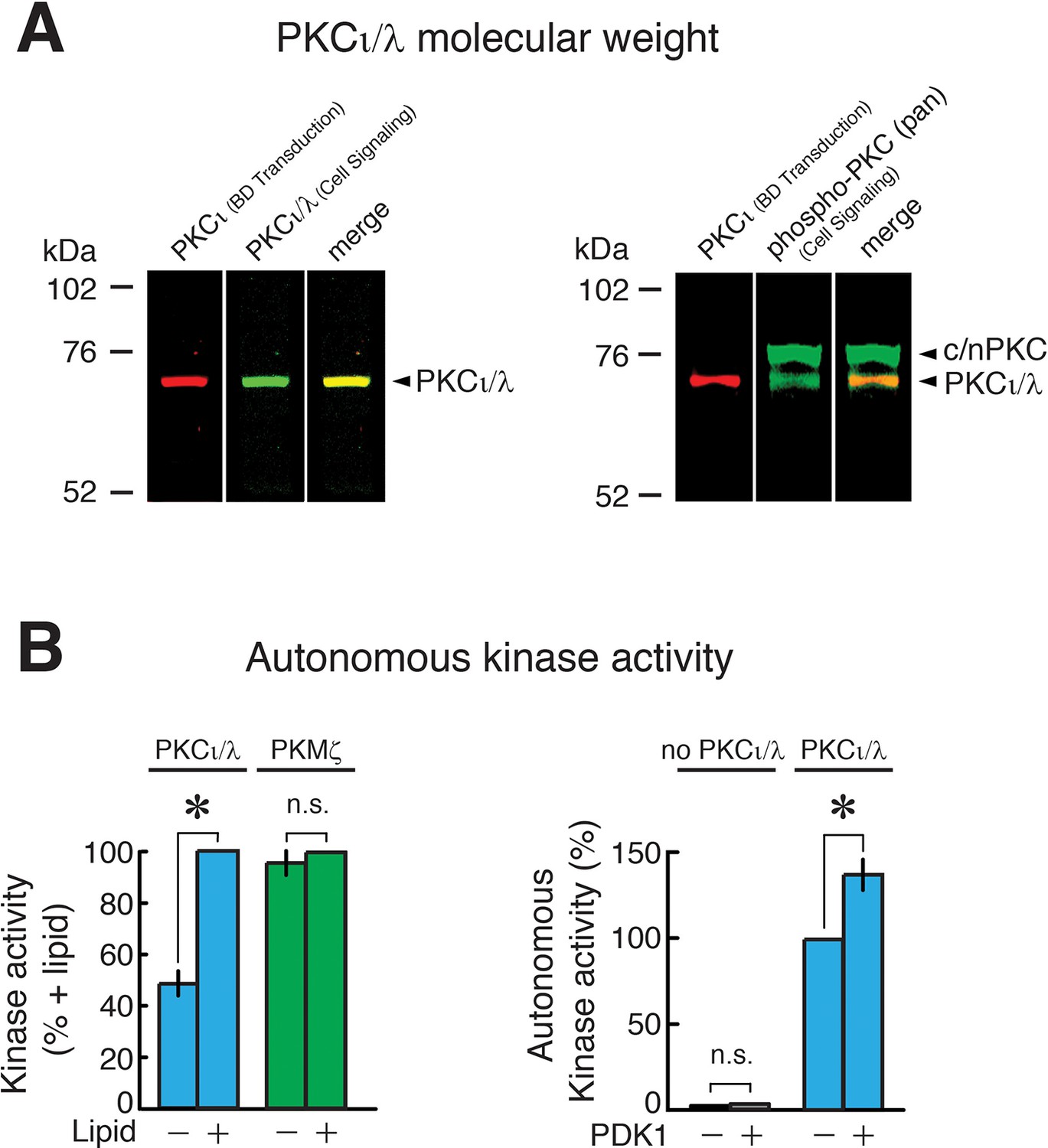
Characterization of PKCι/λ immunoreactivity, molecular weight, and kinase activity.
