Recombination, meiotic expression and human codon usage
- Université de Lyon, Université Claude Bernard, France
- UnivLyon, ENS de Lyon, Univ Claude Bernard, CNRS UMR 5239, INSERM U1210, Laboratoire de Biologie et Modélisation de la Cellule, France
Figures
Figure 1

Variation in synonymous codon usage and in GC3 among functional categories.
(A) Factorial map of the principal-component analysis of synonymous codon usage in GO functional categories in the human genome. Each dot corresponds to a GO gene set, for which the relative synonymous codon usage (RSCU) was computed. GO categories that are associated with ‘differentiation’ or with ‘proliferation’ are displayed in blue and in red, respectively. (B) Correlation between the RSCU of GO gene sets (first PCA axis) and their average GC-content at third codon position (GC3). (C) Distribution of GC3 of human protein coding genes. Red: ‘proliferation’ genes (N = 1,008); blue: ‘differentiation’ genes (N = 2,833); grey: other genes (N = 12,129). (D) Correlation between the GC3 of mono-isoacceptor amino acids and multi-isoacceptor amino acids. For each GO gene set, the average GC3 was computed separately for amino acids decoded by multiple tRNA isoacceptors (N = 14 multi-isoacceptor amino acids), and for those decoded by one single tRNA isoacceptor (mono-isoacceptor amino acids: Phe, Asp, His, Cys). Amino-acids encoded by a single codon (Met, Trp) were excluded.
Figure 2 with 1 supplement
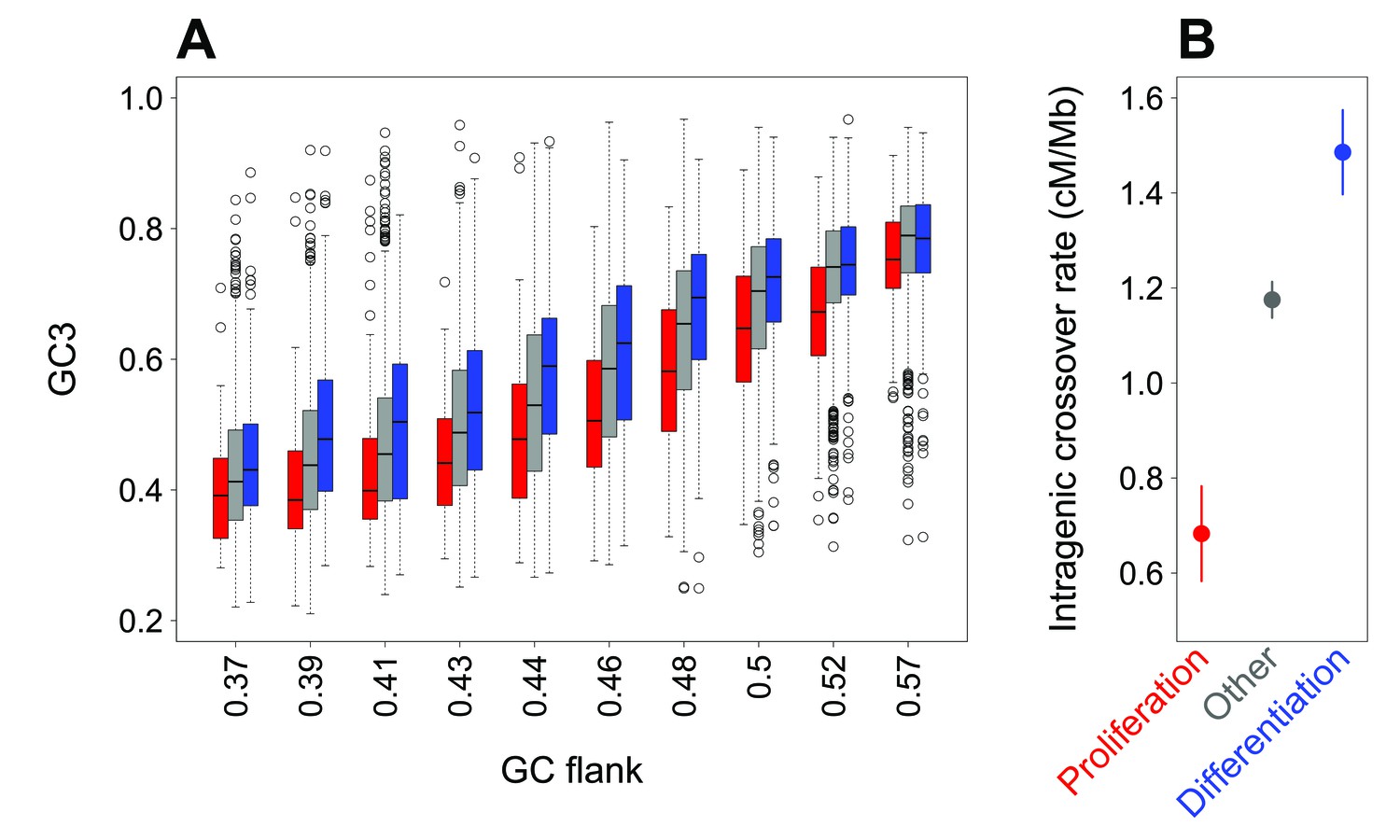
Difference in SCU between ‘proliferation’ and ‘differentiation’ genes is linked to variation in intragenic crossover rate, and not to their isochore context.
(A) Variation in gene GC3 according to the GC content of their flanking region (GC-flank) in each functional category. Genes were first binned into 10 classes of equal sample size according to their GC-flank, and then split into three sets according to their functional category: ‘proliferation’ (red), ‘differentiation’ (blue), and ‘other’ genes (grey). Boxplots display the distribution of GC3 for each functional category within each GC-flank bin. (B) Mean sex-averaged intragenic crossover rate (HapMap) in each functional category. Error bars represent the 95% confidence interval of the mean.
