Splicing repression allows the gradual emergence of new Alu-exons in primate evolution
- UCL Institute of Neurology, United Kingdom
- MRC-Laboratory of Molecular Biology, United Kingdom
- Institute de Biologie de l’ENS (IBENS), CNRS UMR 8197, France
- University College London Genetics Institute, United Kingdom
- Goethe University Frankfurt, Germany
- Institute of Molecular Biology (IMB), Germany
Figures
Figure 1 with 4 supplements
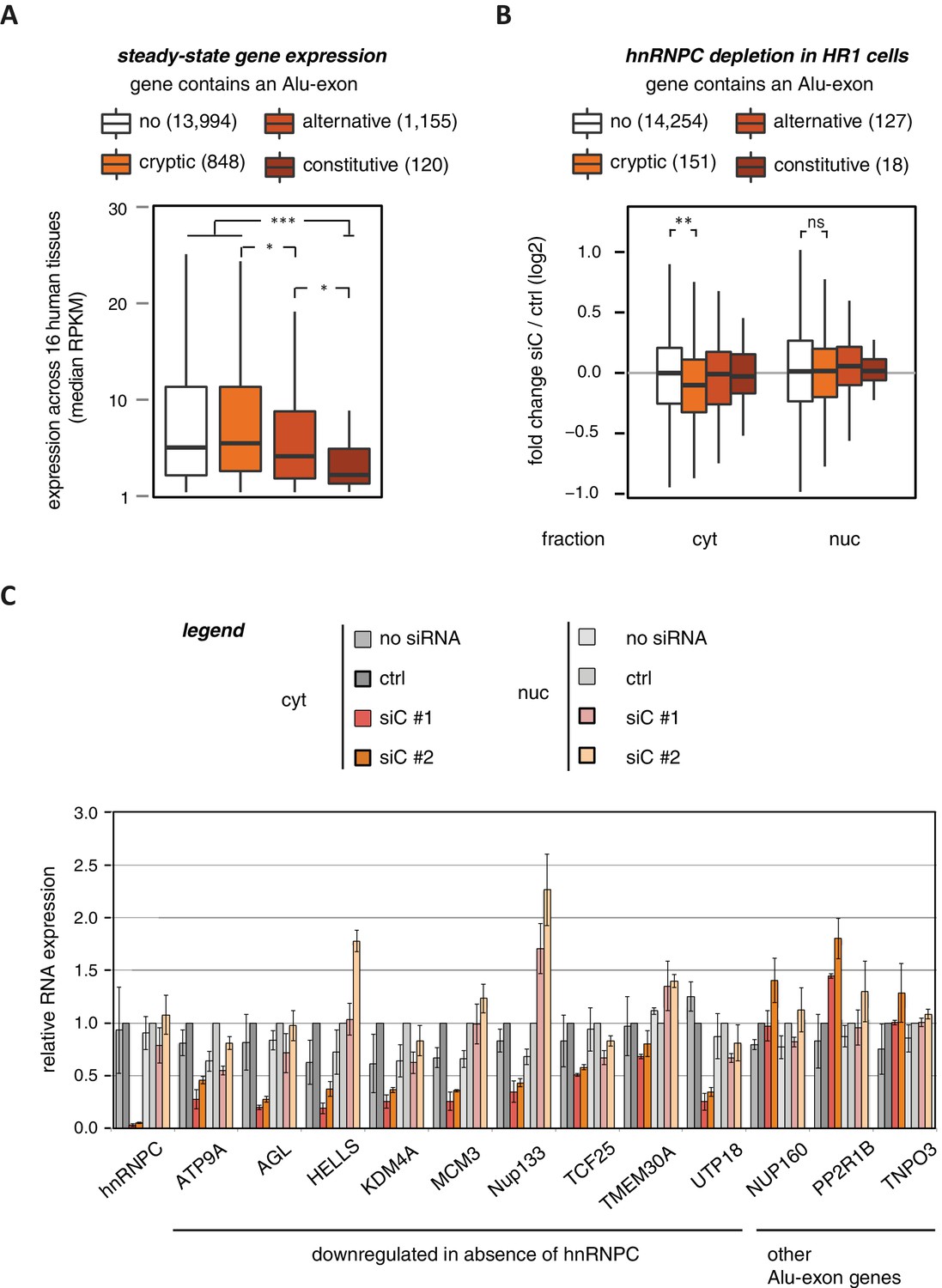
Alu-exons are associated with reduced gene expression.
(A) Expression levels of genes containing a cryptic, alternative or constitutive Alu-exon across a panel of 16 human tissues. For each gene, the median expression level was calculated from the Illumina BodyMap 2.0 dataset, considering only widely expressed genes (median RPKM ≥ 1). Expression in each individual tissue is shown in Figure 1—figure supplement 1. Differences in the distribution were tested by an ANOVA design, for details see Materials and methods. (B) RNAseq data of cytoplasmic and nuclear RNA from HR1 cells transfected with siRNAs against hnRNPC (siC #1 and #2) or controls (‘no siRNA’: mock transfection; ctrl: unspecific control oligonucleotide) was used to test if inclusion of Alu-exons impacts on gene expression. Protein-coding genes with or without cryptic, alternative or constitutive Alu-exons were grouped, considering only Alu-exons with sufficient coverage (minimal five reads/million including one junction-spanning read; 550 Alu-exons). Shown is the distribution of expression fold changes (log2). To estimate the expression fold changes, we compared cells depleted of hnRNPC (siC #1 and #2) against controls (no siRNA and control oligonucleotide) using DESeq (Anders and Huber, 2010). Differences between Alu-exon gene abundance across groups were tested by Kruskal-Wallis Rank Sum test within each RNA fraction (cytoplasmic RNA, p-value = 0.01819; nuclear RNA, p-value = 0.3646). Indicated pairwise comparisons were tested with a two-sided Wilcoxon Rank Sum test. ** indicates p-value < 0.01. (C) RNA expression levels of 12 Alu-exon genes identified from the cytoplasmic and/or nuclear RNAseq datasets. Loss of transcripts from Alu-exon genes in hnRNPC-depleted cells was generally restricted to cytoplasmic RNA. Gene expression was quantified by quantitative RT-PCR from HR1 cells transfected with siRNAs against hnRNPC (siC #1 and #2) or controls (‘no siRNA’: mock transfection; ctrl: unspecific control oligonucleotide), error bars represent standard deviation of the mean (s.d.m.; n = 3 or 4). Figure 1—figure supplement 2 shows the expression of Alu-exon genes in the individual tissues, which is summarised in Figure 1A. Figure 1—figure supplement 2 provides quality control on the nuclear-cytoplasmic fractions by Western blot and quantitative RT-PCR. Figure 1—figure supplement 3 presents semi-quantitative RT-PCR quantifications of individual Alu-exon transcripts compared to the Alu-exon-free transcripts. This validates that Alu-exon transcripts are depleted from the cytoplasmic RNA pool. Figure 1—figure supplement 4 demonstrates that cytoplasmic depletion of Alu-exon transcripts is not the result of a lack of mRNA export from the nucleus.
Figure 1—figure supplement 1
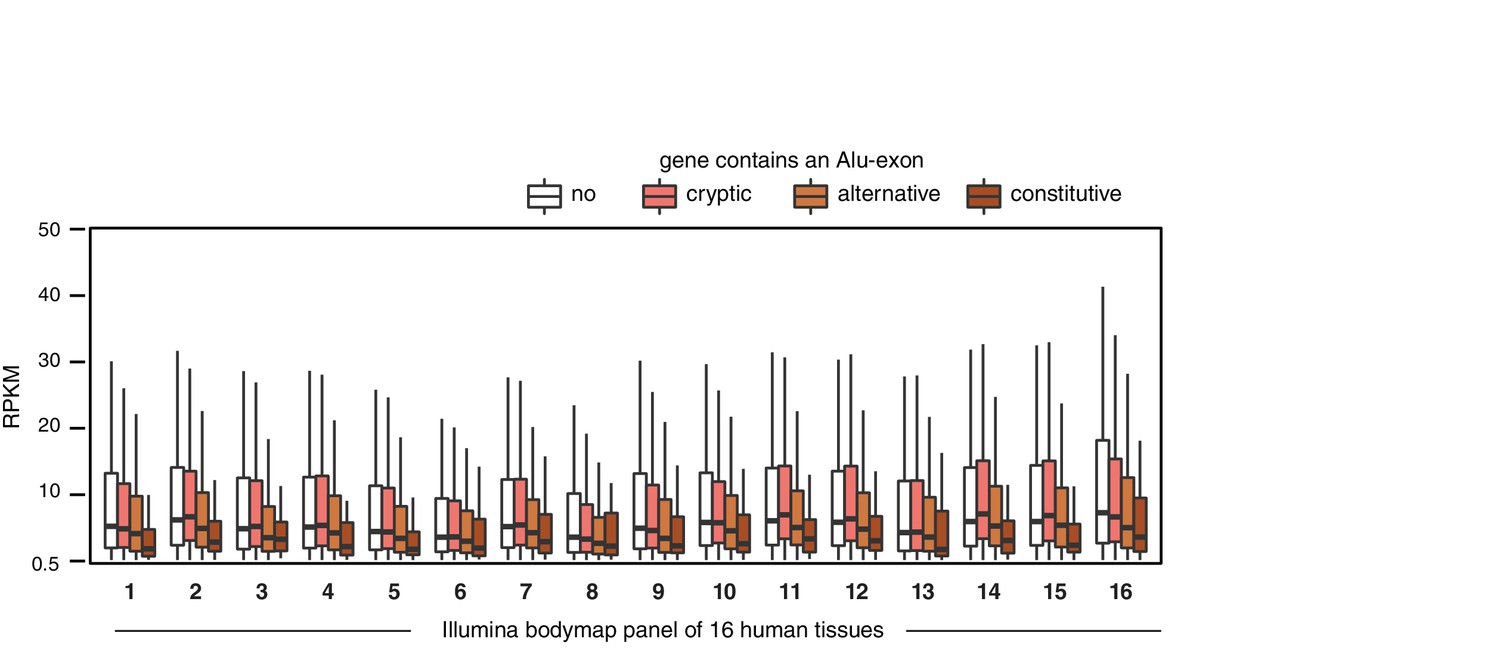
Alu-exon gene expression in various human tissues.
Expression levels of genes containing a cryptic, alternative or constitutive Alu-exons across a panel of human tissues. For each tissue, only expressed genes were selected (RPKM ≥ 1). Tissues are adipose, adrenal glands, brain, breast, colon, heart, kidney, liver, lung, lymph nodes, ovary, prostate, skeletal muscles, testes, thyroid to white blood cells, labelled (1) to (16).
Figure 1—figure supplement 2
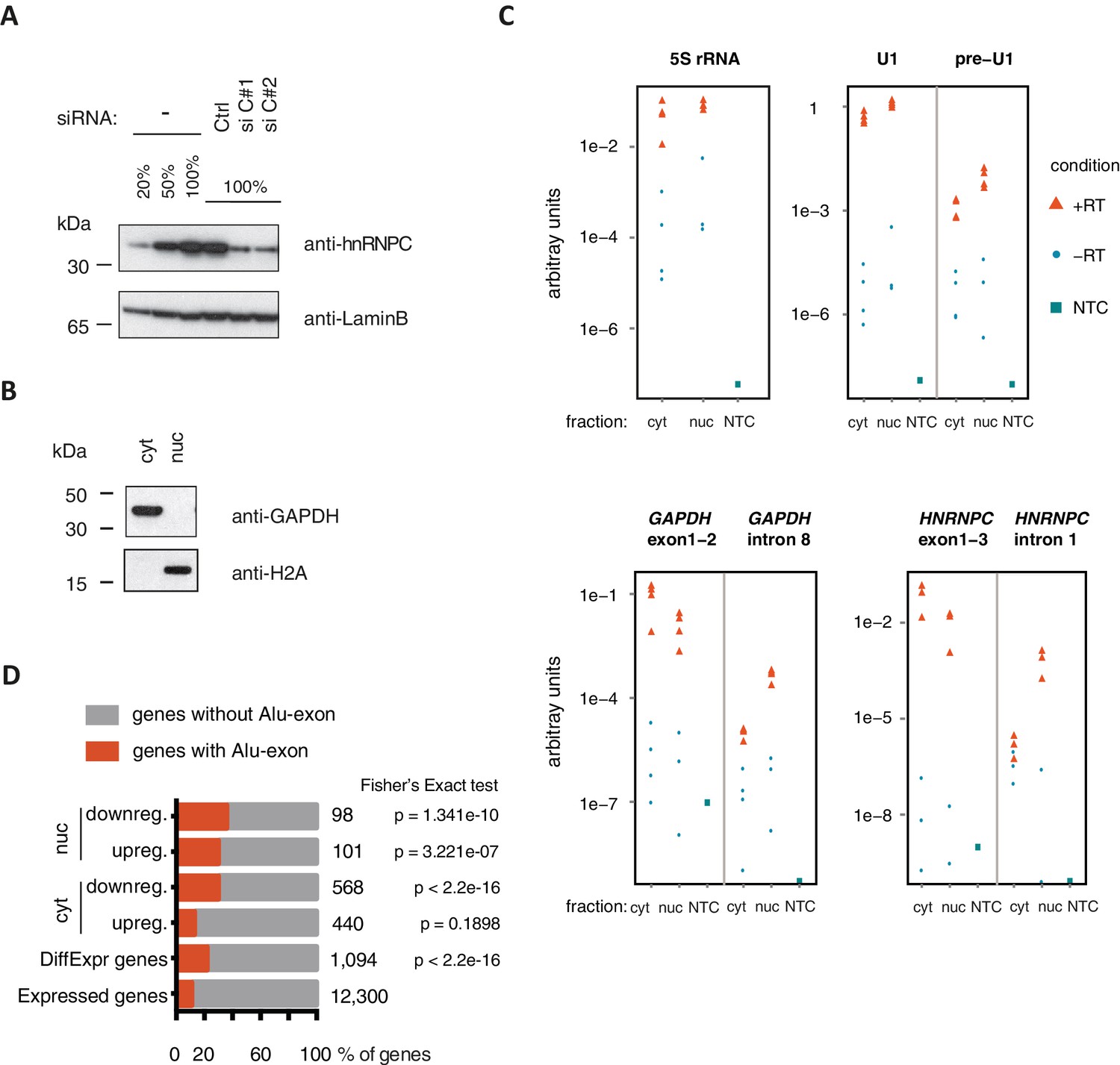
Quality control of cytoplasmic and nuclear RNA fractions.