(A) Left, immunoblot of extract of mouse hippocampus shows two different antisera to PKCι/λ detect a protein band with a molecular weight of 72 kDa (yellow shows merged image of red and green). Right, the antisera to the activation loop of atypical PKC (Cell Signaling) recognizes both PKCι/λ (comigrating with the band recognized by the BD Transduction antiserum) and a broad band at 76–80 kDa, which are the molecular weights of conventional/novel PKCs (c/nPKC) (Naik et al., 2000) that have similar activation loop phosphorylation sequences. The 72 kDa molecular weight we observe for PKCι/λ is consistent with the molecular weight originally described for mouse PKCλ (74 kDa) (Akimoto et al., 1994) and with data from Lee et al. (72 kDa) (Lee et al., 2013). We note that Volk et al., who had not observed increases in PKCι/λ in hippocampus or changes in PKCι/λ during LTP in either wild-type or PKMζ-null mice, reported PKCι/λ’s molecular weight detected by a single antiserum as ~80–82 kDa (Volk et al., 2013). The reason for the discrepancies between the data of Volk et al. on the molecular weight of PKCι/λ and its response to tetanization vs. the data from other laboratories is unclear. (B) Characterization of PKMζ and PKCι/λ enzymatic activity. Left, PKCι/λ is stimulated by lipids and has autonomous activity in the absence of lipids (with and without phosphatidylserine, n's = 4, t3 = 9.35, p = 0.0026, d = 4.68; baseline vs. without phosphatidylserine, t3 = 4.37, p = 0.022, d = 2.18). PKMζ is entirely autonomously active, showing no significant difference in activity with or without lipids (n’s = 4, t3 = 1.33, p = 0.28, d = 0.67). Right, phosphoinositide-dependent kinase-1 (PDK1) phosphorylation of PKCι/λ’s activation loop, which is measured by phospho-specific antiserum in Figure 3A and Figure 3—figure supplement 1A, enhances PKCι/λ’s autonomous activity (n’s = 8, t7 = 4.88, p = 0.0018, d = 1.72). PDK1 does not phosphorylate the standard ε-peptide substrate used to assay atypical PKCs.