Figure 2—figure supplement 1
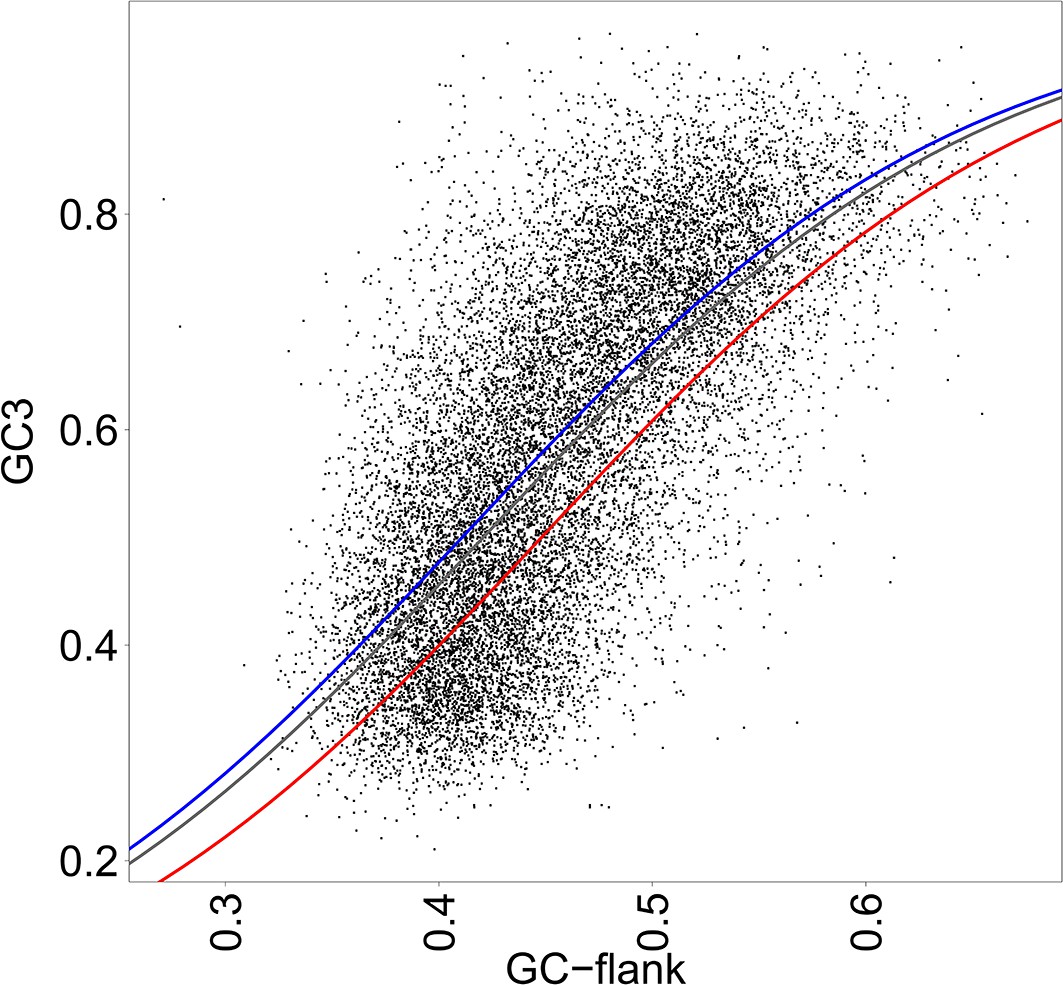
Correlation between the GC3 of genes and the GC content of their flanking regions (GC-flank).
Each dot corresponds to one gene. GC-flank was measured in 10 kb upstream and 10 kb downstream of the transcription unit. The curves show a generalized linear model (glm), predicting GC3 according to GC-flank and gene functional category. Glm is performed with a binomial logistic regression. The curves corresponding to ‘differentiation’ genes (blue), ‘proliferation’ genes (red) and other genes (grey), differ significantly (LRT of glm with and without gene function, p-values<2.10−16). Correlation coefficients were computed on logit transformed values, independently for ‘differentiation’ genes (N = 2,833, R2 = 0.46), ‘proliferation’ genes (N = 1,008, R2 = 0.48), other genes (N = 12,129, R2 = 0.49) and all genes (N = 15,970, R2 = 0.48). All p-values<2.10−16.
Figure 3 with 3 supplements
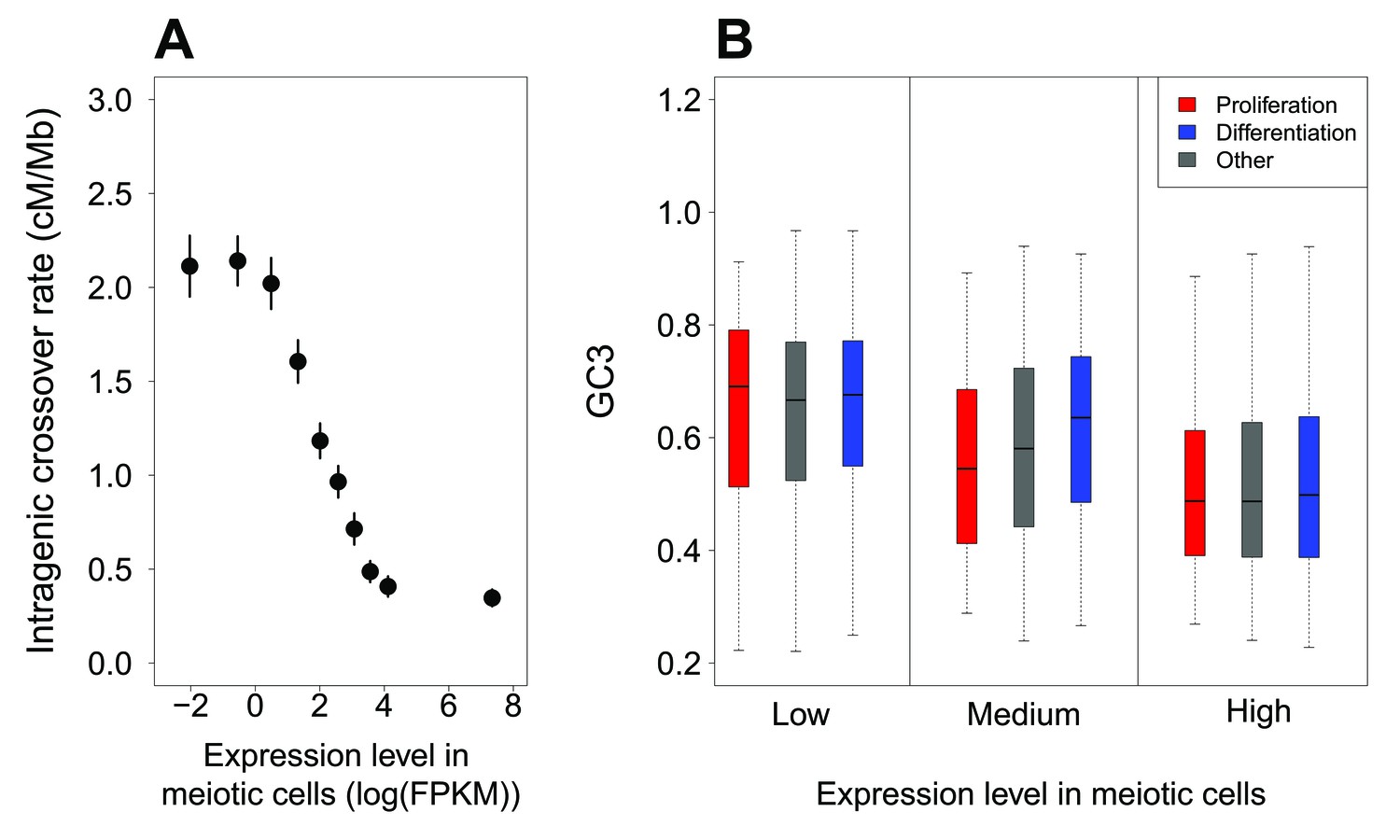
Variation in intragenic crossover rate and GC3 according to expression levels in meiotic cells.