(A) The efficiency of hnRNPC depletion was tested by semi-quantitative Western blot. For comparison, a dilution series of lysate from control cells was included. (B) Cytoplasmic and nuclear protein lysates were collected together with RNA lysates, and probed for marker proteins by Western blot. GAPDH and H2A were chosen as abundant cytoplasmic and nuclear marker proteins, respectively. No cross-contamination of the cytoplasmic lysate with nuclear lysate was detectable, nor vice versa. (C) Quantitative RT-PCR was used to measure the abundance of a representative set of RNAs, in cytoplasmic and nuclear RNA or control reactions without template cDNA (NTC). Top: 5S rRNA and processed U1 snRNA were used as proxy for total RNA abundance. Unprocessed U1 snRNA is expected to be more abundant in the nucleus than in the cytoplasm (pre-U1). Bottom: To test for leakage of intronic sequences into the cytoplasm, two abundant mRNAs (GAPDH, HNRNPC) were quantified by two distinct sets of primers amplifying either an exon-exon junction or an intronic sequence. Intronic RNA was readily amplified from nuclear RNA but was close to negative control levels in which the RT enzyme was omitted (‘-RT’) in cytoplasmic RNA. This indicated that leakage of intronic RNA due to disruption of nuclei was negligible. (D) RNAseq data of cytoplasmic and nuclear RNA of HR1 cells depleted of hnRNPC was used to identify genes differentially expressed upon hnRNPC depletion, using DESeq (Anders and Huber, 2010). For each group, enrichment for genes containing Alu-exons was tested against all expressed genes using Fisher’s exact test. Enrichment of Alu-exon genes among genes with expression changes in cytoplasmic RNA explain the enrichment within all differentially expressed genes. ‘DiffExpr genes’ are all genes with padj < 0.01, combining differentially expressed genes from the cytoplasm and nucleus.
Figure 1—figure supplement 3
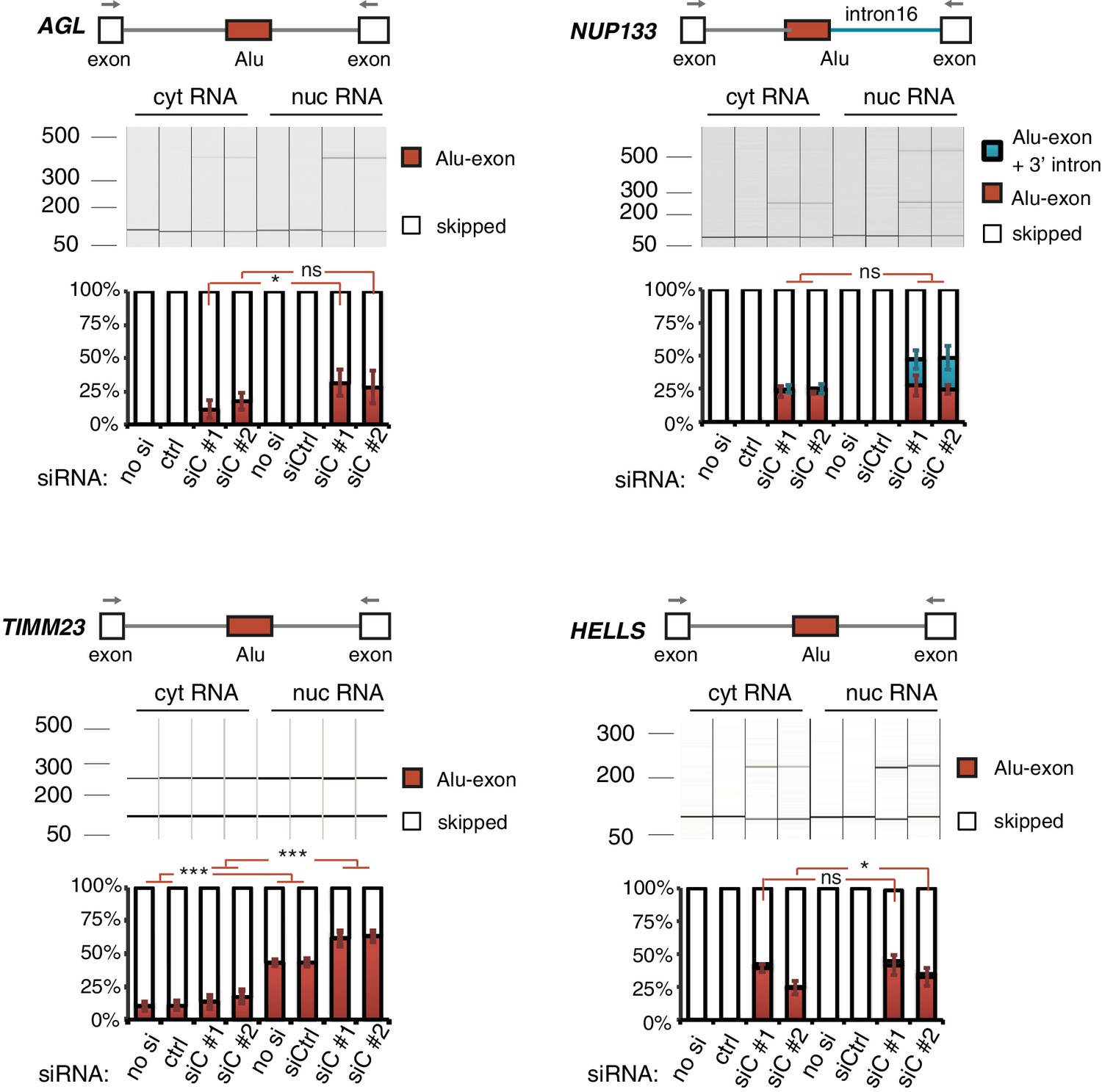
Alu-exon transcripts are frequently depleted from the cytoplasmic RNA pool.
The relative abundance of the Alu-exon transcripts of AGL, NUP133, TIMM23 and HELLS was measured in cytoplasmic and nuclear RNA of cells depleted of hnRNPC (siC #1 and #2), or control cells (‘no si’: no siRNA; ctrl: unspecific control oligonucleotide). Overall, we observed a tendency for Alu-exon transcripts to be present in the nucleus but lost in the cytoplasm. The relative abundance of Alu-exon transcripts was significantly higher in the nucleus than in the cytoplasm in case of AGL and TIMM23. Note that NUP133 generates two transcripts that contain the Alu-exon sequence, an Alu-exon transcript and a larger 3' intron intron-retaining Alu transcript. The later was not present in the cytoplasm, while the Alu-exon transcript was of equal abundance in both subcellular fractions. The Alu-exon transcript of HELLS did not show consistent depletion in the cytoplasm, but we later found that HELLS produces multiple Alu-exon isoforms (Figure 2—figure supplement 2). Abundance was measured relative to the constitutively spliced transcript by semi-quantitative RT-PCR. Shown are the averages across independent replicates (n = 3–4), error bars represent standard deviation of the mean. To test for significance, two-way ANOVA was performed treating the RNA fraction as confounding factor. Multiple comparison correction was done according to Tukey’s HSD.
Figure 1—figure supplement 4
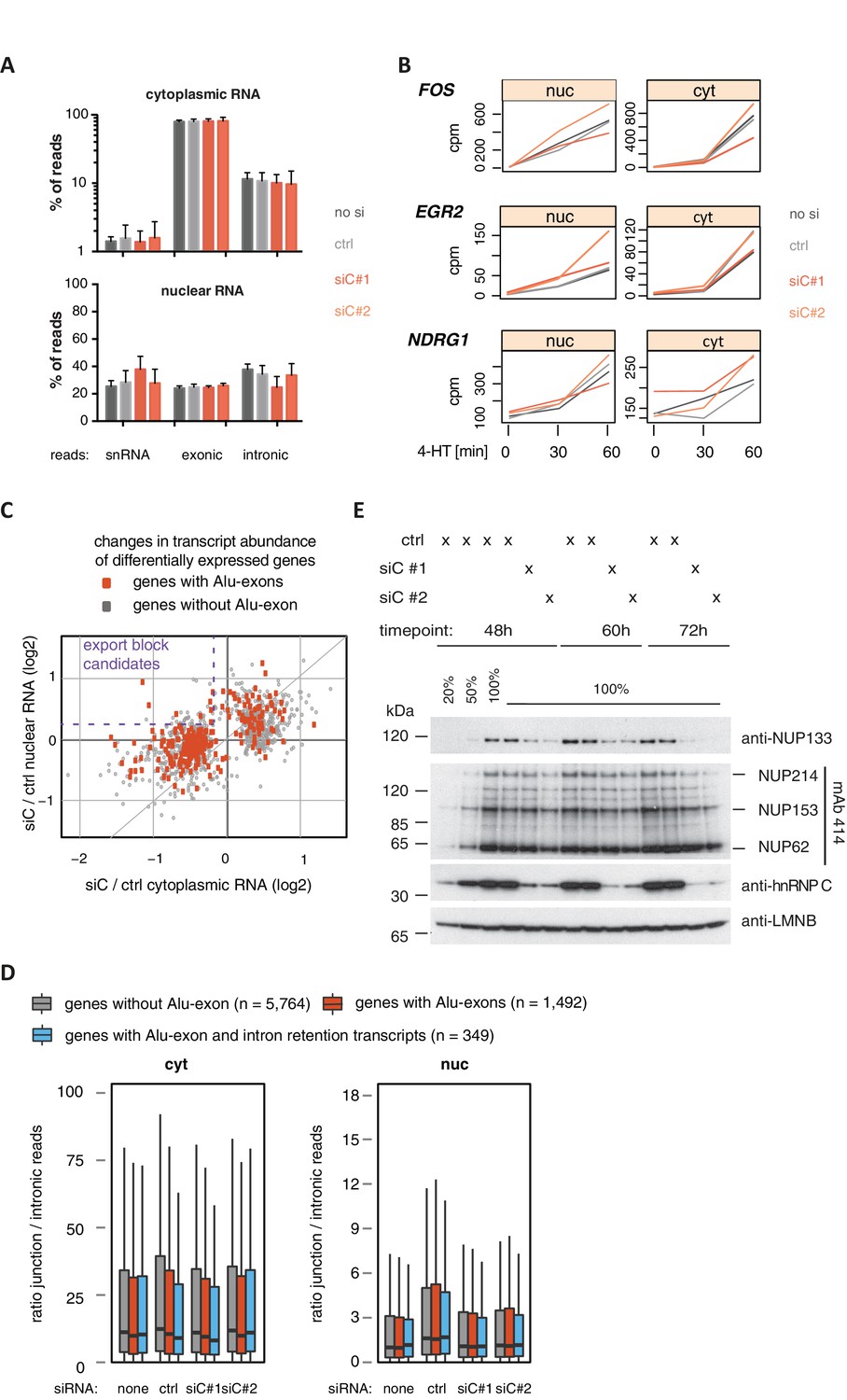
Cytoplasmic depletion of Alu-exon genes is not caused by a defect in mRNA export upon hnRNPC depletion.