Figure 4 with 1 supplement
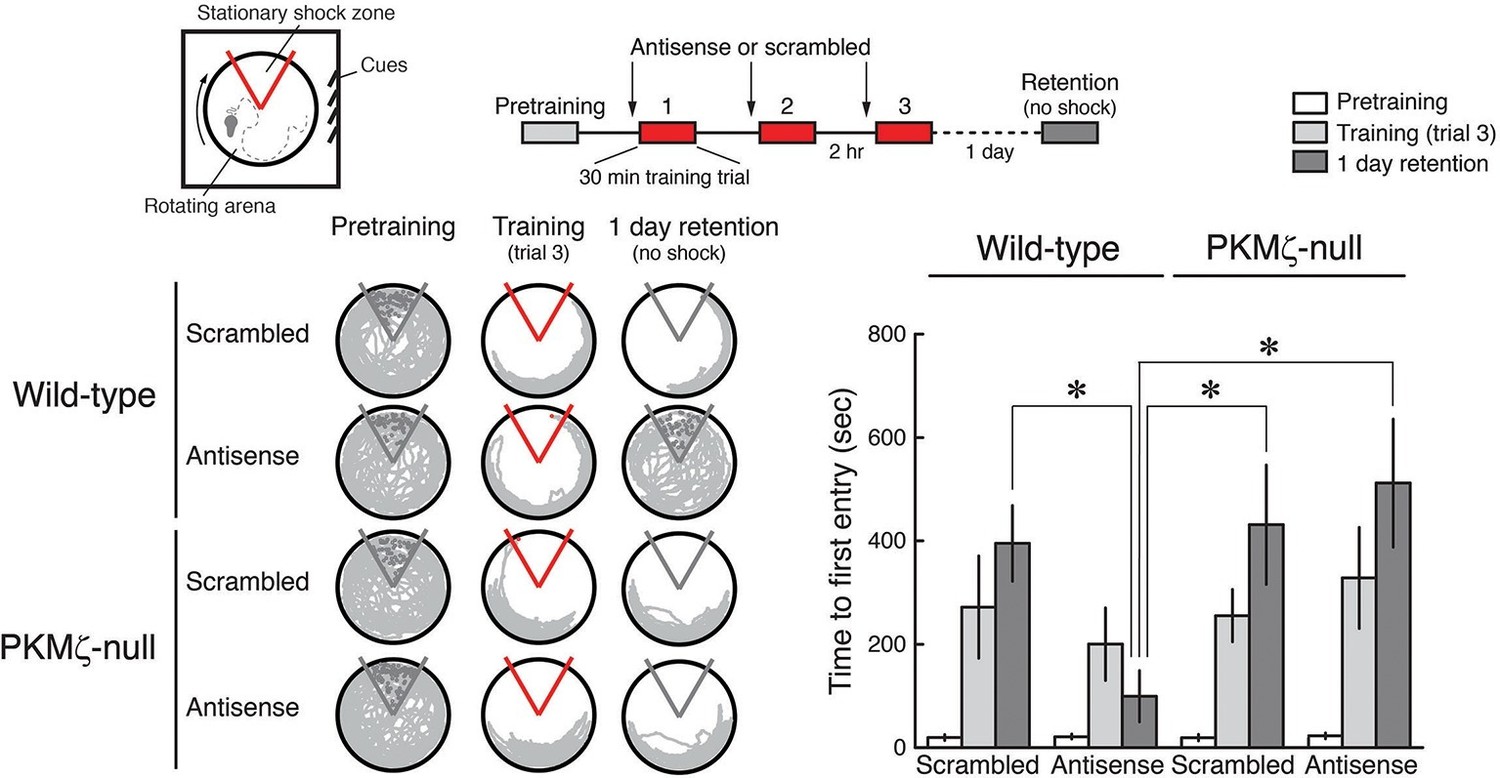
PKMζ is essential for spatial long-term memory in wild-type mice, and compensation accounts for spatial long-term memory in PKMζ-null mice.
PKMζ-antisense blocks spatial long-term memory in wild-type mice but has no effect on long-term memory in PKMζ-null mice. Inserts above, (left) schematic diagram of active place avoidance training apparatus and (middle) 1-day training protocol. Intrahippocampal injections were 1 nmol oligodeoxynucleotide in 0.5 µl vehicle/side, 20 min before each training session. Below left, representative paths during pretraining, the trial at the end of training, and during retention testing with the shock off 1 day after training. The shock zone is shown in red with shock on, and gray with shock off. Red circles denote where shocks are received, and gray circles where shocks would have been received if the shock were on. Right, time to first entry measure of active place avoidance memory (mean ± SEM). There is a significant interaction between the effects of genotype and treatment (scrambled, antisense) (F1,39 = 4.14, p = 0.049, η2 = 0.037). The individual effects of genotype and treatment are F1,39 = 5.89, p = 0.02, η2 = 0.053 and F1,39 = 1.37, p = 0.25, η2 = 0.012, respectively. Memory retention in the wild-type mice treated with PKMζ-antisense differs from the other groups (*, significant post-hoc tests; wild-types, n’s = 12, PKMζ-nulls, scrambled, n = 8, antisense, 11).
Figure 4—figure supplement 1
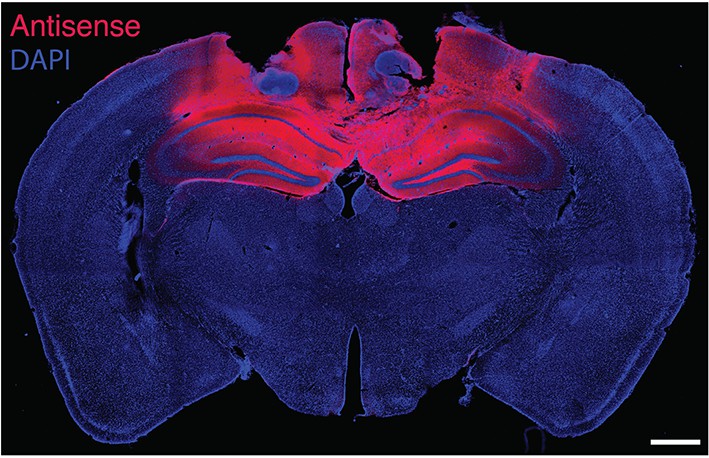
Fluorescence labeling of biotinylated PKMζ-antisense injected bilaterally in mouse hippocampi.
Animal is sacrificed 50 min after the last of three 1 nmol in 0.5-µl vehicle injections per hippocampus, equivalent to the end of training in Figure 4. DAPI is counterstain. Scale bar = 600 µm.