(A) Genes were classified according to their sex-averaged expression level in meiotic cells into 10 bins of equal sample size. The mean sex-averaged intragenic crossover rate (HapMap) was computed for each bin. Error bars represent the 95% confidence interval of the mean. Similar results were obtained when analyzing sex-specific crossover rates and expression levels or when using DSB maps to measure of recombination rate (Figure 3—figure supplement 3). (B) Variation in GC3 according to meiotic expression levels. Genes were first binned into 3 classes of equal sample size according to their sex-averaged expression level in meiotic cells (low:<3.07 FPKM; high:>22.68 FPKM: medium: the others), and then split into three sets according to their functional category: ‘proliferation’ (red), ‘differentiation’ (blue), and ‘other’ genes (grey). Boxplots display the distribution of GC3 for each functional category within each expression bin.
Figure 3—figure supplement 1
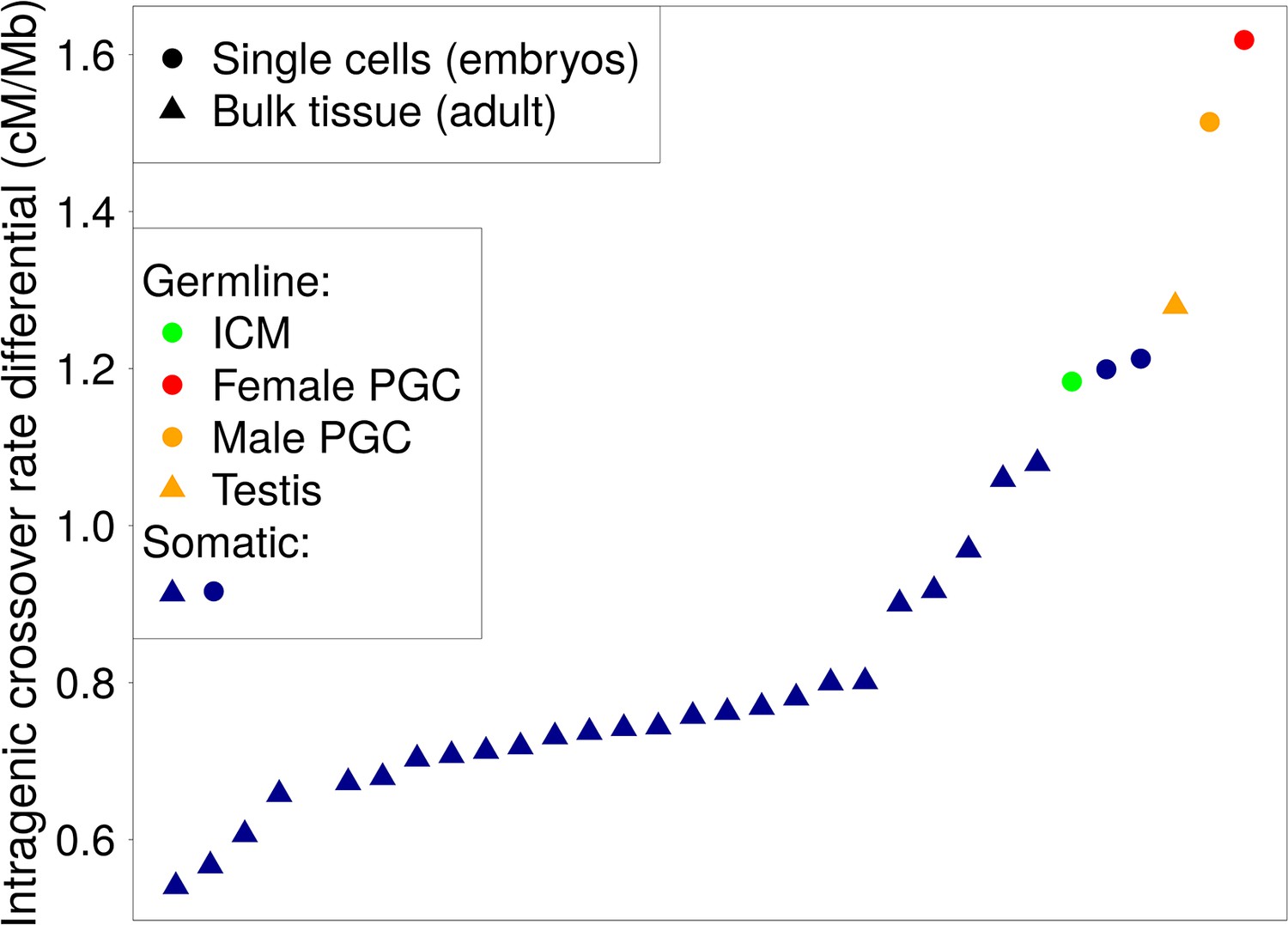
Differential intragenic crossover rate between lowly and highly expressed genes in adult tissues and in individual embryonic cells.
This differential is computed as the difference between the mean sex-averaged intragenic crossover rates (HapMap) of lowly expressed genes (10% most lowly expressed for bulk tissue data or non-expressed genes for single cells data) and the mean of the 10% most highly expressed genes. Dots are ordered by increasing differential values. Rounded dots correspond to data from individual embryonic cells (Guo et al., 2015) and triangles to adult tissues (Fagerberg et al., 2014). Dark blue dots: somatic adult tissues and somatic embryonic cells are in dark blue. Orange dots: male testis tissue and primordial germ cells (between 4 and 19 weeks). Red dot: female primordial germ cells (between 4 and 17 weeks). Green dot: inner cell mass ICM of the blastocysts.