(A) Percentage of reads mapping to snRNAs, introns or exons are shown for each sample. Examined were RNAseq samples of cytoplasmic and nuclear RNA of cells transfected with siRNAs for hnRNPC (siC #1 and #2) or controls (no siRNA, control oligonucleotide). snRNA abundance served as internal reference. (B) Assessment of the cytoplasmic accumulation of the ERK-inducible genes FOS, EGR2 and NDRG1 in control and hnRNPC-depleted cells. Cells were treated with 100 nM 4-HT for 0, 30 or 60 min, and reads mapping to each gene after normalisation for library size (normalised ‘counts’) are shown as line chart. Controls and hnRNPC depletion experiments as in (A). Our time-course showed ERK-induced genes increased first in the nucleus and then with a time-delay in the cytoplasm, in line with a transcriptional burst in gene expression. 4-HT: 4-hydroxytamoxifen. (C) A scatter plot comparing the fold change (log2) of genes upon depletion of hnRNPC in cytoplasmic and nuclear RNA. To estimate expression fold changes, we compared cells depleted of hnRNPC (siC #1 and #2) against controls (no siRNA, control oligonucleotide) using DESeq (Anders and Huber, 2010). Alu-exon genes are marked in red, and the upper-left quadrant is labelled as a pattern considered as consistent with a defect in mRNA export. (D) The ratio of junction-spanning to intronic reads was calculated for each gene in cytoplasmic and nuclear RNA. We separated Alu-exon genes associated with and without significant intron retention (adjusted p-value < 0.01, see also Figure 3). No accumulation of unspliced RNA in the nucleus is observed for either group of genes. (E) The abundance of NUP133 protein was tested by semi-quantitative Western blot in cells depleted of hnRNPC for 48, 60 and 72 hr. NUP133 protein levels gradually decrease in hnRNPC-depleted cells. Other components of the nuclear pore are not affected as demonstrated by the mAb414 antibody, which recognises FGF repeats found in all inner nuclear pore proteins marked at the side. LaminB protein levels served as loading control.
Figure 2 with 2 supplements
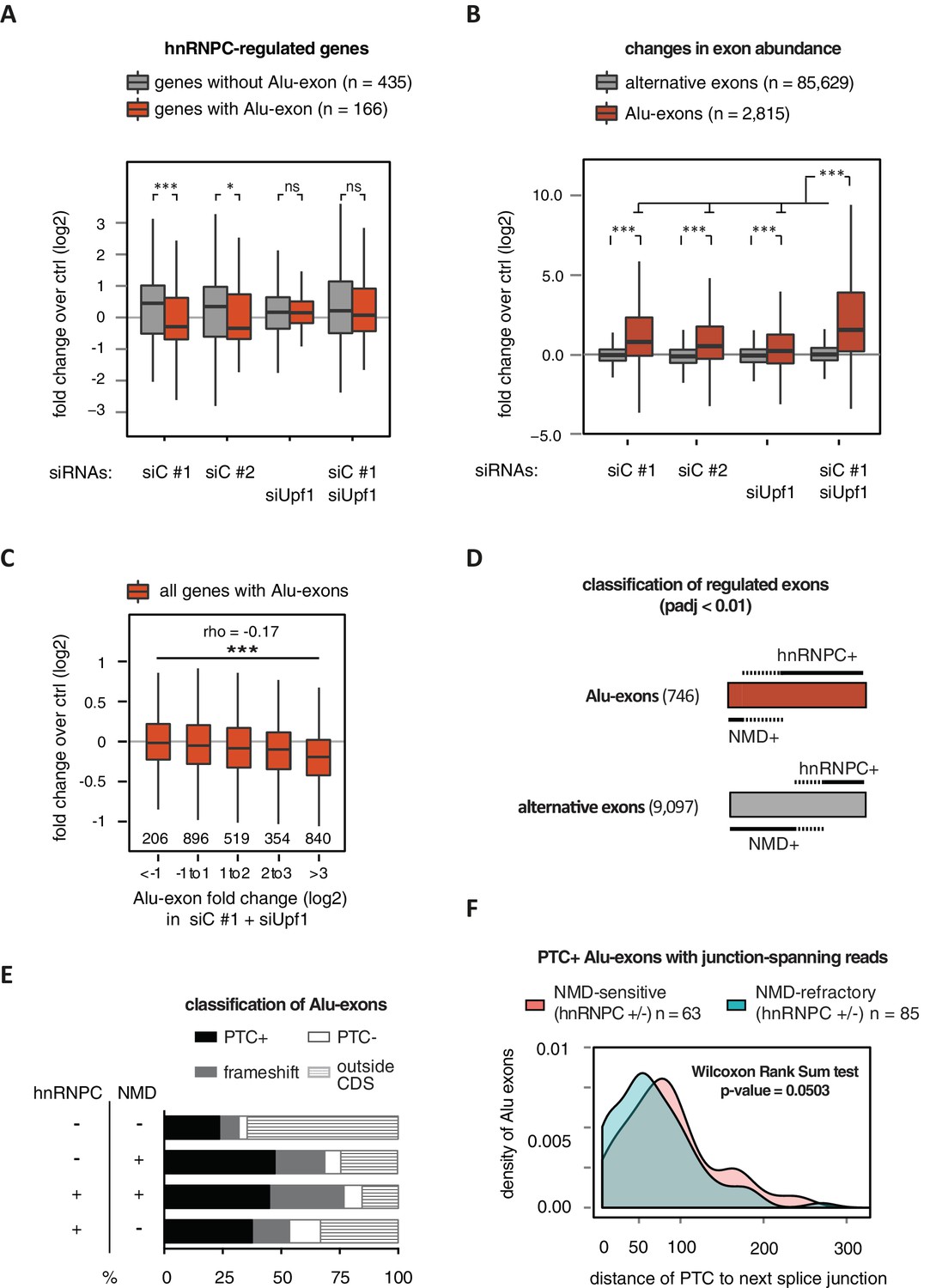
Alu-exons are a novel class of NMD substrates.
(A) Depletion of hnRNPC in total RNA led to significant expression changes in 601 protein-coding genes (p-value < 0.01), of which 166 contain an Alu-exon. Boxplot showing the distribution of expression fold changes (log2) over control (unspecific control oligonucleotide) in cells depleted of hnRNPC (siC #1 and siC #2) and/or UPF1 (siUpf1) as indicated below. Differences between Alu-exon gene abundance across groups were tested by Kruskal-Wallis Rank Sum test (p-value < 2.2e-16), and pairwise comparisons within each condition were tested with a two-sided Wilcoxon Rank Sum test. * and *** indicate p-value < 0.05 and 0.001, respectively. (B) Boxplot showing the changes in exon abundance upon hnRNPC and/or UPF1 depletion as fold changes (log2) over control (unspecific control oligonucleotide), analysed by DEXSeq (Lykke-Andersen et al., 2000). Only alternative exons and Alu-exons in protein-coding genes were considered. Differences between Alu-exon abundance across groups were tested by Kruskal-Wallis Rank Sum test (p-value < 2.2e-16), and pairwise comparisons were corrected according to Siegel-Castellan. *** indicates p-value < 0.001. (C) Boxplot showing relationship between gene expression changes upon hnRNPC depletion and Alu-exon usage. Alu-exons were stratified according to their fold change (log2) in exon abundance in cells depleted of UPF1 and hnRNPC (siC #1), which should be the maximal achievable inclusion of each Alu-exon. Protein-coding genes with Alu-exons were filtered for a minimal RPKM of 1 (2809 genes in total). Significant anti-correlation between Alu-exon inclusion and Alu-exon gene expression levels was tested by Spearman correlation (indicated by ρ coefficient). (D) Depiction of the overlap of hnRNPC- and NMD-sensitive exons that are significantly regulated (adjusted p-value < 0.01) under the conditions in (B). Exon sets as in (B). Dashed lines visualise proportion of exons that are coordinately regulated by both hnRNPC and NMD. hnRNPC+ and NMD+ denote upregulation in hnRNPC-depleted and UPF1-depleted cells, respectively. All proportions are drawn to scale within each group. (E) Bar diagram showing the proportion of Alu-exons outside (grey hatching) and inside the coding sequence (CDS), with the latter being further subdivided into exons introducing a PTC (PTC+, black), without PTC but introducing a frame-shift (grey), or being in frame (PTC-, white) for the following Alu-exon categories: non-regulated (hnRNPC-/NMD-, n = 313), NMD-specific (hnRNPC-/NMD+, n = 72), shared-target (hnRNPC+/NMD+, n = 207), and hnRNPC-specific (hnRNPC+/NMD-, n = 465). (F) Density plot showing the distance of the PTC to the downstream splice site for NMD-sensitive (orange) and NMD-refractory (green) PTC+ Alu-exons. Only Alu-exons that were supported by junction-spanning reads on either side in our RNAseq data were taken into account. In Figure 2—figure supplement 1, the lack of functional NMD due to UPF1 depletion is validated by semi-quantitative and quantitative RT-PCR as well as by analysis of transcriptomic changes in RNAseq of UPF1-depleted cells. In Figure 2—figure supplement 2, we present semi-quantitative RT-PCR quantifications of individual Alu-exon transcripts compared to the Alu-exon free transcripts. This validates that Alu-exon transcripts are depleted from the cytoplasmic RNA pool by NMD.
Figure 2—figure supplement 1
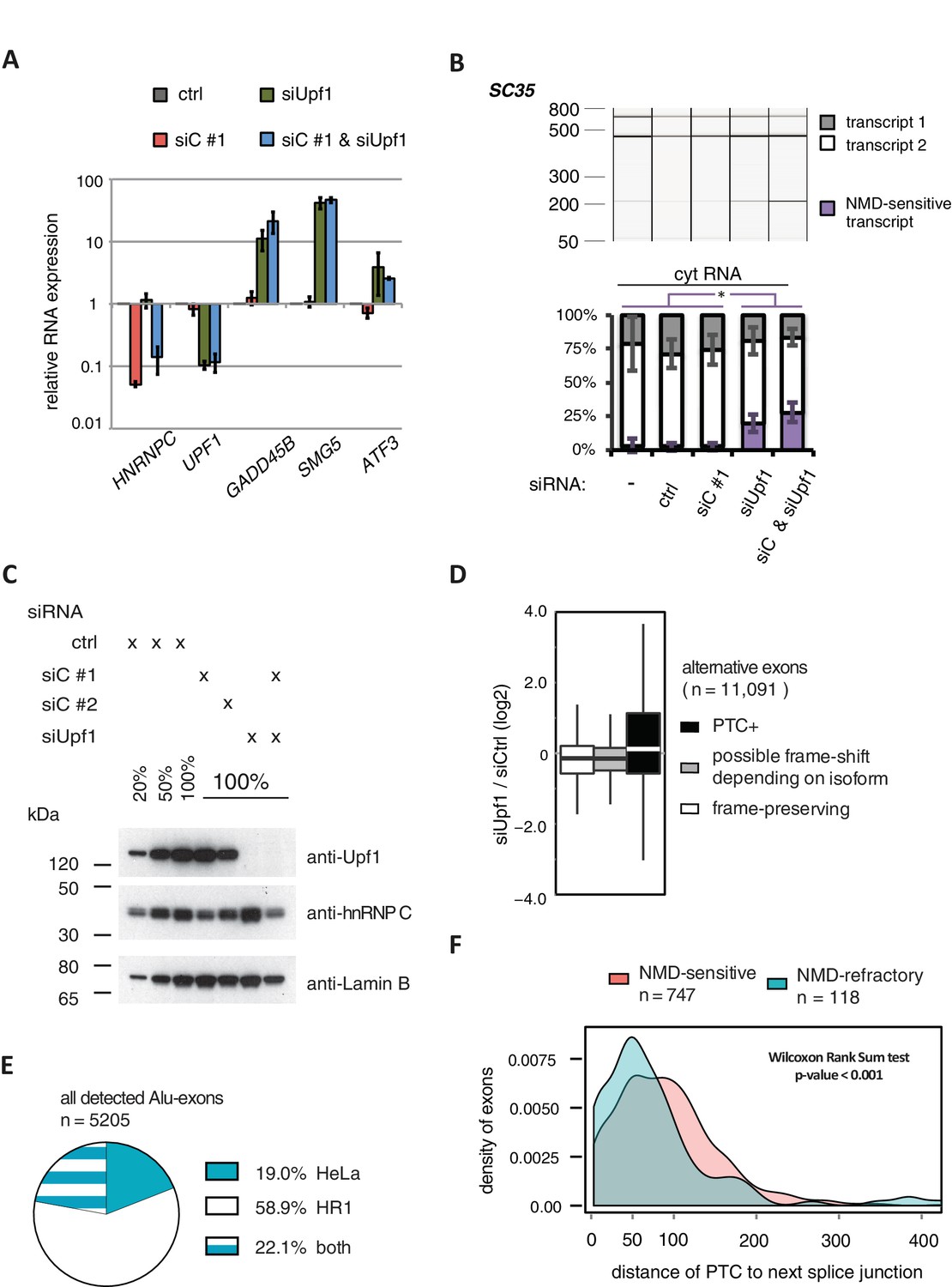
Validation of disenabling NMD by UPF1 depletion.