Figure 5
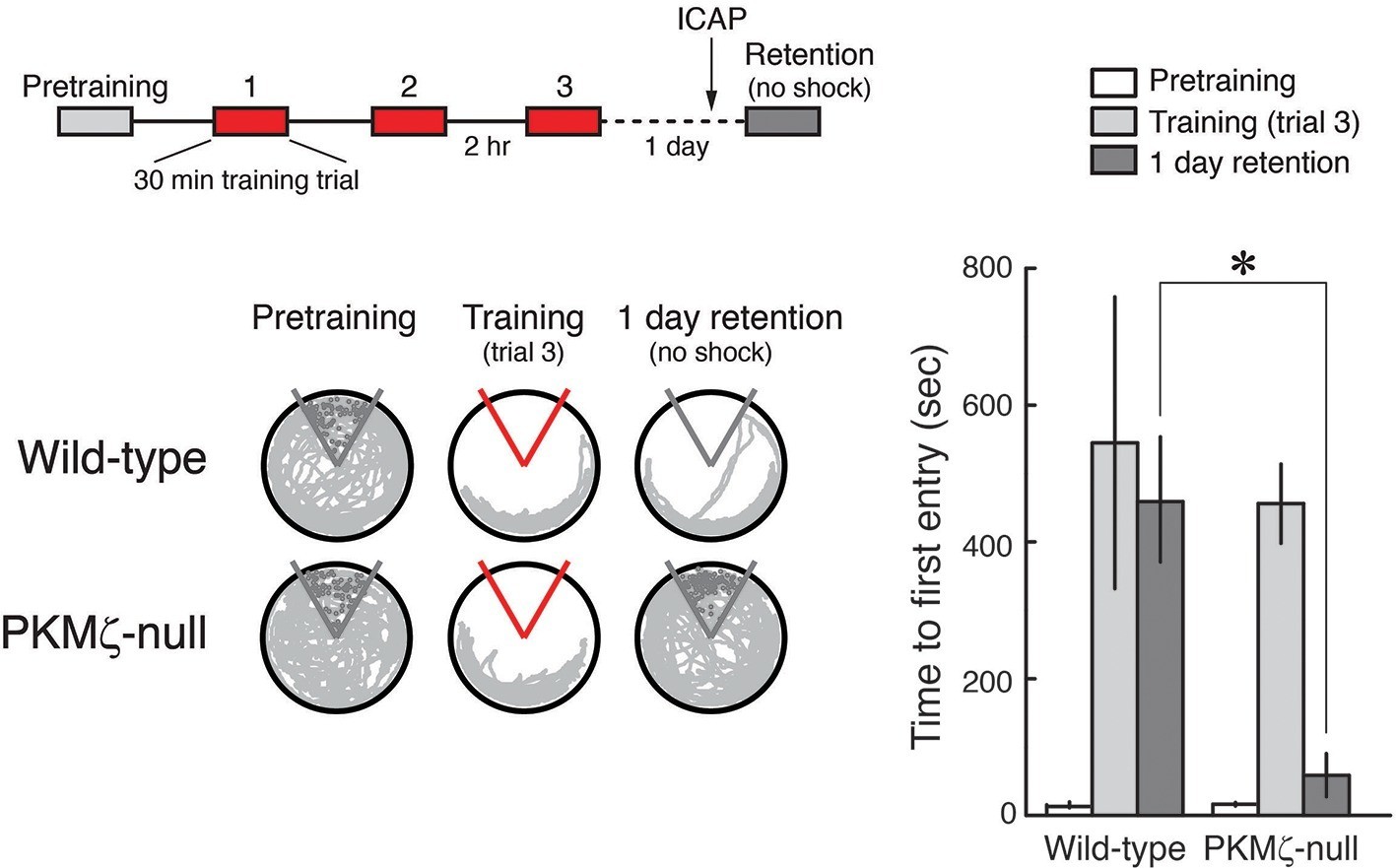
PKCι/λ inhibitor shows spatial long-term memory retention is mediated by distinct molecular mechanisms in PKMζ-null mice and wild-type mice.
Insert above, schematic diagram of 1-day training protocol. Injections were 1 nmol ICAP in 0.5 µl vehicle/side, 2 hr before retention testing. Below left, representative paths during pretraining, the trial at the end of training, and during retention testing with the shock off 1 day after training. The shock zone is shown in red with shock on, and gray with shock off. Gray circles denote where shocks would have been received if the shock were on. Right, time to first entry measure of active place avoidance memory (mean ± SEM). There is a significant difference in 1-day retention between genotype (F1,13 = 14.12, p = 0.0024, d = 1.95); wild-type, n = 8; PKMζ-null, n = 7.