Figure 3—figure supplement 2
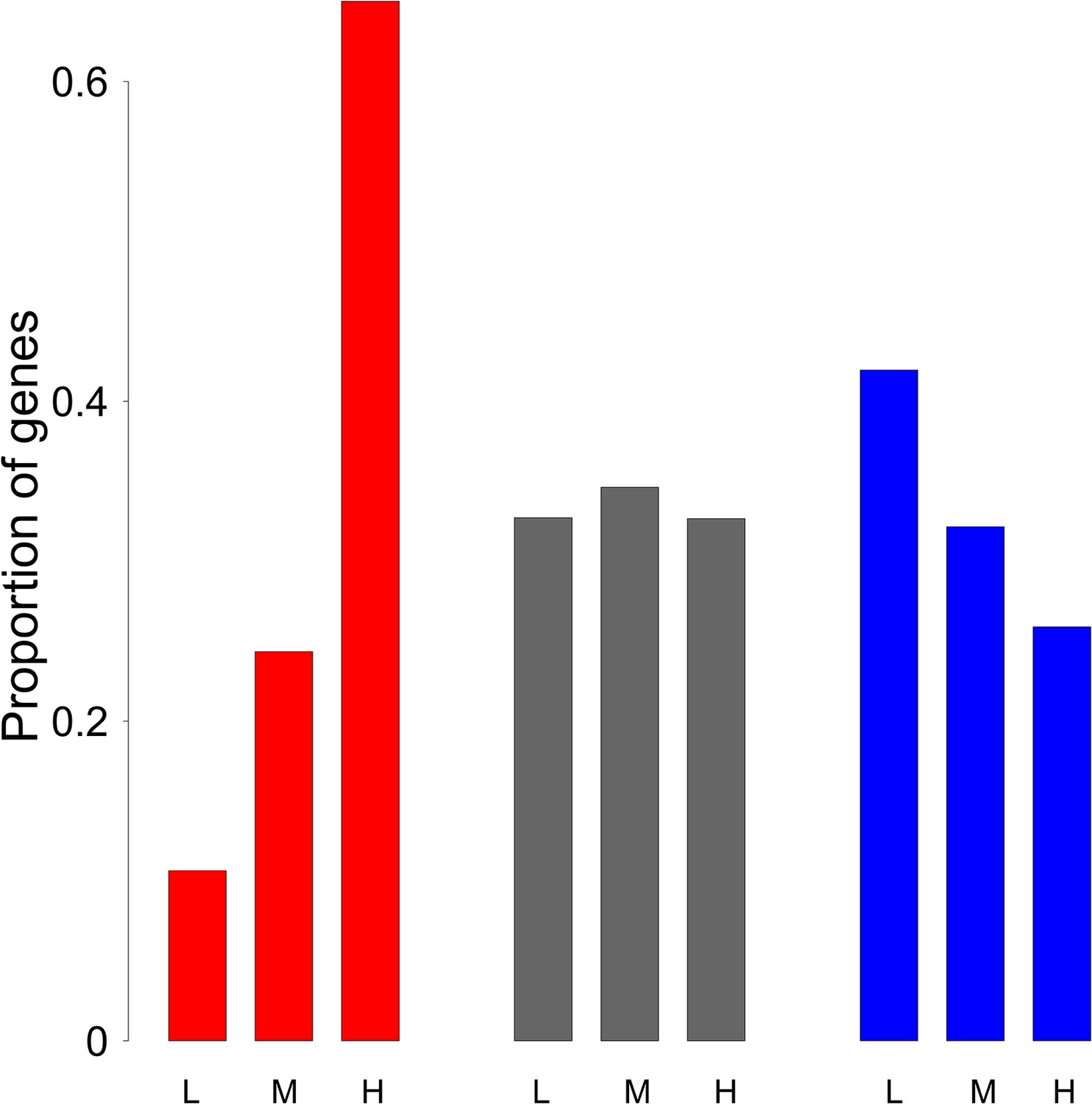
Comparison of the distribution of meiotic gene expression levels for ‘proliferation’, ‘differentiation’ and other genes.
For each functional category (‘proliferation’: red, ‘differentiation’: blue, and ‘other’ genes: grey), barplots display the distribution of genes among the three classes of sex-averaged meiotic expression level (as defined in Figure 3): low (L):<3.07 FPKM; high (H):>22.68 FPKM; medium (M).
Figure 3—figure supplement 3
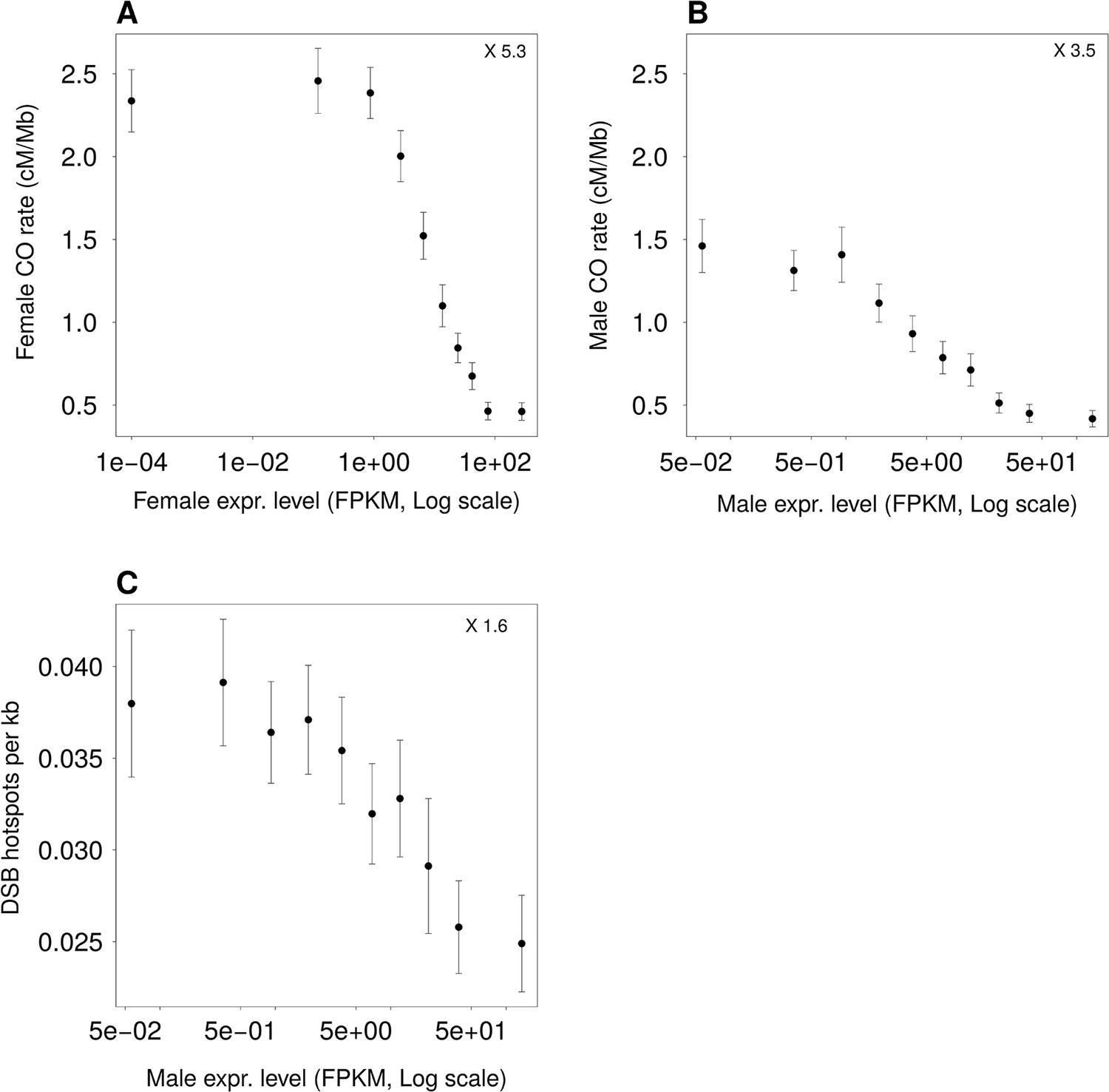
Variation in intragenic recombination rate and GC3 according to expression levels in meiotic cells.