(A) Quantitative RT-PCR was used to measure the abundance of three genes known to be NMD substrates, GADD45B, SMG5 and ATF3 (Chan et al., 2007), as well as HNRNPC and UPF1 for control. Each of the NMD targets was upregulated to similar extent in UPF1-depleted or UPF1 and hnRNPC co-depleted cells (n = 2–3). Error bars represent s.d.m. (B) Depletion of UPF1 led to stabilisation of a known NMD-sensitive isoform of SC35 specifically in the cytoplasmic fraction, measured by semi-quantitative RT-PCR. Data are plotted as average of three independent replicates, error bars represent s.d.m. (C) The efficiency of UPF1 and hnRNPC depletion and co-depletion was tested by semi-quantitative Western blot. (D) To test our PTC prediction, we examined the change in expression of exons with or without PTC, comparing UPF1-depleted cells to control. As exon set, we used all UCSC-annotated alternative exons in protein-coding genes. (E) Comparison of the number of Alu-exons that we were able to annotate from our previously published work in HeLa cells (Zarnack et al., 2013) and the RNAseq data collected in HR1 cells (including UPF1/hnRNPC co-depleted cells). (F) The distribution of distances of the PTC to the downstream splice site is presented for NMD-sensitive and NMD-refractory exons. The analyses are based on all significantly regulated alternative PTC+ exons with junction-spanning reads on both splice sites.
Figure 2—figure supplement 2
Expression analysis of NMD-sensitive and NMD-refractory Alu-exons.
The relative abundance of the Alu-exon transcript of six genes was measured by semi-quantitative RT-PCR in cytoplasmic RNA from control, hnRNPC- and UPF1-depleted cells. Shown are gel visualisations of capillary electrophoresis and quantification of average Alu-exon inclusion (n = 3), error bars represent s.d.m. The upper scheme illustrates the position of the Alu-exon relative to the flanking constitutive and alternative cassette exons (if present, ‘ACE’) and the position of the primers used for RT-PCR. (A) Quantification of the Alu-exon transcripts of the AGL, NUP133, MCM3 and HELLS genes. These four Alu-exon transcripts were found to be significantly upregulated by co-depletion of hnRNPC and UPF1 and are examples of shared-target Alu-exons. Note that the Alu element within intron nine of MCM3 produces two different Alu-exons with alternative 5’ splice sites. (B) Quantification of a second Alu-exon transcript in the MCM3 and HELLS genes. These two Alu-exon transcripts were found to be refractory to NMD based on their abundance in UPF1/hnRNPC co-depleted cells. The same was observed for the Alu-exons in TNPO3 and ZFX, which also did not change in gene expression upon hnRNPC depletion. These represent examples of hnRNPC-specific Alu-exons. For comparison, quantification of the Alu-exon transcripts of TNPO3 and ZFX in nuclear RNA are shown. To test for significant changes in Alu-exon transcript abundance, one-way ANOVA was performed on each dataset from cytoplasmic RNA. Multiple comparison correction was done according to Tukey’s HSD.
Figure 3 with 3 supplements
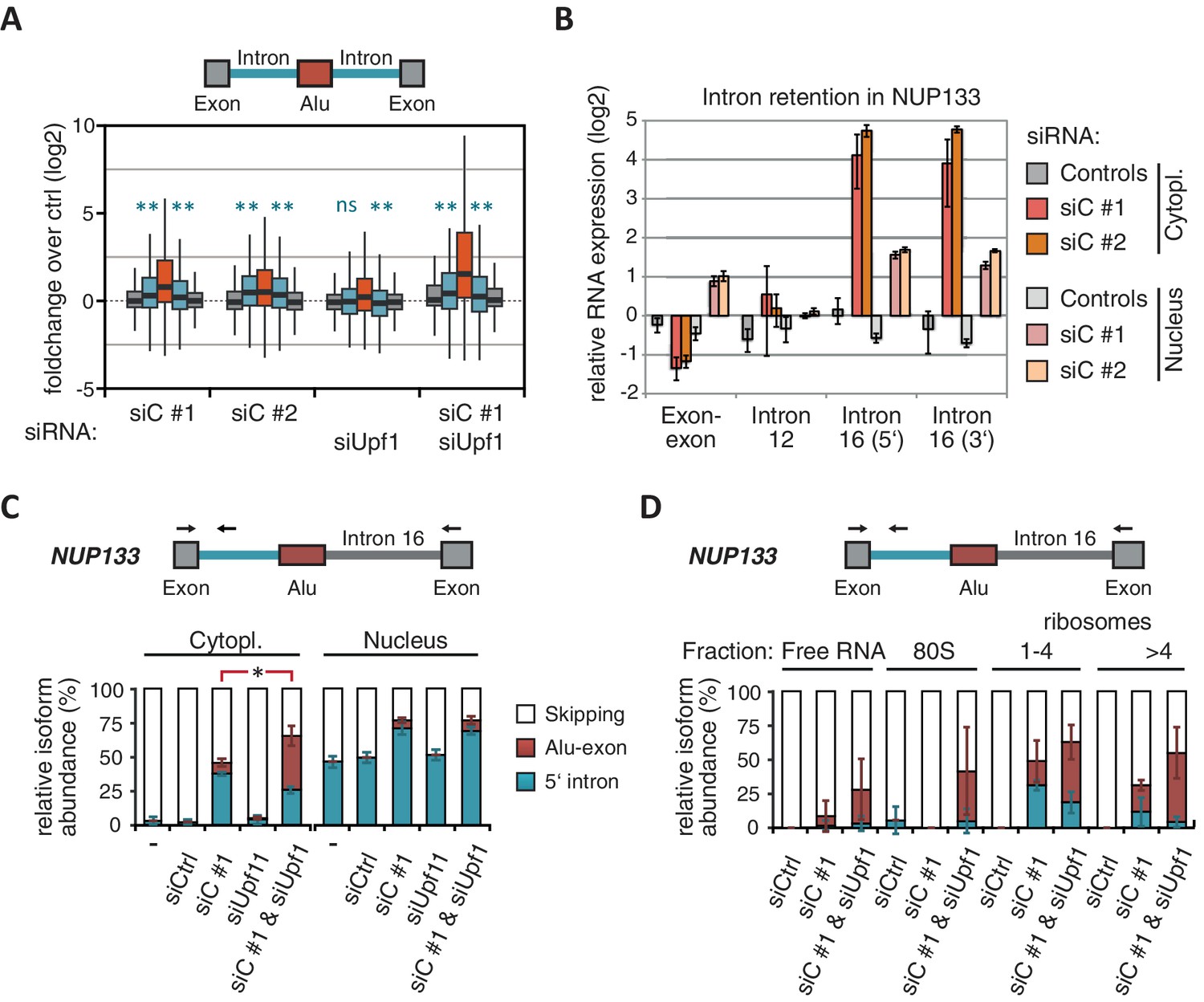
NMD-refractory transcripts are cytoplasmic and polysome-associated.
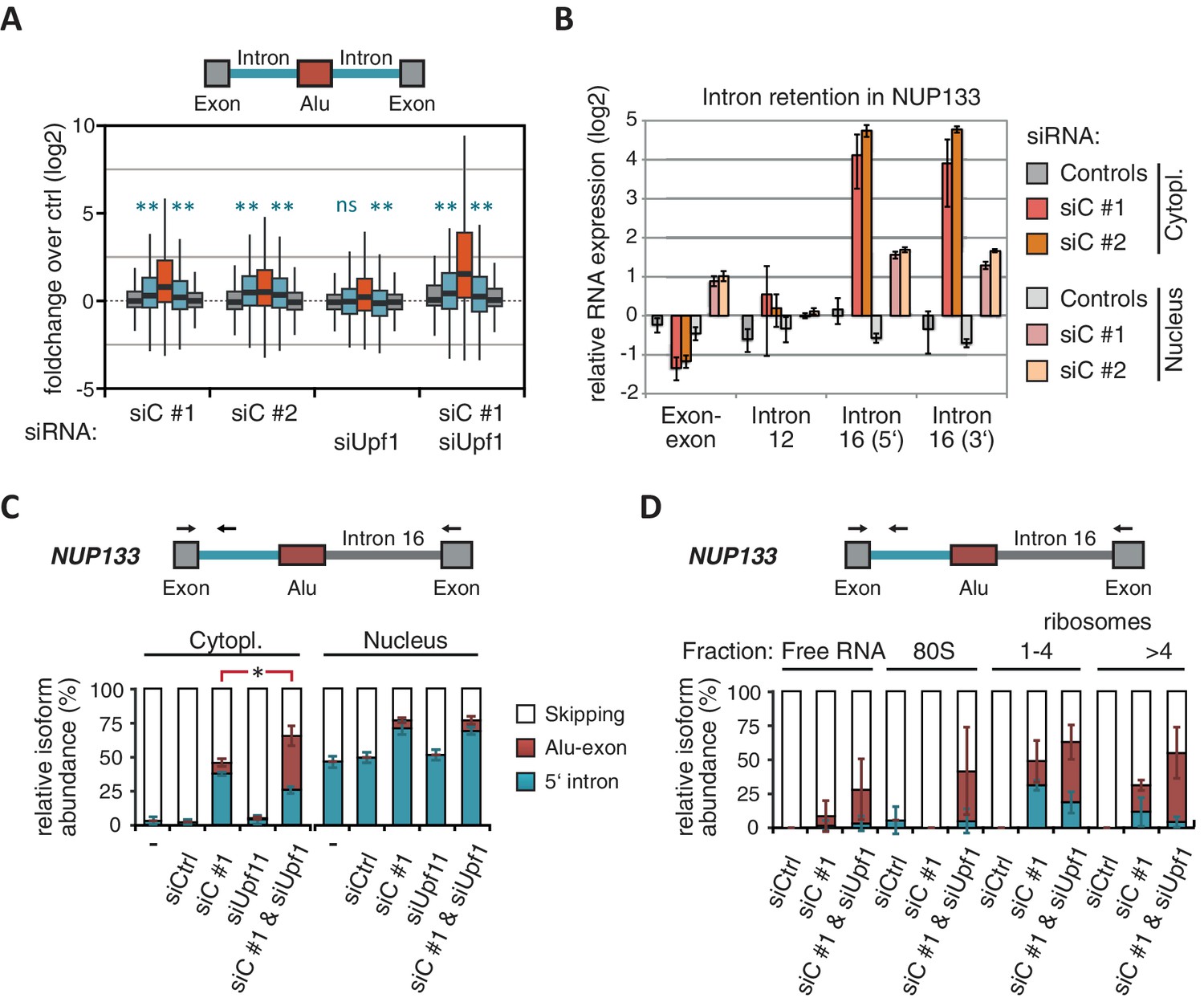
(A) Boxplot presenting the fold changes (log2) of Alu-exons as well as their two flanking introns and downstream and upstream exons upon hnRNPC or UPF1 depletion over control, as analysed by DEXSeq. Only significantly regulated Alu-exons from protein-coding genes were selected (n = 746, adjusted p-value < 0.01). To test for significant differences in intron retention, distribution of log2 fold changes was tested against the null distribution with a two-sided Wilcoxon Rank Sum test. ** indicates p-value < 0.01. Note that the downstream intron in UPF1-depleted samples was significantly less often retained than in control, with median of -0.13 and 99% confidence interval (-0.20,–0.06). For hnRNPC-depleted samples, 99% confidence intervals confirmed a significant increase in retention. (B) Abundance of the introns flanking the Alu-exon in NUP133 was measured by quantitative RT-PCR. As control, the overall mRNA abundance was quantified by an exon-exon-spanning primer, and abundance of the Alu-exon-free intron 12 was measured for comparison. Quantifications were performed for cytoplasmic and nuclear RNA of cells depleted of hnRNPC (siC #1 and #2), or control cells (no siRNA and control oligonucleotide) and normalised to the abundance of control mRNAs (eIF4G and SDH) in control cells (by ΔΔCt). Data are plotted as average of three independent replicates, error bars represent s.d.m. (C) The relative abundance of the intron-retaining Alu transcript of NUP133 was measured in cytoplasmic and nuclear RNA samples as in (B), with abundance being measured relative to the Alu-exon transcript and the constitutively spliced transcript by semi-quantitative RT-PCR. To test for significance, one-way ANOVA was performed separately for cytoplasmic and nuclear samples. Multiple comparison correction was done according to Tukey’s HSD. * indicates p-value < 0.05. Semi-quantitative RT-PCR analysis is averaged across three independent replicates, error bars represent s.d.m. (D) The relative abundance of the intron-retaining Alu transcript of NUP133 was measured in RNA fractions from polysome gradients of cells depleted of hnRNPC (siC #1), or UPF1 and hnRNPC (siC + siUPF1), or control cells (no siRNA and control oligonucleotide). For details on fractions from polysome profile, see Figure 3—figure supplement 2D. Semi-quantitative RT-PCR analysis is averaged across three independent replicates, error bars represent s.d.m. Figure 3—figure supplement 1 presents our analysis on retention of introns, which are flanking Alu-exons, and all other introns. Figure 3—figure supplement 2 shows additional evidence that NMD-refractory transcripts are cytoplasmic and polysome-associated. Figure 3—figure supplement 3 presents RNAseq traces as examples for intron retention in three genes, NUP133, GMPS and C8ORF76.
