Atg9 antagonizes TOR signaling to regulate intestinal cell growth and epithelial homeostasis in Drosophila
- Academia Sinica, Taiwan
- College of Life Science, National Taiwan University, Taiwan
- National Taiwan University College of Medicine, Taiwan
Abstract
Autophagy is essential for maintaining cellular homeostasis and survival under various stress conditions. Autophagy-related gene 9 (Atg9) encodes a multipass transmembrane protein thought to act as a membrane carrier for forming autophagosomes. However, the molecular regulation and physiological importance of Atg9 in animal development remain largely unclear. Here, we generated Atg9 null mutant flies and found that loss of Atg9 led to shortened lifespan, locomotor defects, and increased susceptibility to stress. Atg9 loss also resulted in aberrant adult midgut morphology with dramatically enlarged enterocytes. Interestingly, inhibiting the TOR signaling pathway rescued the midgut defects of the Atg9 mutants. In addition, Atg9 interacted with PALS1-associated tight junction protein (Patj), which associates with TSC2 to regulate TOR activity. Depletion of Atg9 caused a marked decrease in TSC2 levels. Our findings revealed an antagonistic relationship between Atg9 and TOR signaling in the regulation of cell growth and tissue homeostasis.
https://doi.org/10.7554/eLife.29338.001Introduction
Autophagy is a highly regulated lysosomal degradation process by which intracellular components are degraded and recycled for cell viability and homeostasis. There is increasing evidence of the importance of autophagy in a variety of physiological and pathological processes, including differentiation, development, aging, and tumorigenesis (Jiang and Mizushima, 2014; Mizushima and Levine, 2010). The autophagic pathway is controlled by a series of evolutionarily conserved autophagy-related (Atg) proteins to generate double-membraned vesicles, the autophagosomes, which subsequently fuse with lysosomes for the degradation of their contents (Feng et al., 2014). Atg9 is the only multi-spanning transmembrane protein in the family and is involved in promoting lipid transport to autophagosomes during their formation (Tooze, 2010; Yamamoto et al., 2012). In yeast, distinct from other Atg proteins, Atg9 cycles between cytoplasmic membrane pools and the preautophagosomal structure (PAS) (Mari et al., 2010). The Atg9-containing vesicles are recruited to PAS by the Atg1-kinase complex during autophagosome formation (Rao et al., 2016; Suzuki et al., 2015). Similarly, mammalian Atg9 (mAtg9) localizes to the trans-Golgi network, endosomal system, and plasma membrane under normal conditions, whereas it translocates to autophagic membranes upon autophagy induction (Orsi et al., 2012; Popovic and Dikic, 2014; Puri et al., 2013; Young et al., 2006). The trafficking of mAtg9 is important for autophagy induction, and several proteins, including Ulk1, ZIPK, p38IP, TRAPPC8, TBC1D5, and the AP2 complex (Imai et al., 2016; Lamb et al., 2016; Popovic and Dikic, 2014; Tang et al., 2011; Webber and Tooze, 2010; Young et al., 2006), regulate the spatio‐temporal distribution of mAtg9 during autophagy.
In addition to autophagy, mAtg9 can modulate dsDNA-induced innate immune responses by regulating the STING-TBK1assembly (Saitoh et al., 2009). Recently, Imagawa et al showed that mAtg9 also plays a role in necrotic programmed cell death during bone morphogenesis (Imagawa et al., 2016). Our previous study in Drosophila showed that Atg9 functions not only as an essential component of autophagy, but also interacts with Drosophila tumor necrosis factor receptor-associated factor 2 (dTRAF2) to regulate ROS-induced c-Jun N-terminal kinase (JNK) signaling, including JNK-mediated autophagy activation and intestinal stem cell (ISC) proliferation (Tang et al., 2013). Moreover, oxidative stress-induced autophagy can inhibit JNK activity through a negative feedback mechanism to prevent the over-activation of JNK-mediated stress responses, thereby helping the maintenance of midgut homeostasis. However, the molecular regulation and physiological function of Atg9 remain largely unknown.
Target of rapamycin (TOR), a serine/threonine kinase, functions as a central player in the regulation of cell growth and metabolism in response to various environmental stimuli, including nutrient status, growth factors, and amino acids (Saxton and Sabatini, 2017). Under nutrient-rich conditions, TOR promotes protein synthesis and energy metabolism while suppressing autophagy (Russell et al., 2014). Under nutrient deprivation conditions, TOR is inhibited, leading to the induction of autophagy. TOR negatively regulates autophagy by phosphorylating and inhibiting Atg1/Unc51-like kinase 1 (Ulk1) complex activity (Alers et al., 2012). The Atg1/Ulk1 kinase is thought to act as the most upstream autophagy regulator for the initiation of autophagosome formation (Itakura and Mizushima, 2010). Atg1/Ulk1 recruits downstream Atg proteins to the phagophore assembly site and phosphorylates several Atg proteins, including the Ambra1/Beclin1/Vps34 complex and Atg9 (Cheong et al., 2008; Di Bartolomeo et al., 2010; Papinski et al., 2014; Russell et al., 2013). Interestingly, recent studies have shown that Atg1/Ulk1 can negatively regulate TOR signaling in Drosophila and mammals (Lee et al., 2007; Scott et al., 2007), suggesting a tight interplay between Atg1/Ulk1-dependent autophagy and TOR-mediated cell growth.