Autosomal genes (>5 kb) were classified into 10 bins of equal sample size according to their expression level in female (A) or male (B, C, D) meiotic cells. (A) Mean intragenic crossover rate in female meiosis. (B) Mean intragenic crossover rate in male meiosis. (C) Mean density in intragenic DSB hotspots in male meiosis. Error bars represent the 95% confidence interval of the mean.
Figure 4 with 2 supplements
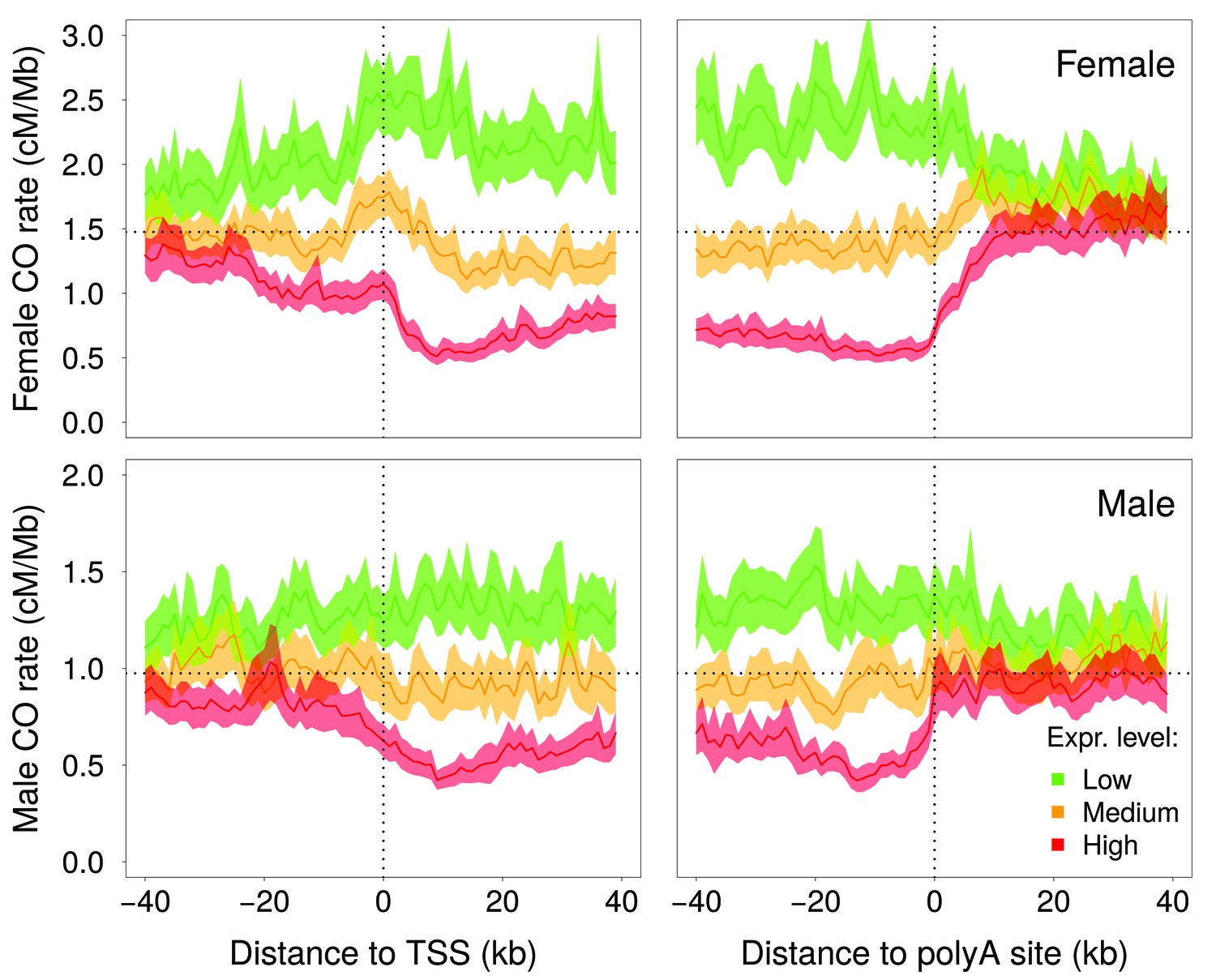
Variation in crossover rate as a function of the distance to transcription start site (TSS) and to the polyadenylation site, and according to meiotic expression level.
Autosomal genes longer than 5 kb (N = 15,055) were classified into three bins of equal sample size according to their expression level in female (top panels) or male meiosis (bottom panels): low (green), medium (orange) and high (red) expression level. Sex-specific crossover rates were measured in 1 kb-long non-overlapping windows. Shaded area represent the 95% confidence interval of the mean.