Figure 3—figure supplement 1
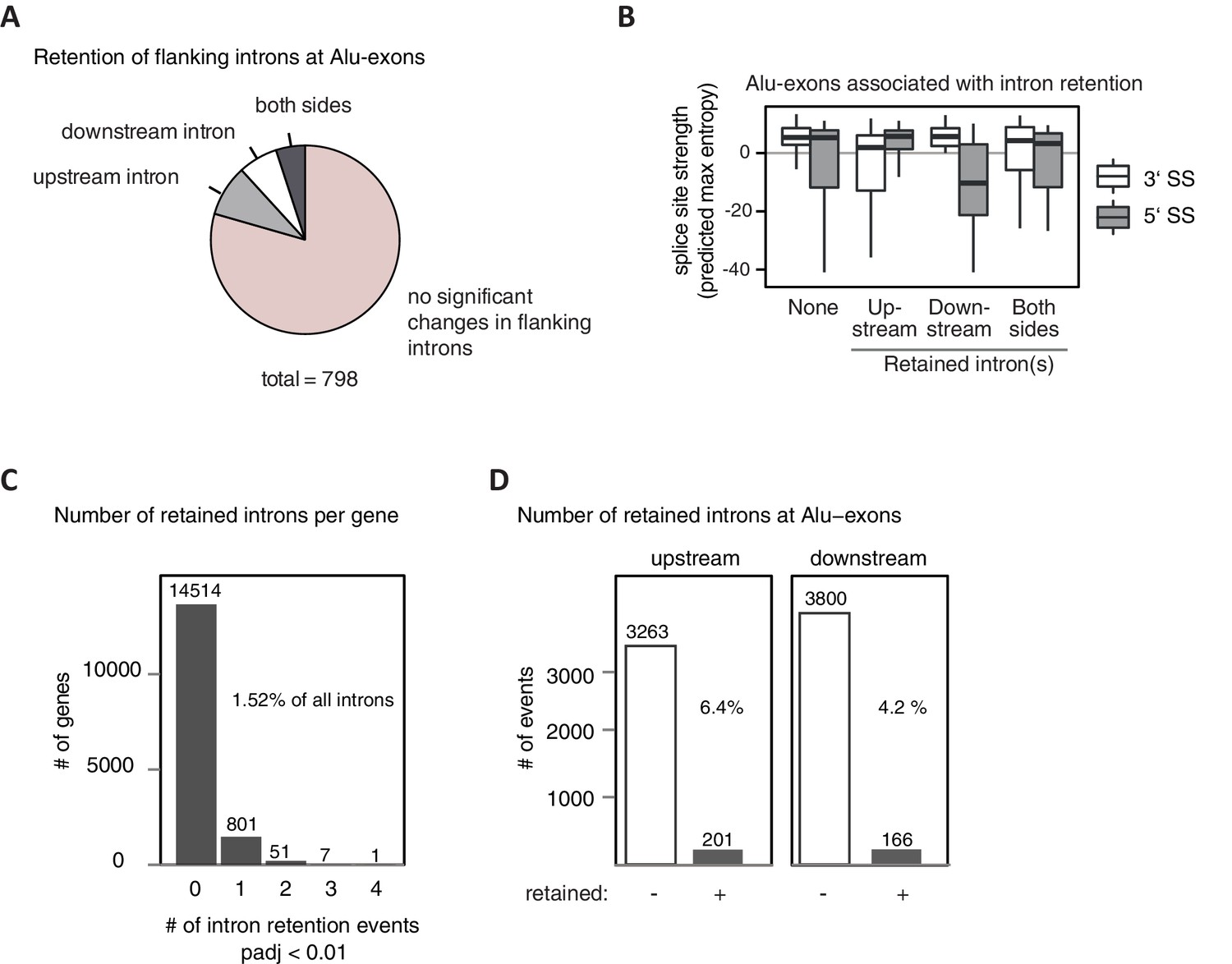
Alu-exons with weak splice sites cause formation of intron transcripts.
A modified DEXSeq approach was used to test for significant intron retention (for details, see Methods) in hnRNPC/UPF1 co-depleted cells, covering specifically introns flanking Alu-exons (A and B) or any intron in cells depleted of hnRNPC (C and D). (A) We selected Alu-exons with junction-spanning reads and tested if the intron flanking the Alu-exon is significantly retained in cells co-depleted of hnRNPC and UPF1 (adjusted p-value < 0.01). (B) All Alu-exons were according to which of the flanking introns was retained (upstream or downstream intron, both or none). Shown is the maximum entropy score of the 5' and 3' splice site (SS) of each Alu-exon within the four groups, predicted based on nucleotide sequence (Yeo and Burge, 2004). Similar results were observed if Alu-exons were filtered for junction-spanning reads (data not shown). (C) Retention of all introns not flanking an Alu-exon was monitored and the number of genes with a given number of significantly retained introns is shown. In total, our approach detected sequence retention for 1.52% of introns in hnRNPC-depleted cells. padj: adjusted p-value. (D) All introns flanking an Alu-exon (upstream or downstream) were tested as in (C). Introns flanking Alu-exons were much more likely to be retained than introns not flanking an Alu-exon. Retention is scored based on adjusted p-value < 0.01.
Figure 3—figure supplement 2
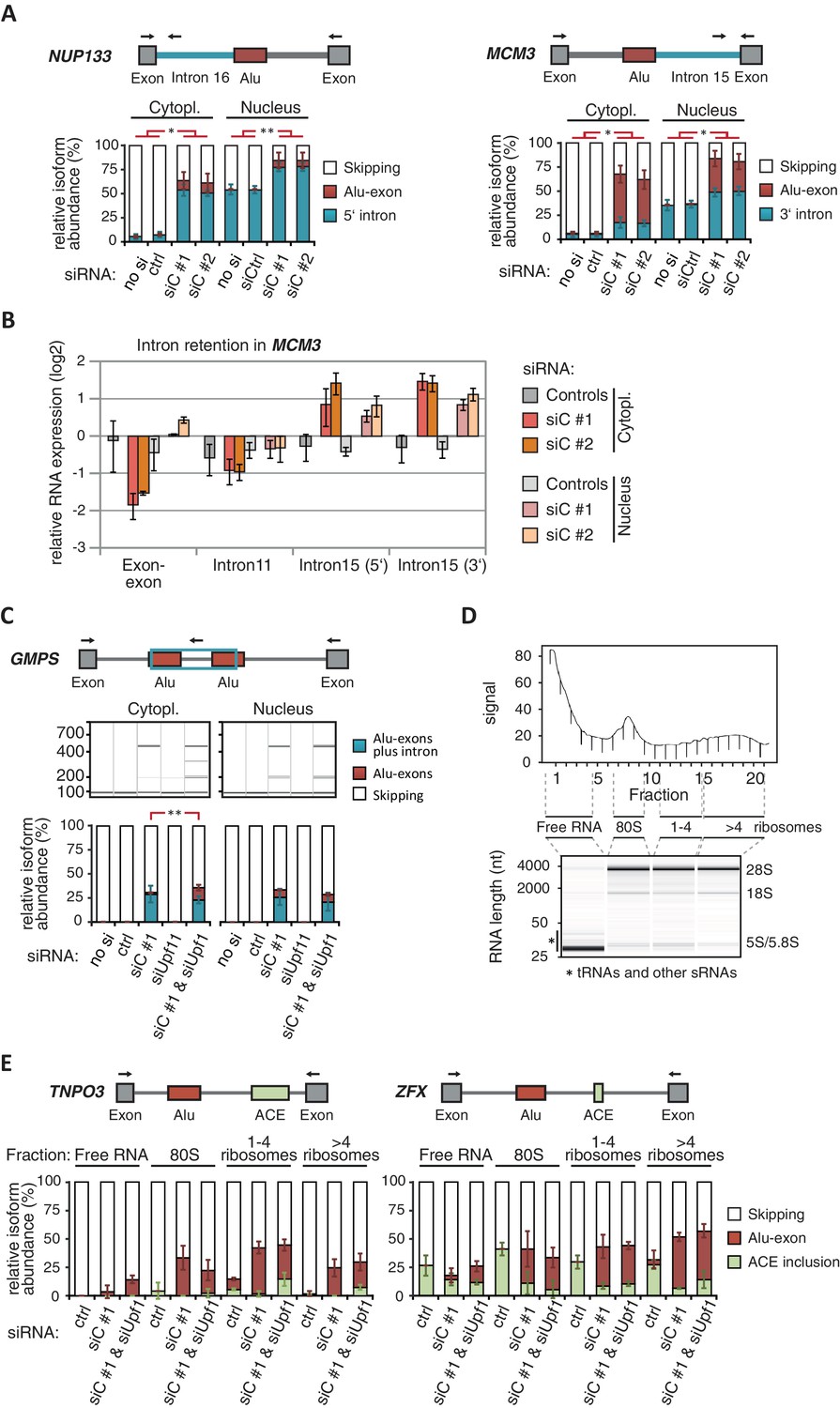
Cytoplasmic NMD-refractory transcripts.
(A) The relative abundance of the intron-retaining Alu transcripts of MCM3 and NUP133 were measured in cytoplasmic and nuclear RNA from cells depleted of hnRNPC (siC #1 and #2), or control cells (‘no si’, no siRNA; siCtrl, control oligonucleotide). Semi-quantitative RT-PCR analysis is averaged across independent biological replicates (MCM3: n = 5, NUP133: n = 2), error bars represent s.d.m. (B) RNA abundance of the introns flanking the Alu-exon in MCM3 was measured by quantitative RT-PCR. As control, the overall mRNA abundance was quantified by an exon-exon spanning primer, and the abundance of the Alu-exon free intron 11 was measured in parallel. Cytoplasmic and nuclear RNA of cells depleted of hnRNPC (siC #1 and #2), or control cells (no siRNA and control oligonucleotide), and normalised to abundance of abundant RNAs (using ΔΔCt, control RNAs were eIF4G and SDH mRNAs). Shown are the averages across independent replicates (n = 3–4), error bars represent s.d.m. (C) The relative abundance of the intron-retaining Alu transcript of GMPS was measured by semi-quantitative RT-PCR in cytoplasmic RNA from control, hnRNPC- and UPF1-depleted cells. Shown are gel visualisations of capillary electrophoresis and quantification of average Alu-exon inclusion across independent biological replicates (n = 3), error bars represent s.d.m. (D) A representative image of absorption at 230 nm is shown for the polysome profiles used in Figure 3D and in (E). Fractions from the polysome sedimentation were pooled as indicated. Bioanalyzer traces run with 100 ng RNA of each pool are shown below, demonstrating that ‘free RNA’ contains very little rRNA. (E) The relative abundance of the NMD-refractory Alu-exons in TNPO3 and ZFX were measured in RNA fractions from polysome gradients as in Figure 3D.
Figure 3—figure supplement 3
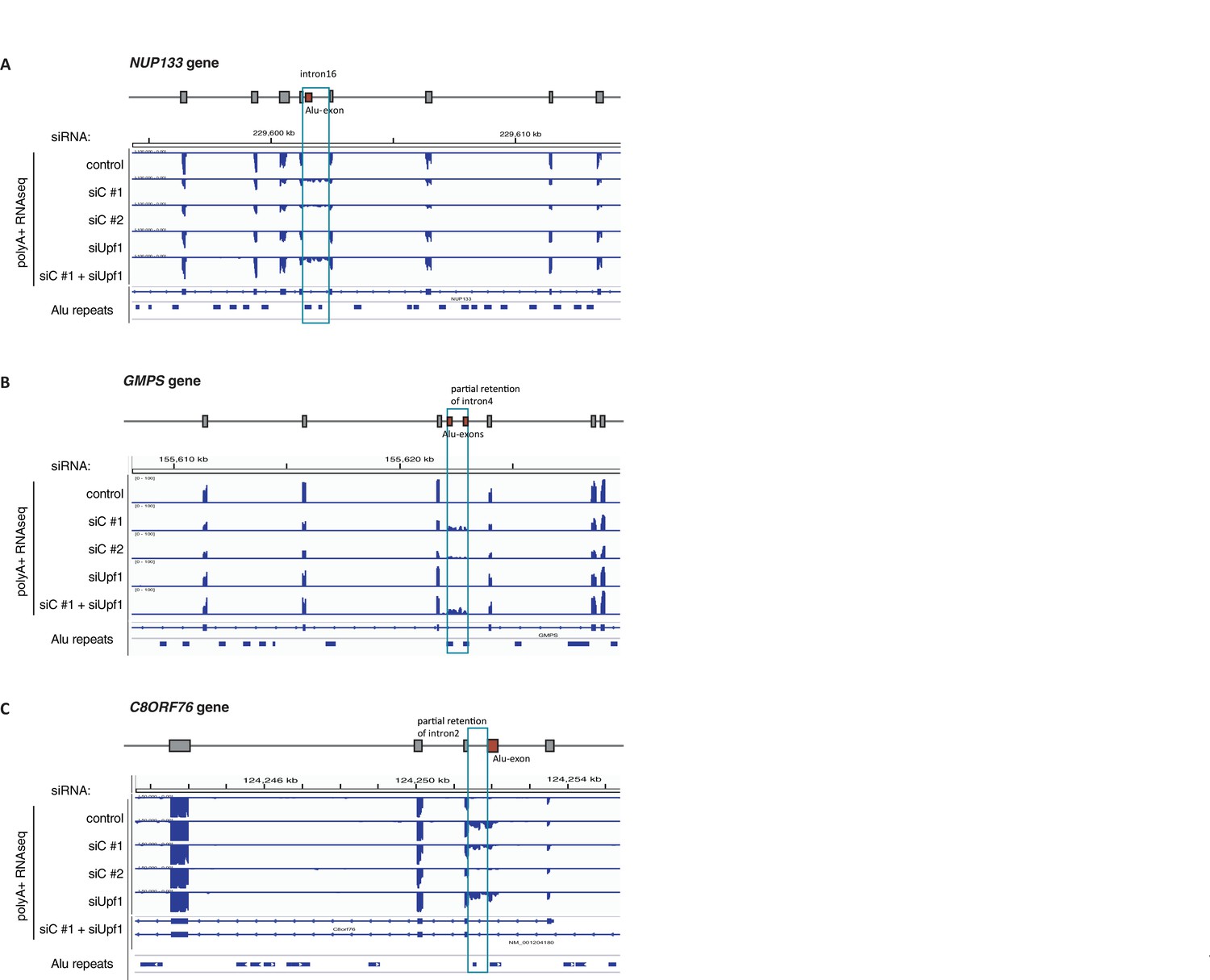
Examples of intron-retaining Alu transcripts.