Figure 6 with 1 supplement
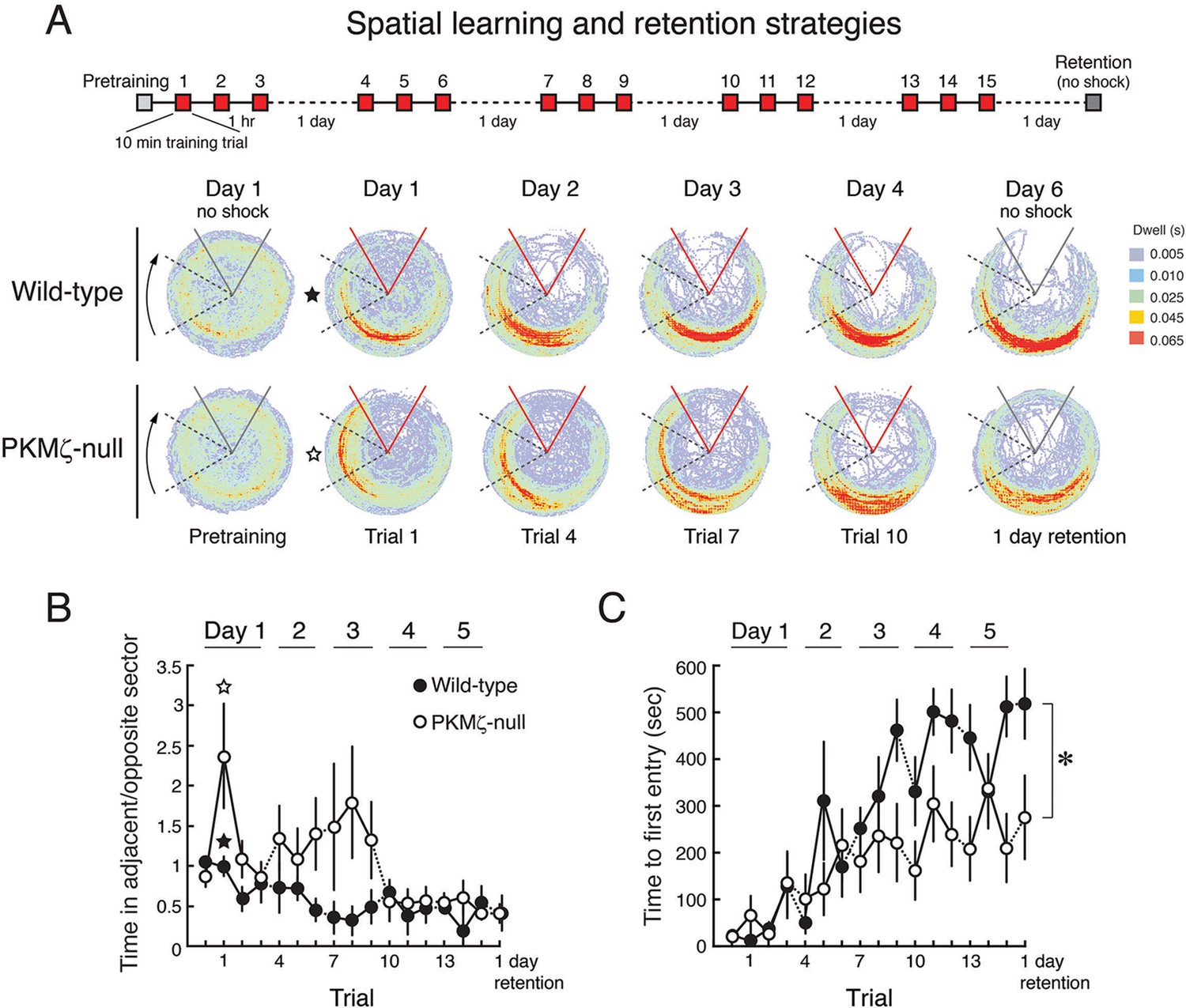
With a weaker training protocol, PKMζ-null mice show inefficient spatial learning and deficits in spatial memory.