Figure 4—figure supplement 1
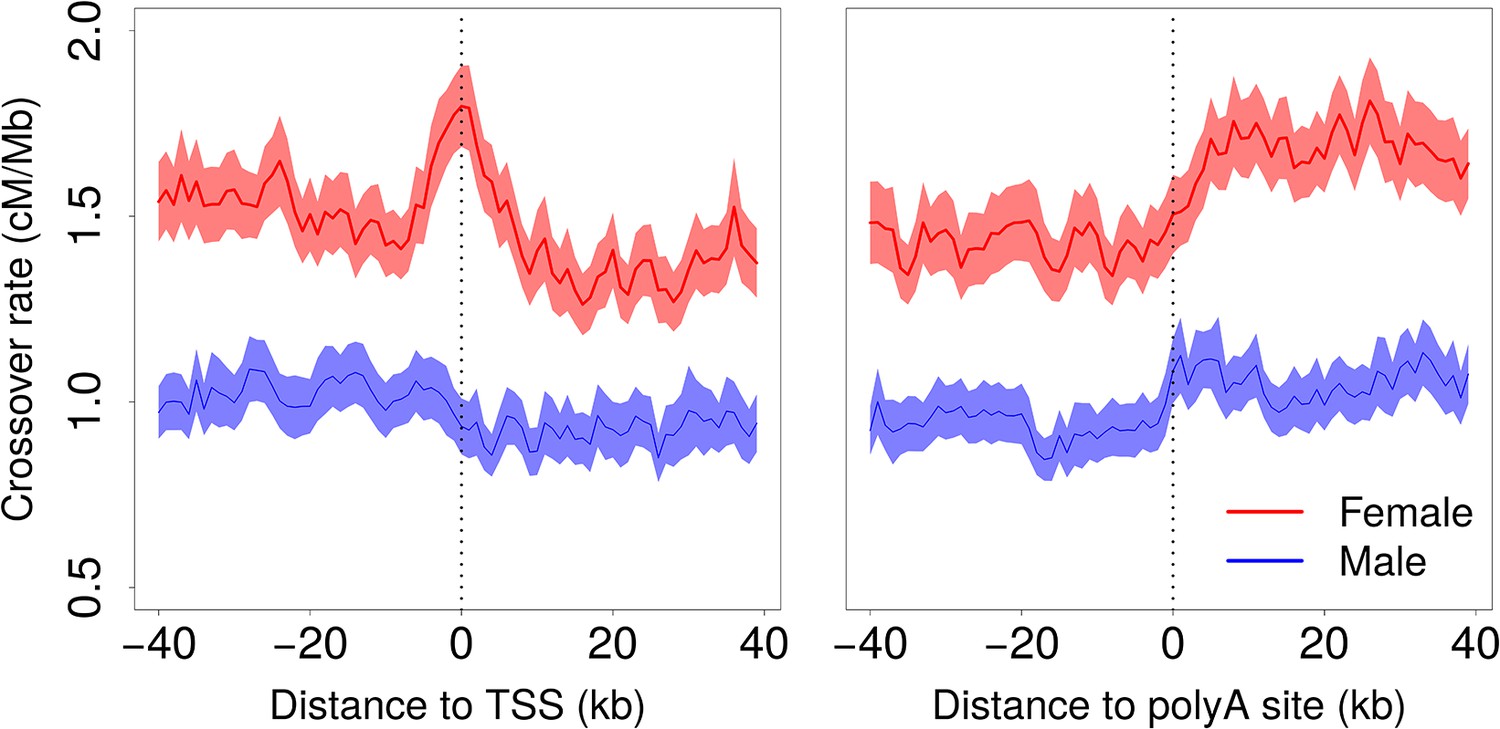
Variation in crossover rate as a function of the distance to transcription start site (TSS) and to the polyadenylation site.
Autosomal genes longer than 5 kb (N = 15,055). Male (blue) and female (red) crossover rates were measured in 1 kb-long non-overlapping windows. Shaded areas represent the 95% confidence interval of the mean.
Figure 4—figure supplement 2
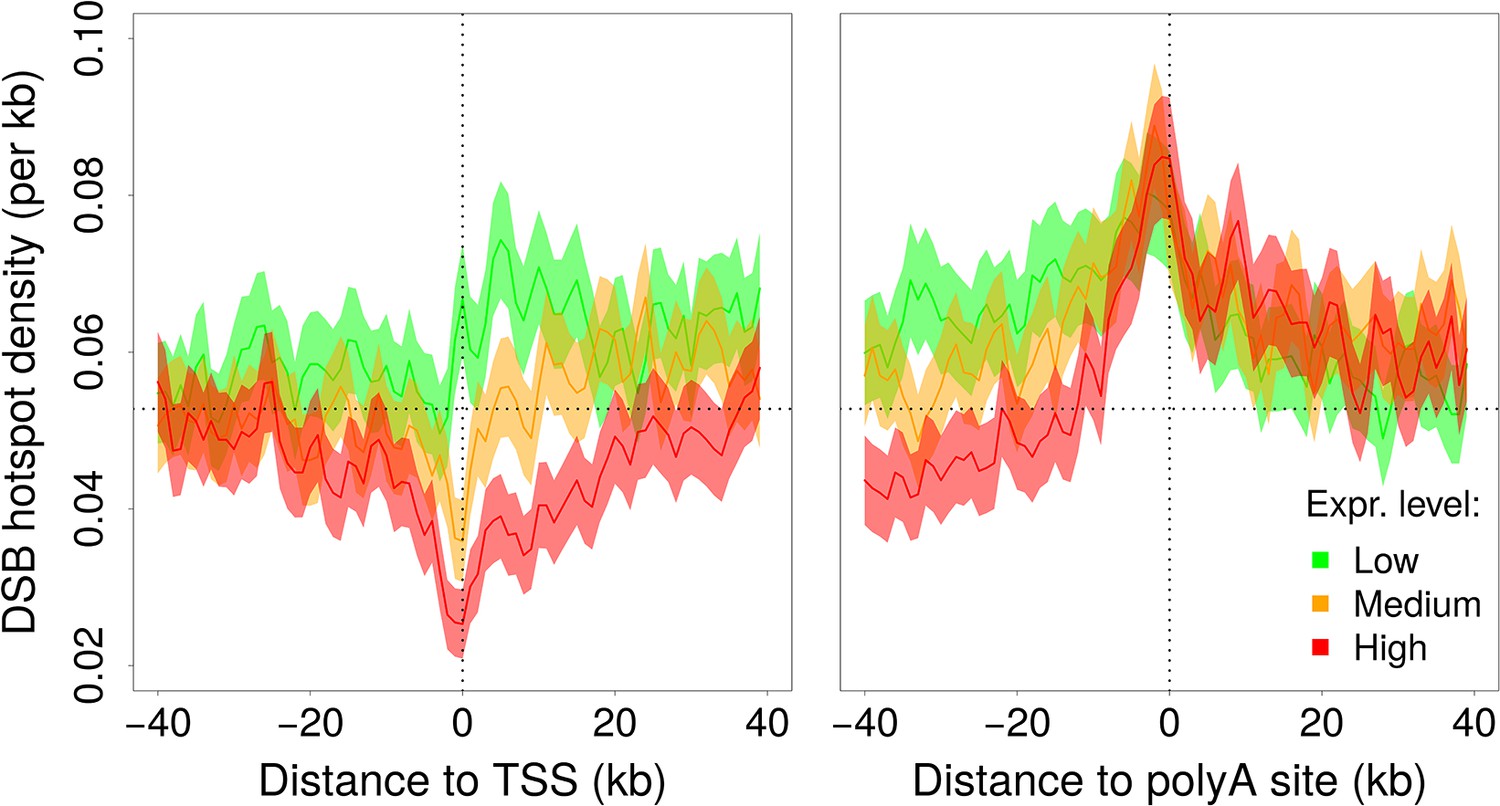
Variation in DSB hotspot density as a function of the distance to transcription start site (TSS) and to the polyadenylation site, and according to meiotic expression level.