RNAseq data were visualised with IGV. Data were generated from polyA-selected RNA from cells depleted of hnRNPC (siC #1 and #2), UPF1 (siUPF1), or UPF1 and hnRNPC, or control cells (unspecific control oligonucleotide). All traces are scaled according to the total number of reads in each sample. (A) An Alu-exon in intron 16 of the NUP133 gene is associated with intron. The Alu-exon is validated by junction-spanning reads on both sides. Note that although hnRNPC/UPF1 co-depletion restores overall expression of NUP133 in hnRNPC-depleted cells, the coverage of the intron region is not changing compared to the flanking exons, confirming that the intron-retaining Alu transcript itself is not depleted by NMD. (B) Two Alu-exons in intron four of the GMPS gene are associated with partial intron. Both Alu-exons are validated by junction-spanning reads at the 3' splice site while junction-spanning reads at the 5’ splice sites of the Alu-exons are absent. Both exons can be detected by PCR (see Figure 3—figure supplement 2C). Note that although hnRNPC/UPF1 co-depletion restores overall expression of GMPS in hnRNPC-depleted cells, the coverage of the intron region is not changing compared to the flanking exons, confirming that the intron transcript itself is not depleted by NMD. (C) An Alu-exon in intron two of the C8ORF76 gene is associated with intron. The Alu-exon is validated by junction-spanning reads at the 3' splice site but not at the 5' splice site.
Figure 4 with 2 supplements
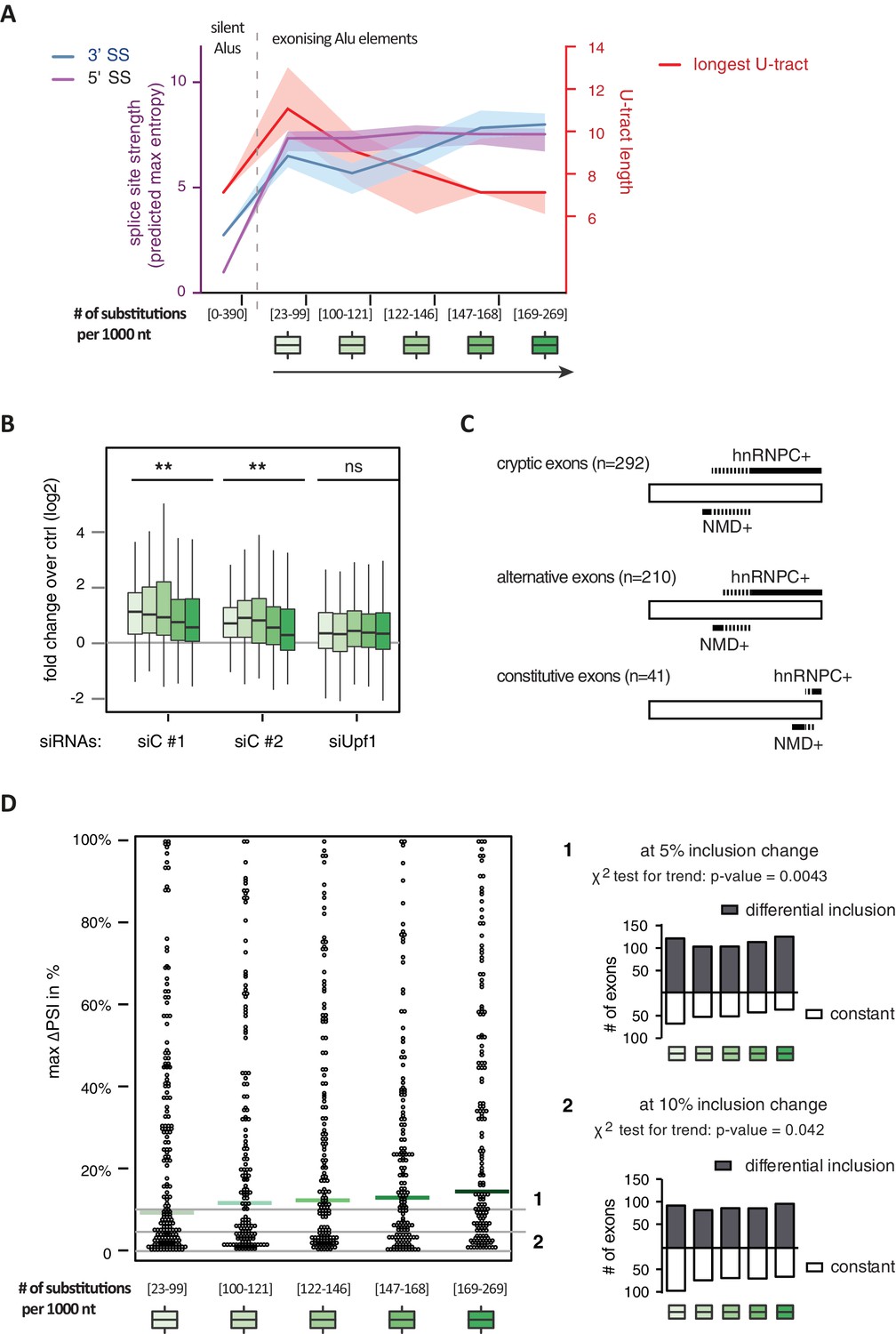
Alu-exon age correlates with loss of U-tracts and repression by hnRNPC.
All 798 Alu-exons validated by junction-spanning reads at either splice site were stratified by the number of substitutions from the Alu consensus sequence provided by RepeatMasker (Xiao et al., 2009), as proxy for their evolutionary age, into five groups of roughly equal size (156–162 exons) with [23-99], [100-121], [122-146], [147–168] and [169–269] substitutions per 1000 nucleotides. (A) The median splice site strength and U-tract length of the different age groups of Alu-exons are shown on the left and right y-axis, respectively (purple, blue and red lines). 95% confidence intervals of the median were estimated by bootstrapping, and are plotted as area above and below the medians. The distributions of the splice site strengths and U-tract lengths in each group are shown in Figure 4—figure supplement 2A and B. For comparison, the median splice site strengths and U-tract lengths of all non-exonising (‘silent’) Alu elements are shown. (B) Changes in Alu-exon abundance of each group upon hnRNPC or UPF1 depletion as log2 fold changes (log2fc) over control, analysed by DEXseq. The correlation between substitutions in the Alu element and log2fc of each exon was tested by a linear model. ** indicates p-value < 0.01. (C) Depiction of the number of hnRNPC- and NMD-sensitive exons. Dashed lines visualise proportion of exons regulated by both hnRNPC and NMD. Only Alu-exons with an adjusted p-value < 0.01 are shown as hnRNPC+ or NMD+, Alu-exons with an adjusted p-value > 0.1 are assigned as non-regulated exons, and all other Alu-exons are ignored. (D) To test for differential splicing of Alu-exons across human tissues, we analysed GTEx expression data (GTEx Consortium, 2015) and calculated percent exon inclusion (PSI) values. For each exon with sufficient coverage by junction-spanning reads (n = 1039), the difference between the tissue with the lowest and highest inclusion is shown (max. ΔPSI), stratified by Alu-exon divergence. The median is shown as coloured line in each group. To illustrate the differences between Alu-exon groups, two arbitrary thresholds for ‘differential inclusion’ (5% and 10%) were used to set the number of exons that are differentially included or constant (right side). The substitution groups were then treated as contingency tables to test for differences in the number of differentially included exons with a χ2 test for trend. In Figure 4—figure supplement 1, we quantify the relationship between U-tract length and repression by hnRNPC on Alu-exons. Figure 4—figure supplement 2 presents the complete distribution of the data on 3' and 5' splice site strength, U-tract lengths and log2 fold changes summarised in Figure 4A–C.
-
Figure 4—source data 1
Percent exon inclusion of Alu-exons in different human tissues.
We list percent exon inclusion for each Alu-exon with sufficient coverage across the GTEx data (889 exons with at least 4000 junction-spanning reads across 8555 individual samples). In addition, the data table includes chromosome position of the exon, mean skipping counts, the maximum and minimum inclusion across the tissues, the maximum difference in exon inclusion across the tissues, strength of the 3' splice site. In human, the length of the longest U-tract, substitutions in the Alu elements and substitution group. If no junction-reads were detected in a tissue extending from the upstream directly to the downstream exon ('skipping junctions'), we report 100% exon inclusion in this tissue.
- https://doi.org/10.7554/eLife.19545.015
Figure 4—figure supplement 1
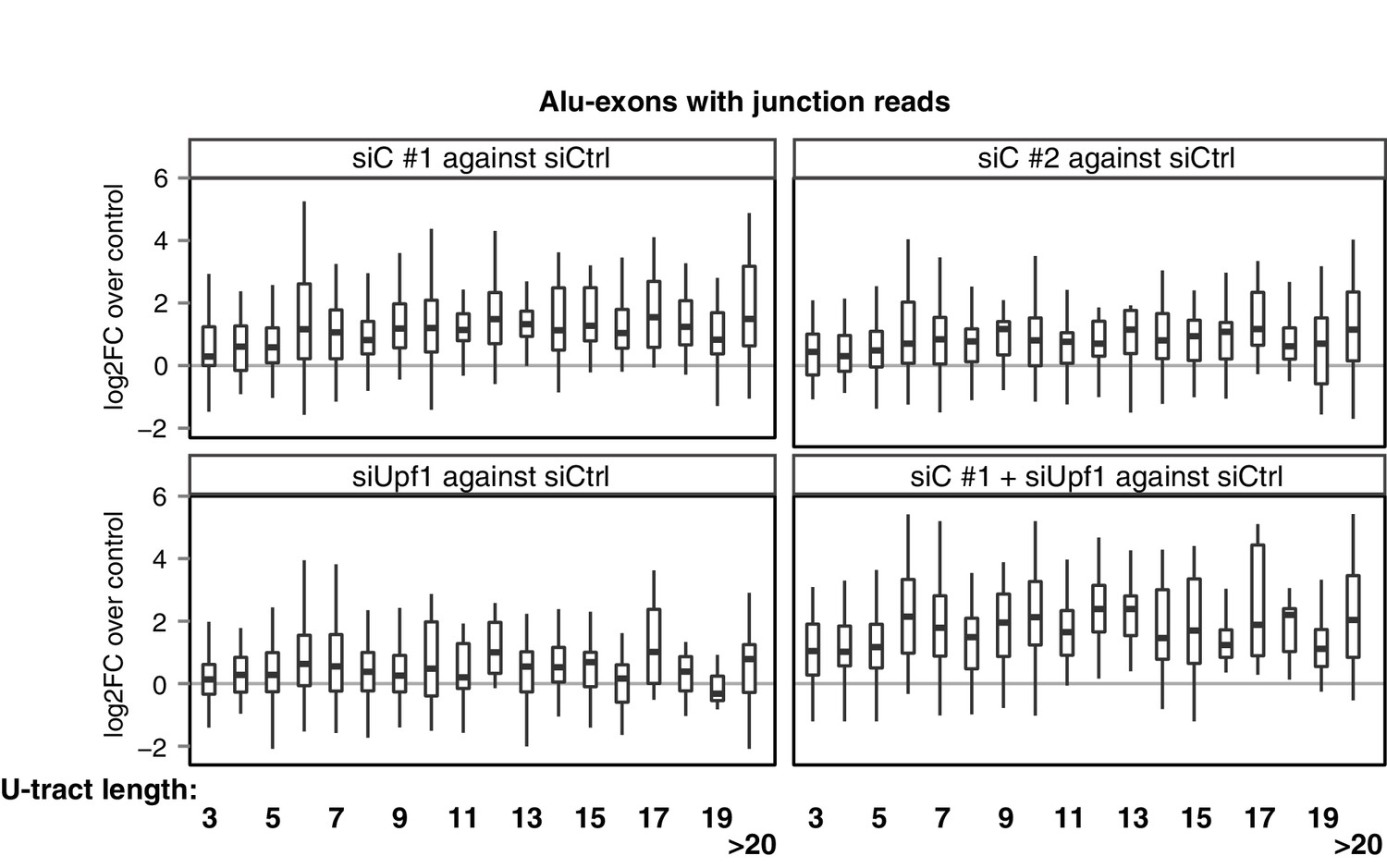
Repression of Alu-exons in dependence of U-tract length.