(A) Above, schematic diagram of 5-day training protocol. Below, color-coded time-in-location maps for wild-type and PKMζ-null mice during pretraining, beginning of training, midpoint of training, asymptote levels of performance, and 1-day memory retention without shock. In the first training trial, wild-type mice move to areas of the arena opposing the shock zone, whereas PKMζ-null mice remain in the adjacent quadrant about to enter the shock zone. Only after multiple days of training do the PKMζ-null mice show the normal avoidance behavior. N’s = 10. (B) Plotting the ratio of time spent in the adjacent quadrant about to enter the shock zone and the safe quadrant opposite the shock zone for each trial shows PKMζ-null mice remain in the adjacent quadrant more than wild-type mice for ~9 trials over 3 days of training. Mean ± SEM; data are analyzed by genotype x trial 2-way ANOVA followed by post-hoc tests as appropriate. The PKMζ-null mice prefer being in the least efficient place for avoiding shock (genotype: F1,237 = 16.6, p = 6.30 X 10–5, η2 = 0.047). Effects of trial and the genotype x trial interaction are not significant. Pretraining response is denoted as trial 0. (C) Time to first entry into the shock zone increases to an asymptote over 3–4 days of training in both wild-type and PKMζ-null mice. Whereas the asymptote for wild-type mice is over 500 s, it is about half that for PKMζ-null mice. The main effects and interactions are all significant (genotype: F1,255 = 22.4, p = 3.67 X 10–6, η2 = 0.053; trial: F16,255 = 7.3, p = 2.35 X 10–14, η2 = 0.28; interaction: F16,255 = 1.8, p = 0.031, η2 = 0.068). Post-hoc tests do not distinguish PKMζ-null memory on any trial from the pretraining and first training trials when there is no avoidance memory, whereas wild-type memory is significantly better as early as day 3, trial 3, and is superior to pretraining and PKMζ-null estimates of 1-day memory from day 4 through the final retention test on day 6.
Figure 6—figure supplement 1
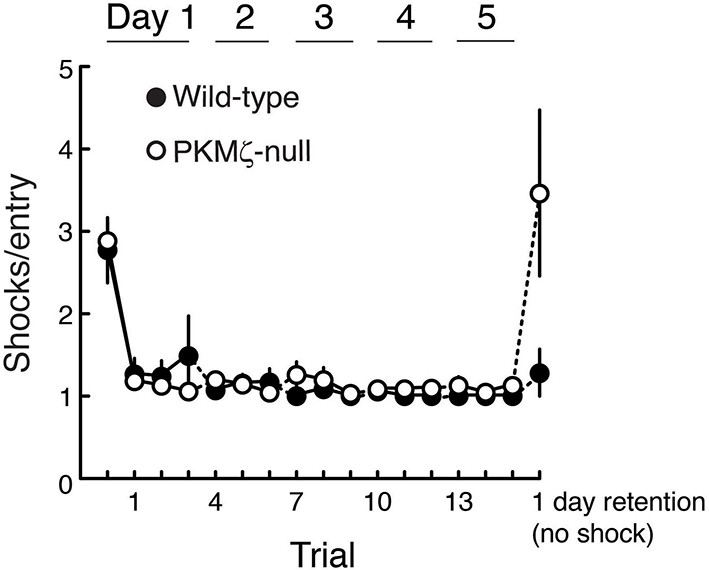
Wild-type and PKMζ-null mice are indistinguishable in their motivation to escape shock during active place avoidance training.
The shock level is 0.2 mA for all animals, which is determined to be the minimum that elicited an escape response. After entering the shock zone, shocks are repeated every 1.5 s until the mouse leaves the zone. The ability to escape shock is estimated by counting the number of shocks the mouse received before it left the shock zone. Within the first training trial both the wild-type and PKMζ-null mice learn to leave the shock zone so they only rarely receive more than 1 shock. This indicates good and equivalent ability to escape in the two groups throughout the training. These observations are confirmed by a significant genotype X trial interaction (F16,255 = 4.8, p = 1.54 X 10–8, η2 = 0.11). Post-hoc test detects that for both genotypes during pretraining with shock off, the number of shocks the mice would have received for each shock zone entrance (had the shock been on) is greater than during all the training sessions with shock on, as expected. During retention with the shock off, post-hoc test detects that the PKMζ-null animals would have received more shocks for each entrance into the shock zone than the wild-type mice. This difference is consistent with worse retention of avoidance memory in the PKMζ-null mice and may indicate that the place avoidance memory is less persistent in the PKMζ-null mice because it rapidly extinguishes (n’s = 10).