Autosomal genes longer than 5 kb (N = 15,055) were classified into three bins of equal sample size according to their expression level in male meiosis: low (green), medium (orange) and high (red) expression level. DSB hotspot density (detected by DMC1 ChipSeq in males) were measured in 1 kb-long non-overlapping windows. Shaded areas represent the 95% confidence interval of the mean.
Figure 5

Correlation between expression level and GC3 in a panel of tissues and cell types.
(A) Bulk adult tissues data (Fagerberg et al., 2014) and (B) early embryo single-cell data (Guo et al., 2015). These two subsets were obtained via very different protocols, which prevents direct cross-comparisons. Samples are sorted by increasing correlation coefficient (R2) between expression levels and GC3 (NB: all correlations are negative). Samples containing somatic cells are shown in blue; male germ cells in orange (testis or single cell) and female germ cells in red (PGC: primordial germ cells). The green point corresponds to cells from the inner cell mass (ICM) of the blastocysts, i.e. pluripotent cells from an early stage of development preceding the differentiation of germ cells.
Figure 6 with 1 supplement
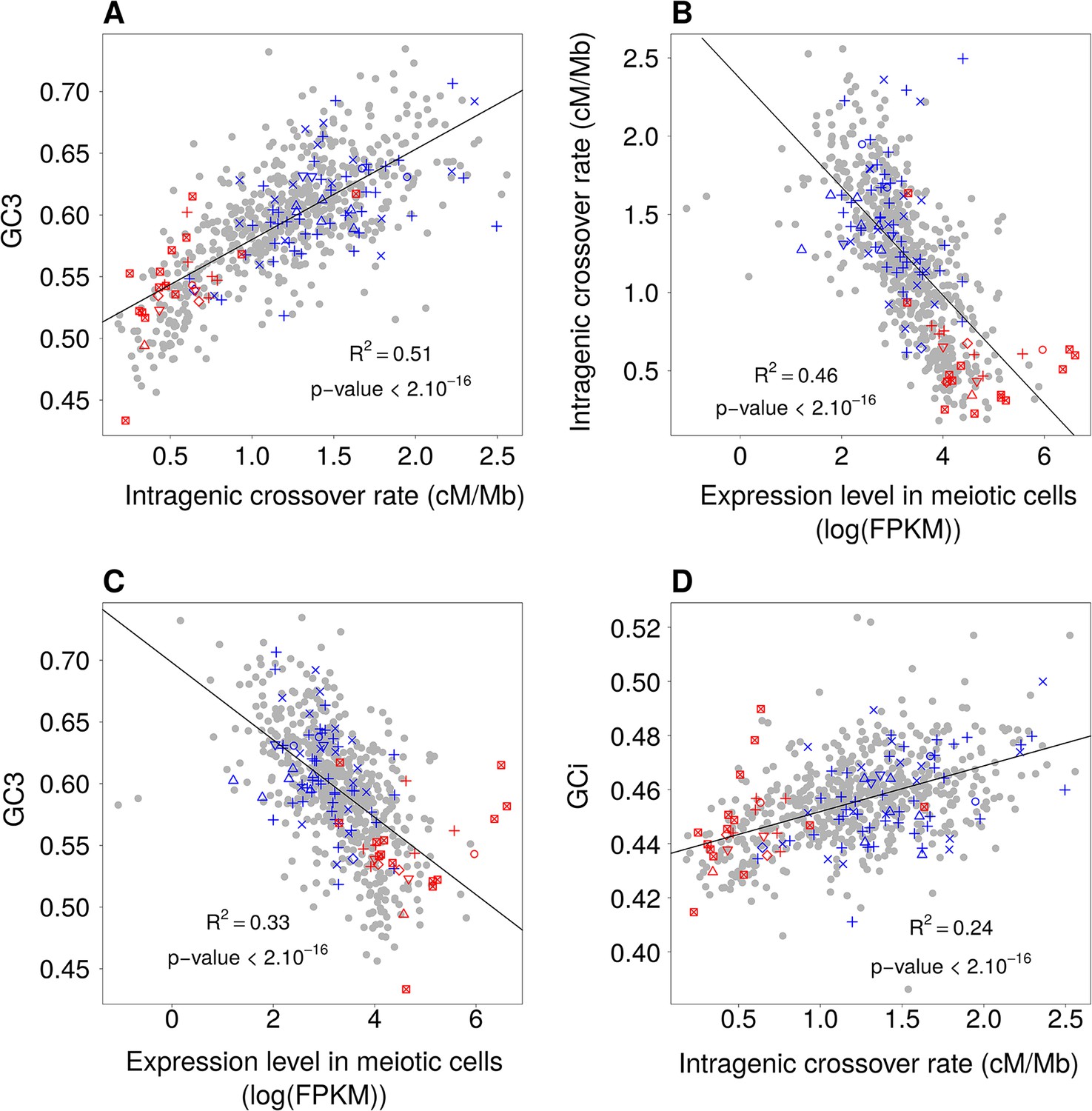
Relationships between GC-content, intragenic crossover rates and meiotic expression levels (sex-averaged) among functional gene categories.