All Alu-exons in protein-coding genes that are validated by junction-spanning reads at both the 5' and the 3' splice site (SS) were selected. Boxplots showing the changes in exon abundance upon hnRNPC and/or UPF1 depletion as fold changes (log2; log2FC) over control, in dependence of the length of the U-tract preceding the 3' splice site. U-tracts ranged from 3 to 30 continuous uridine nucleotides, with exons preceded by U-tracts of 20 or more uridines grouped together.
Figure 4—figure supplement 2
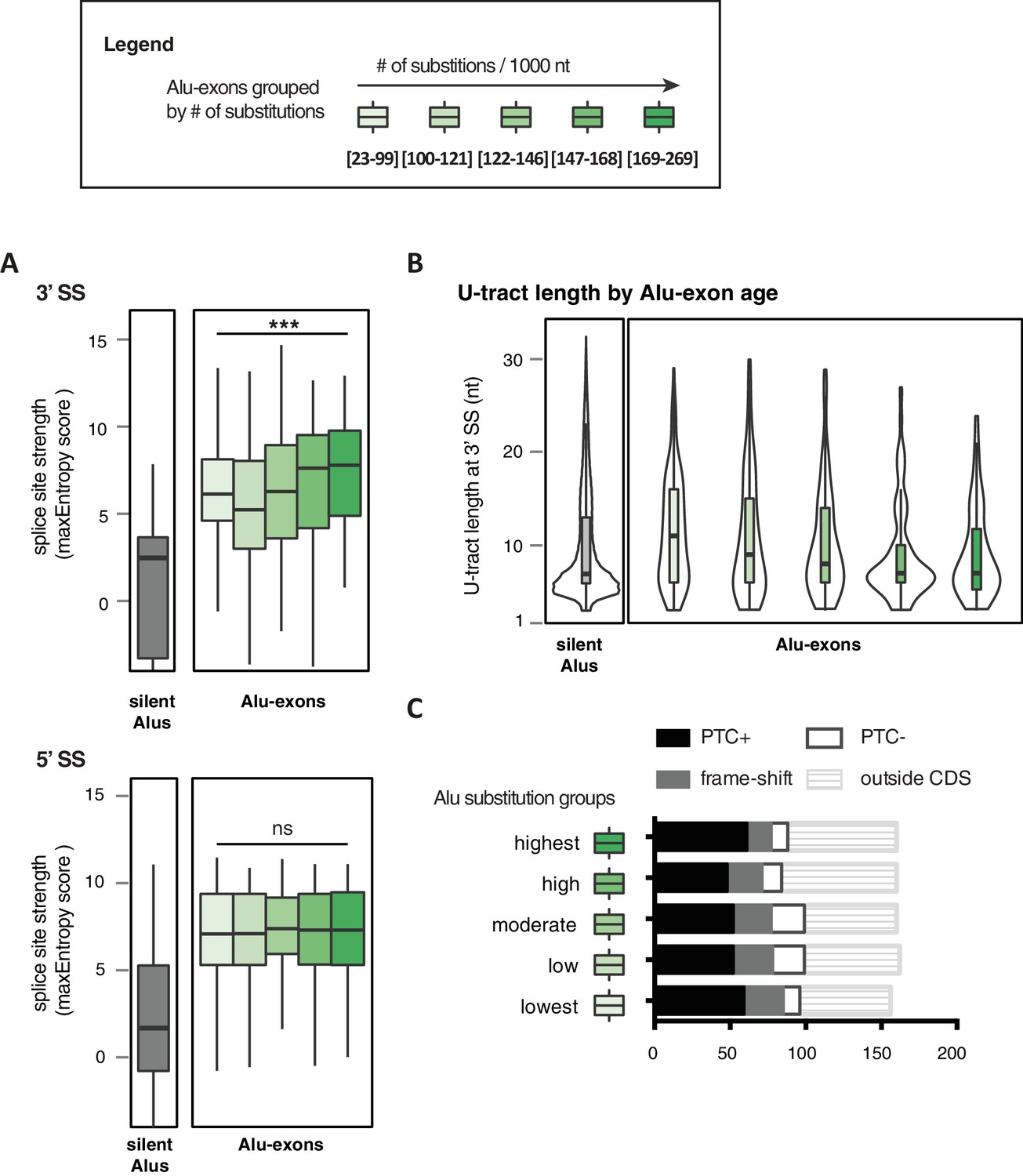
Features of Alu-exons grouped by substitution rate.
All Alu-exons in protein-coding genes that were validated by junction-spanning reads at both the 5' and the 3' splice site (SS; 798 exons in total, were stratified by the number of substitutions from the Alu consensus sequences annotated by RepeatMasker (Smit et al., 1996-2010), as proxy for evolutionary age (A) The maximum entropy score of 5' and 3' splice sites of each exon was predicted based on nucleotide sequence (Yeo and Burge, 2004). The correlation between substitutions and splice site score was tested by a linear model, *** indicates p-value < 0.001. For comparison, the distribution of silent Alu elements is shown in grey; for visualisation, the lower whisker of the silent Alu elements is cut-off at −4. Actual lower whisker values are −7.9 at 5' SS and −13.2 at 3' SS. (B) The length of the U-tract at the 3' splice site of each Alu-exon is shown. For comparison, we show the length of the longest U-tract in silent Alu elements (‘silent’ Alu elements, i.e. non-exonising). Only U-stretches within Alu elements were considered. Thirteen of 798 Alu-exons were removed in (C) because no U-tract of at least three nucleotides was found in the Alu element sequence upstream of the 3' splice site. These exons are likely to utilise a polypyrimidine tract in the close-by genomic sequence. (C) Alu-exons arising from highly diverged Alu elements are not depleted of PTCs. Shown is the number of Alu-exons in each group, which do or do not contain a PTC, or potentially cause a frame-shift, as well as those which are outside of the coding region of the transcript.
Figure 5 with 1 supplement
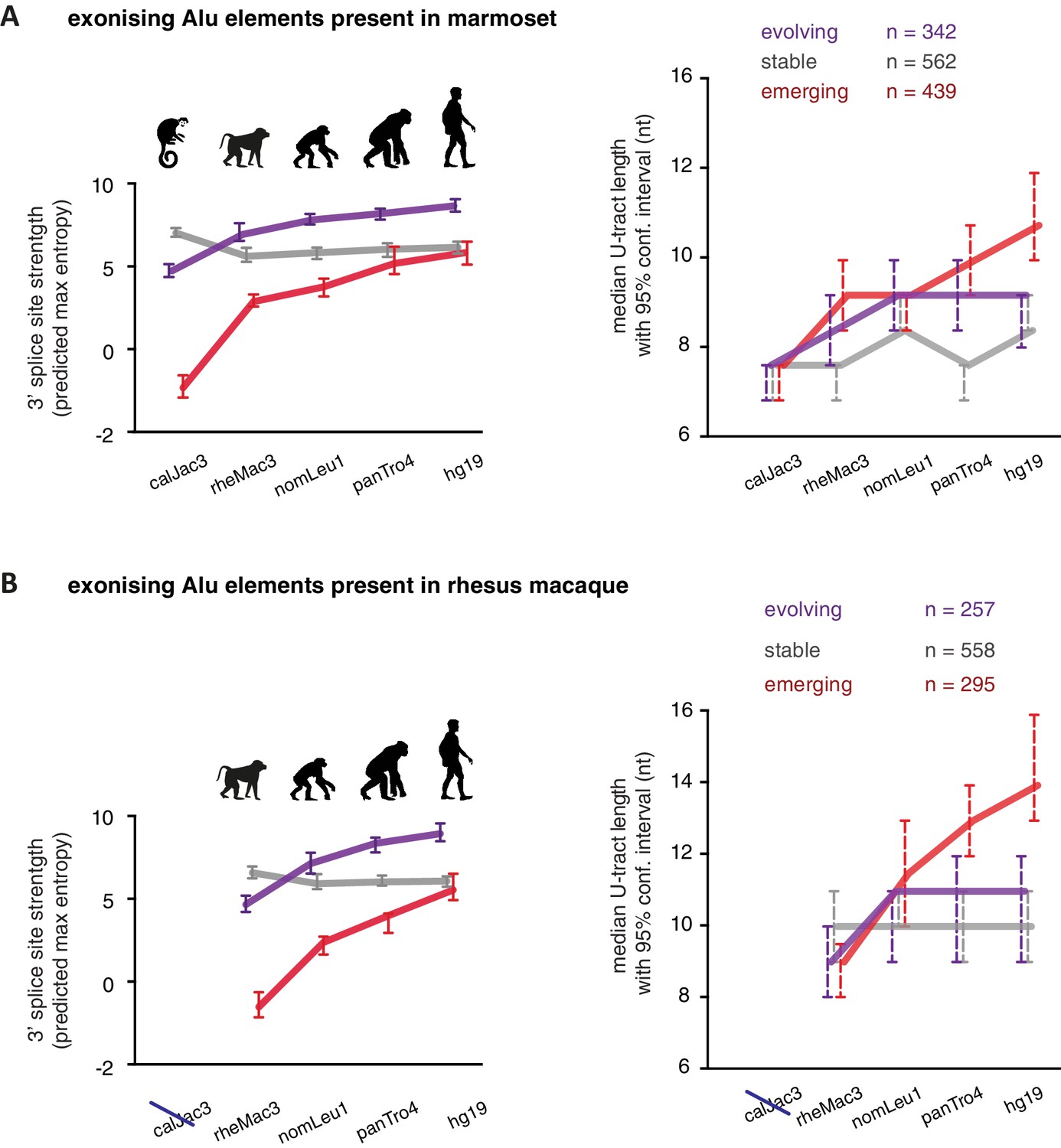
U-tracts of emerging Alu-exons lengthen in parallel to an abrupt gain of 3' splice site sequences.
For all known Alu-exons found in this study or annotated in UCSC, orthologous regions were identified in four other primate genomes and scanned for the presence of an Alu element and a 3' splice site therein. We used the following genomes: hg19 (human), panTro4 (chimpanzee), nomLeu1 (gibbon), rheMac3 (rhesus macaque) and calJac3 (marmoset). Alu-exons were split into three groups depending on the time point of 3' slice site emergence: Exons with an existing 3' splice site and little change in splice site strength across the five species were considered as ‘stable’, exons with an existing 3' splice site that gained strength towards the human lineage were considered as ‘evolving’, and exons in which the 3' splice site emerged after the split of Old and New World monkeys were considered as ‘emerging’. (A) Alu-exons with orthologues present in marmoset (calJac3) were classified as described. Progression in predicted 3' splice site strength of the three groups of Alu-exons is shown on the left. Median lengths of the longest U-tract of the Alu elements in each species are shown on the right. Emerging Alu-exons were present in marmoset but only acquired a 3' splice site later in the evolutionary history of the primates. All data are presented as 95% confidence intervals of the median estimated by bootstrapping. (B) Alu-exons with orthologues present in rhesus macaque (rheMac3) but not in marmoset were classified as described. Predicted 3' splice site strength and median U-tract length in each species as in (A). Emerging Alu-exons were not present in marmoset but in rhesus macaque, and acquired a 3' splice site later in the evolutionary history of primates. All data are presented as 95% confidence intervals of the median estimated by bootstrapping. Figure 5—figure supplement 1 presents the complete distribution of U-tract lengths summarised in Figure 5A and B, as well as the data on the evolutionary youngest Alu-exons specific to the hominoidae lineage.
-
Figure 5—source data 1
List of Alu-exons across our datasets and UCSC annotation, including cross-species annotation.
We merged all Alu-exons identified from RNAseq data and annotated in UCSC (6731 exons), and filtered for non-overlapping exons. If any two exons overlapped, we retained the larger exon. This created a list of 6309 Alu-exons for which we selected the 3' splice site and mapped orthologues positions in four primate genomes. For each 3'splice site (if present), we predicted the splice site strength using MaxEntScan (Yeo and Burge, (2004) and searched for the longest U-tract within the Alu element. The table lists in order: Alu-exon co-ordinates (in hg19), and the annotation by UCSC as alternative or constitutive exon, if not present the exon is annotated as cryptic exon. Next, the furthest species in which we could identify the orthologous position, the exonisation group of the 3' splice site as defined in Figure 5 (see Materials and methods for details), the coordinates, repeat family, substitution group of the Alu element the exon arises from (in hg19), as well as the position, predicted strength of the 3' splice site and longest U-tract of the Alu element in human (hg19), in chimpanzee (panTro4), in gibbon (nomLeu1), in rhesus macaque (rheMac3) and in marmoset (calJac3). All 3' splice site positions start with the −2 position of the canonical 3' splice site (the AG nucleotide) consensus.