Figure 7
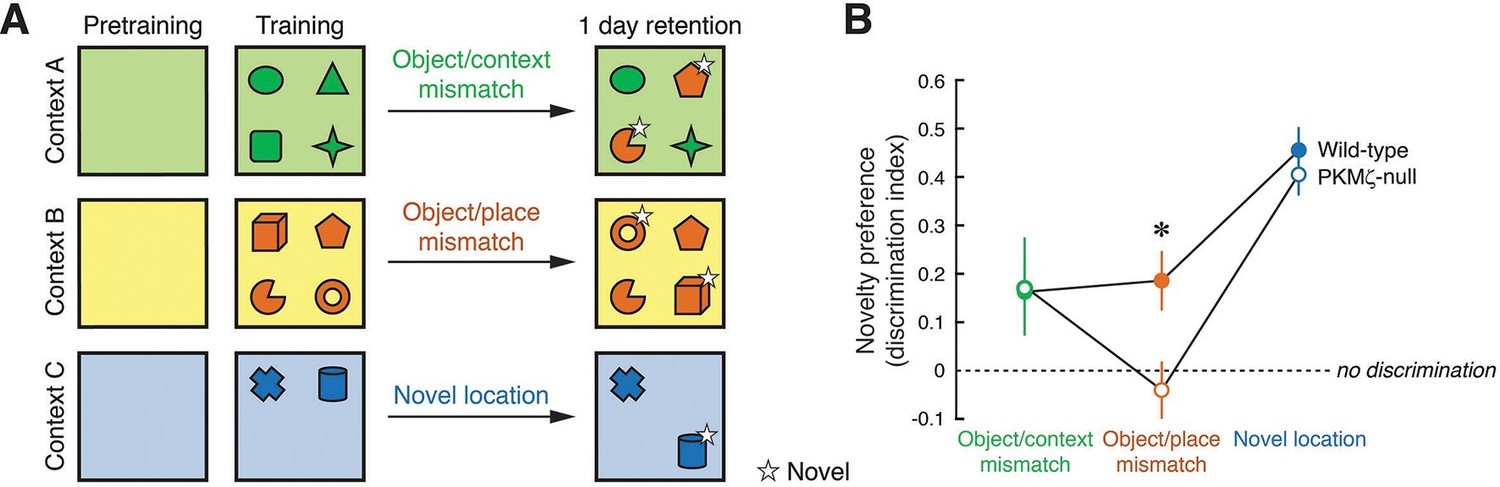
Long-term memory for where objects are encountered during exploration is tested in three unconditioned memory tests, revealing poor memory in PKMζ-null mice when the places of multiple objects are exchanged.
(A) Mice explore distinctive boxes (contexts) during two 5 min trials per day for 3 days (4 objects) and during three 5 min trials for 1 day (2 objects). Counterbalanced object/context mismatch and object/place mismatch tests, and a novel object location test are given a day after the last exploration trial, one test per day. For clarity, only a single version of the counterbalanced tests is shown. (B) Both wild-type (n = 8) and PKMζ-null (n = 8) mice express similar memory discrimination when two of four objects are encountered in the incorrect context, and when one of two objects is encountered in a novel location. However, despite similar levels of investigating the objects (32–36% of the time; effect of group F1,14 = 3.2; p = 0.09, η2 = 0.18; effect of task F2,14 = 0.004; p = 0.95, η2 = 0.00049; group X task interaction F1,14 = 0.18; p = 0.68, η2 = 0.014), only wild-type mice express discrimination memory when the places of two of four objects are exchanged. PKMζ-null mice do not discriminate between objects that are encountered in the familiar and unfamiliar places. The interaction between genotype and memory test is significant (F2,13 = 4.49, p = 0.03, η2 = 0.06). The individual effects of genotype and memory test are F1,14 = 2.23, p = 0.16, η2 = 0.0014 and F2,13 = 47.3, p = 10–6, η2 = 0.025, respectively. *Significant post-hoc tests distinguish wild-type and PKMζ-null memory on the object/place mismatch test.
Videos
Video 1
Inefficient place avoidance in the PKMζ-null mouse.
These videos show place avoidance behavior during the first training trial. The video on the left shows a wild-type mouse and the video on the right a PKMζ-null mouse. The wild-type mouse rapidly learns to move opposite to the shock zone to better avoid shock, whereas the mutant mouse avoids shock inefficiently by remaining in the area adjacent to the shock zone, which is the most vulnerable place.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice
eLife 5:e14846.
https://doi.org/10.7554/eLife.14846




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}