Average values of these parameters were computed for each GO gene set. We then measured correlations between these parameters: (A) Mean GC3 vs. mean sex-averaged intragenic crossover rate (HapMap). (B) Mean intragenic crossover rate vs. mean expression level in meiotic cells. (C) Mean GC3 vs. mean expression level in meiotic cells. (D) Mean intronic GC-content (GCi) vs. mean intragenic crossover rate. GO gene sets associated to ‘proliferation’ (red) or ‘differentiation’ (blue) are displayed as in Figure 1. Similar results were obtained when analyzing separately expression levels in female or male meiosis (Figure 6—figure supplement 1).
Figure 6—figure supplement 1
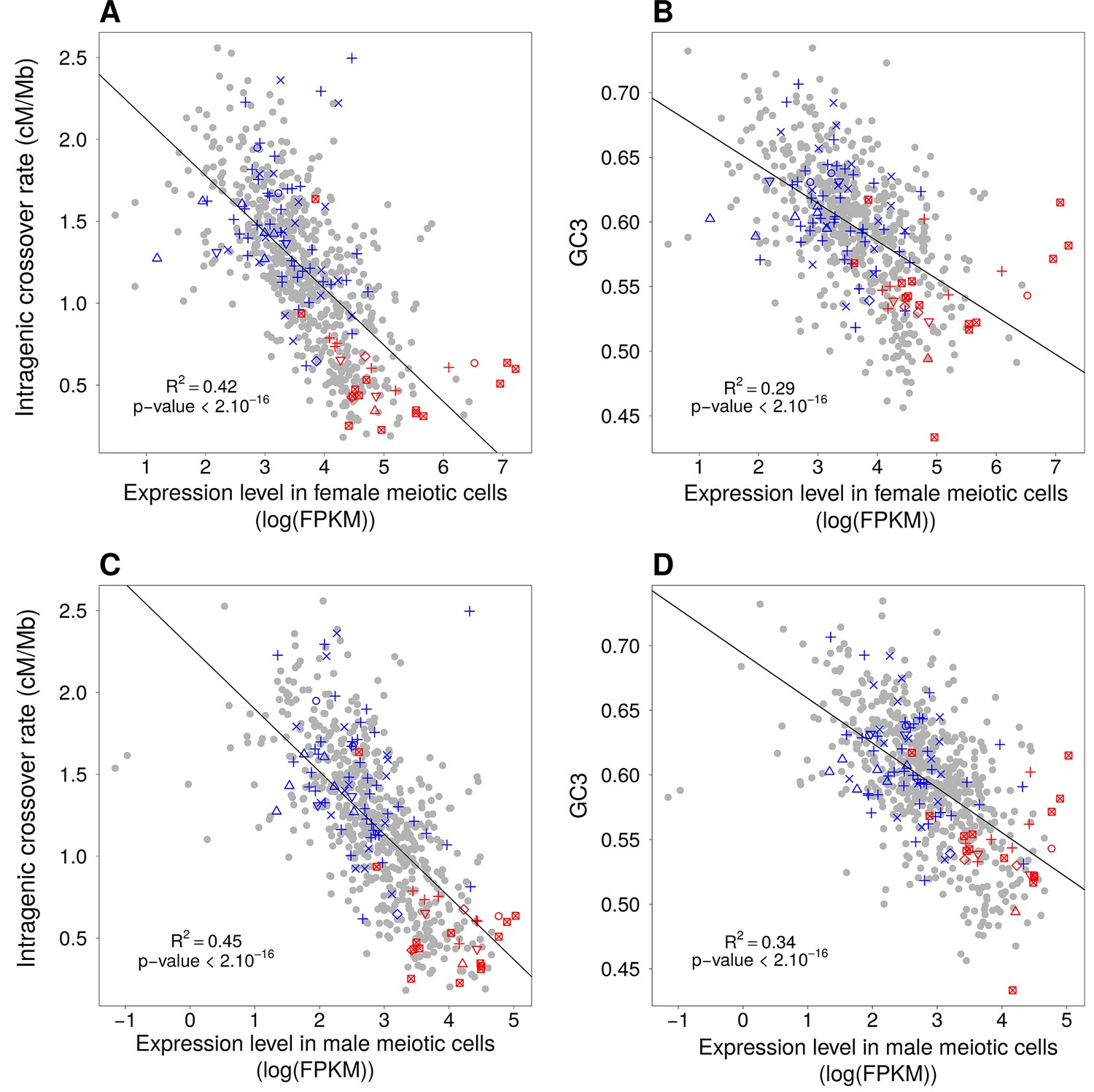
Relationships between expression levels in female or male meiotic cells and GC3 and intragenic crossover rates.
(A, B) Same as Figure 6B and C, but with expression level measured by single-cell analysis of female primordial germ cells at 17 weeks (Guo et al., 2015). (C, D) Same as 6B and C, but with expression level measured in male meiotic cells (Lesch et al., 2016). Expression levels are expressed in log(FPKM).
Author response image 1

Correlation between between intragenic crossover rate and GC3 (measured in the first or last 50 codons of genes), among functional gene categories.
Tables
Table 1
Analysis of the variance of GC3 among individual genes.
Variables included in the linear model are: GC-content of introns (GCi), GC-content of flanking regions (GC-flank), HapMap sex-averaged intragenic crossover rate (log scale), sex-averaged meiotic gene expression level (log scale) and functional category (‘differentiation’, ‘proliferation’ and ‘other’). Pairwise correlations (pairwise R2) were computed between GC3 and each of the other variables. Correlations of the model (model R2) were computed by adding variables sequentially.
| GC3 predictors | Pairwise R2 | p-value | Model R2 | F statistic | p-value |
|---|---|---|---|---|---|
| GCi | 62.7% | <2.10−16 | 62.7% | 30232.4 | <2.10−16 |
| GC-flank | 48.1% | <2.10−16 | 62.9% | 126.8 | <2.10−16 |
| Intragenic crossover rate | 12.8% | <2.10−16 | 66.8% | 1453.3 | <2.10−16 |
| Expression level in meiosis | 8.3% | <2.10−16 | 68.2% | 875.7 | <2.10−16 |
| Functional category | 1% | <2.10−16 | 68.3% | 30.43 | <2.10−16 |
Additional files
-
Transparent reporting form
- https://doi.org/10.7554/eLife.27344.016
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Recombination, meiotic expression and human codon usage
eLife 6:e27344.
https://doi.org/10.7554/eLife.27344




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}