- https://doi.org/10.7554/eLife.19545.019
Figure 5—figure supplement 1
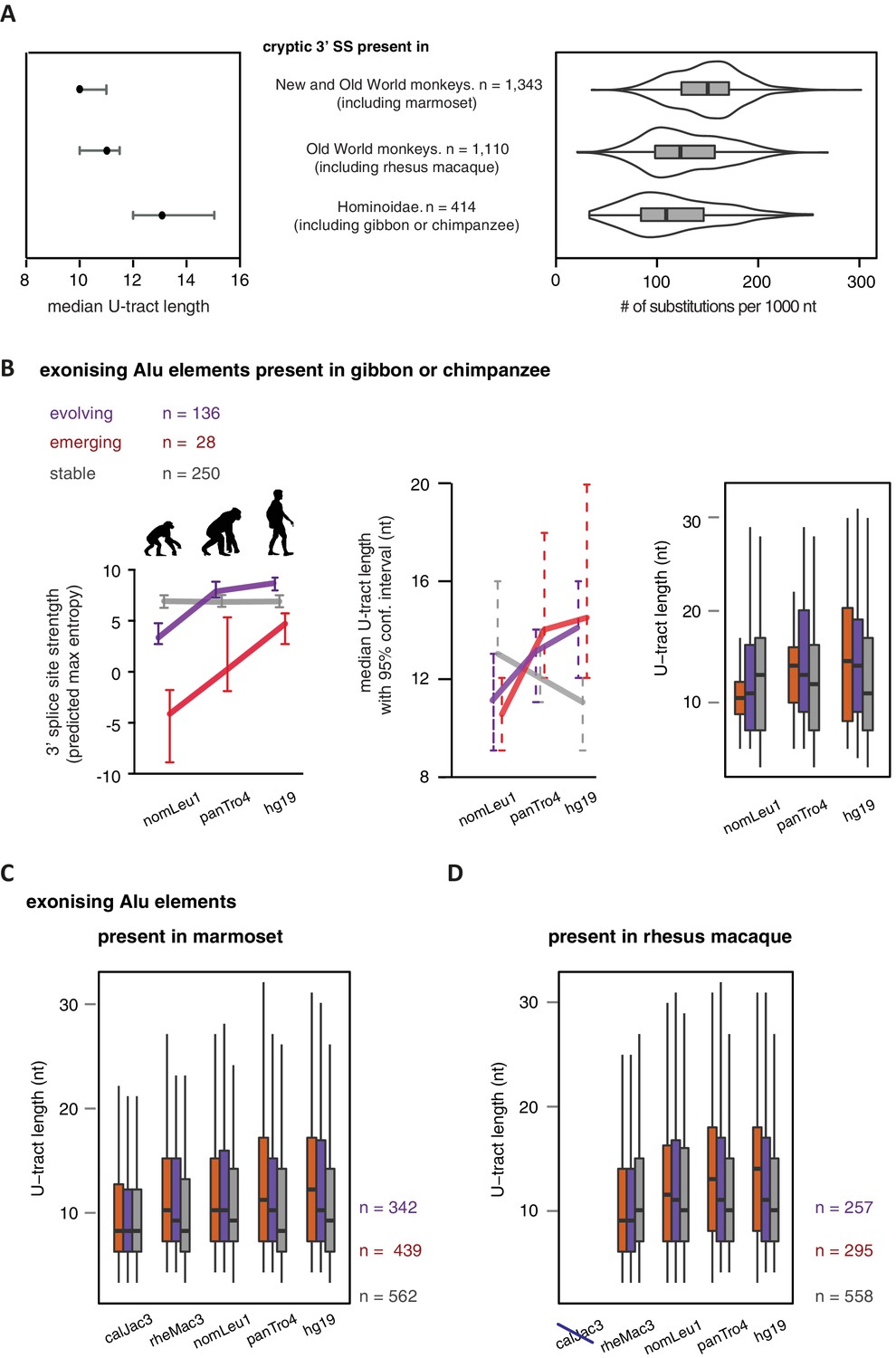
U-tract length of Alu-exons traced throughout primate orthologues.
We searched for the orthologues of all Alu-exons with a strong 3' splice site in human, either annotated in UCSC or described in this study (2867 Alu-exons in total). We used the following genomes: hg19 (human), panTro4 (chimpanzee), nomLeu1 (gibbon), rheMac3 (rhesus macaque) and calJac3 (marmoset). (A) Human Alu-exons were classified according to the time of emergence of their 3' splice site: (Deininger and Batzer, 2002) before Old and New World monkeys, (Lev-Maor et al., 2003) before split of hominoidea superfamily from other Old World monkeys, or (Sorek et al., 2004) only found in hominoidea. Left: The median length of the longest U-tract is shown with 95% confidence intervals estimated by bootstrapping. Right: Substitutions of the Alu elements from consensus is shown. (B) Alu-exons with orthologues present in gibbon or chimpanzee (nomLeu1 or panTro4, respectively) were classified as described in Figure 5, and progression in predicted 3' splice site strength of the three groups of Alu-exons is shown on the left. The longest U-tract of the Alu elements in each species was identified. U-tract length is shown in each species, as 95% confidence intervals of the median estimated by bootstrapping (middle) similar to Figure 5A and B and the underlying distribution of U-tract lengths as a boxplot (right side). (C) Shown is the distribution of U-tract lengths underlying the summarised data presented in Figure 5B. Alu-exons with orthologues present in marmoset (calJac3) were classified as described in Figure 5, and the longest U-tract of the Alu elements in each species was identified. (D) Shown is the distribution of U-tract lengths underlying the summarised data presented in Figure 5C. Alu-exons with orthologues present in rhesus macaque (rheMac3) were classified as described in Figure 5, and the longest U-tract of the Alu elements in each species was identified. Supplemental Dataset 1. List of exons with sufficient coverage for DEXSeq analysis in our RNAseq data. Deposited at the Dryad repository, doi:10.5061/dryad.7h81d. Source Data of Figure 4D. Percent exon inclusion of Alu-exons in different tissues. Source Data of Figure 5. List of Alu exons across our datasets and UCSC annotation, including cross-species annotation.
Figure 6
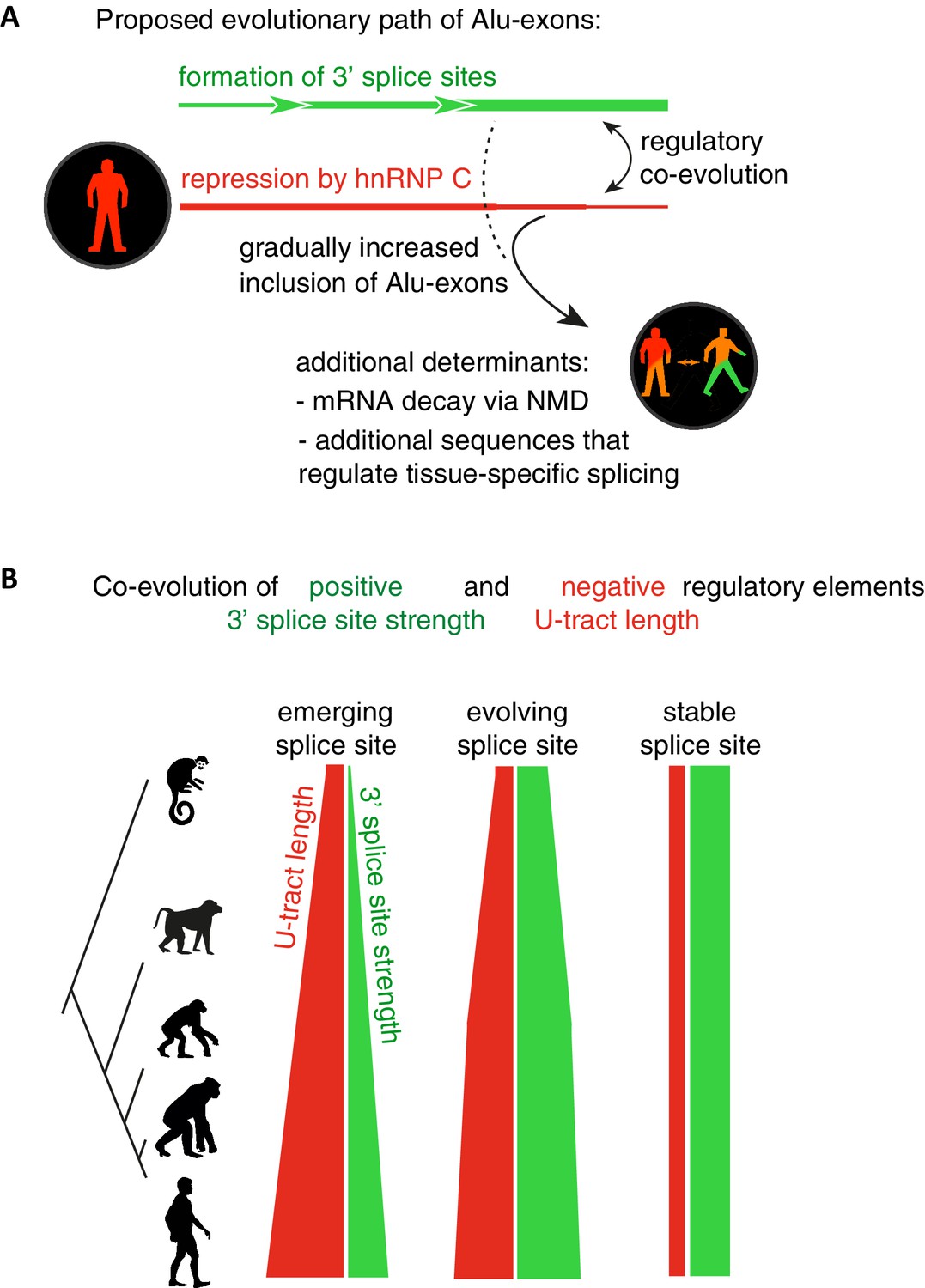
Repressive elements co-evolve with splice site sequences at cryptic exons.
Alignment within primate genomes of Alu elements that contain human Alu-exon reveals a tight coupling between repressive U-tracts and the variation of 3' splice site strength. (A) As the strength of 3' splice sites increases, the repressive U-tracts are lengthened, which recruits hnRNPC to prevent splicing of Alu-exons. Hence, the splice site sequence and the repressive U-tract undergo an evolutionary dynamic that we refer to as regulatory co-evolution. Splice site sequences without a nearby repressive element are depleted, likely due to strong negative selection. Selection pressure for long U-tracts decreased for as-yet unknown reasons at the more ancient Alu-exons (those that contain a splice site in a distant primate species, or their sequence diverges from the Alu consensus), and these exons are less repressed by hnRNPC and have an increased incidence of tissue-specific splicing. At this stage, abundance of the Alu-exon isoform is determined also by its ability to trigger NMD, and likely other factors such as tissue-specific splicing factors. (B) Based on the variation of the 3' splice sites between primate species, we characterised three evolutionary groups of Alu-exons. Most human Alu-exons have stronger 3' splice sites in human compared to New World monkeys, and these exons also have longer repressive U-tracts in human. These exons are split according to the evolutionary trajectory of their 3' splice site sequence into emerging, evolving or stable 3' splice sites. The most ancient Alu-exons, which have a strong and stable 3' splice site, lack any trend towards longer U-tracts. This demonstrates that tight coupling between positive and negative splicing elements establishes a balanced regulatory environment at the newly emerging exons.
Author response image 1
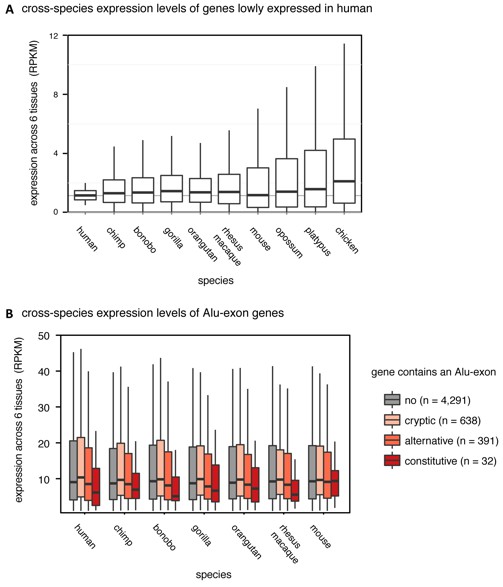
(A) We selected the ~15% most lowly expressed genes across six human tissues (RPKM of 0.5-2, n=2,199), and compared their expression levels (RPKM) in other species. Data from Brawand et al. (Nature 2011). (B) We compared expression levels (RPKM) of Alu-exon genes and their mammalian orthologues in primates and mouse. Species abbreviated as in Figure 1.
Additional files
-
Supplementary file 1
List of RT-PCR and RT-qPCR primers used in this study.
- https://doi.org/10.7554/eLife.19545.022
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Splicing repression allows the gradual emergence of new Alu-exons in primate evolution
eLife 5:e19545.
https://doi.org/10.7554/eLife.19545




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}