Here, we generated null mutants for Drosophila Atg9, and showed that loss of Atg9 severely impairs starvation-induced and developmental autophagy. Atg9 null mutant flies exhibited dramatically reduced lifespans, climbing defects, and hypersensitivity to stress. Surprisingly, ablation of Atg9 also caused increased TOR activity and aberrant enlargement of intestinal epithelial cells in the adult Drosophila midgut. Similar intestinal defects were observed in Atg1, Atg13 and Atg17/Fip200 depletion mutants. We further identified PALS1-associated tight junction protein (Patj) as an Atg9-interacting protein. In mammals, the polarity protein Patj interacts with tuberous sclerosis complex 2 (TSC2), a negative regulator of TOR signaling, to regulate TOR activity (Massey-Harroche et al., 2007). Strikingly, overexpression of Patj and TSC1-TSC2 suppressed adult midgut defects of Atg9 mutants. Depletion of Atg9 resulted in a dramatic decrease in TSC2 levels. Our findings revealed a novel negative feedback loop by which Atg9 inhibits TOR signaling to regulate cell growth and tissue homeostasis.
Results
Generation of Drosophila Atg9 mutant fly
Our previous studies showed that Drosophila Atg9 interacts with dTRAF2 to regulate JNK activation, autophagy induction, and midgut homeostasis under oxidative stress conditions (Tang et al., 2013). To investigate the physiological and developmental functions of Atg9, we generated Atg9 null mutants using two different approaches. First, we replaced the Atg9 open reading frame with a Gal4 knock-in cassette (Atg9Gal4KO) using the ends-out homologous recombination approach (Figure 1A) (Chan et al., 2011). The Gal4 knock-in can be used for gene expression under Atg9 endogenous regulatory elements in the Atg9 mutant background. Second, we employed the CRISPR/Cas9 gene editing approach to replace a short coding region in the first exon of Atg9 with the attPX-3-frameStop-floxed 3xP3-RFP cassette (Kondo and Ueda, 2013), which leads to a pre-maturely truncated Atg9 mutant (Atg9d51) (Figure 1A). The homozygous Atg9Gal4KO and Atg9d51 flies and trans-heterozygous Atg9Gal4KO/Atg9d51 flies are semi-lethal, with a few escapers. Interestingly, the escapers produce no offspring, suggesting fertility defects in Atg9 mutants. We next compared Atg9 expression in wild-type and mutant flies. We confirmed the lack of Atg9 expression in the mutants by RT-PCR and Western blot analysis (Figure 1B and C). Importantly, the gene expression and semi-lethality of Atg9 mutants can be fully rescued by a 5.8 kb genomic construct encompassing the Atg9 transcript and its endogenous regulatory regions (Figure 1A–C). These results demonstrated that Atg9Gal4KO and Atg9d51 specifically disrupt Atg9 function and act as null mutants.
Figure 1 with 1 supplement see all

Generation of mutations in Drosophila Atg9.
(A) Schematic view of Atg9Gal4KO and Atg9d51mutations relative to the Atg9 transcripts. For the Atg9Gal4KO mutation, the complete Atg9 open reading frame was replaced with a Gal4 knock-in cassette. For Atg9d51 mutation, the 52–102 bp after the Atg9 start codon was replaced with the attPX-3-frameStop-floxed 3xP3-RFP cassette. (B) RT-PCR analysis of Atg9 mRNA expression level in control, mutant and Atg9 genomic rescue adult flies. Atg9 mRNA levels were undetectable in the Atg9 mutant. (C) Western blots show the endogenous Atg9 protein in control and Atg9 genomic rescue flies but fail to detect the protein in mutants. (D) LysoTracker Green staining reveals that starvation-induced autophagy is strongly reduced in Atg9 mutant fat bodies, compared with controls. Scale bar: 5 μm. (E) Quantification of data shown in (D). n ≥ 10, data are mean ±s.e.m. *p<0.05, **p<0.01. ns, not statistically significant. (F) Western blots show markedly increased Ref(2)P and ubiquitinated protein levels in Atg9 mutants. (G) Immunostaining of Drosophila thoracic muscles with anti-Ub (FK2) and anti-Ref(2)p antibodies showed an accumulation and colocalization of polyubiquitin protein aggregates and Ref(2)p (arrowheads) in Atg9 mutant flies. Scale bar: 10 μm. Df refers to Df(2R)Exel7142, which removes Atg9 and flanking genes.
-
Figure 1—source data 1
Quantification of lysotracker dots.
- https://doi.org/10.7554/eLife.29338.004
Atg9 mutants have impaired autophagy and increased ubiquitination
RNAi-mediated knockdown of Atg9 inhibits starvation-induced autophagy and developmental autophagy in the larval fat body (Bader et al., 2015; Tang et al., 2013). To determine whether the newly generated Atg9 null mutants also exhibit autophagic defecs, we first stained the larval fat body with the pH-sensitive fluorescent dye LysoTracker, which has been widely used to detect acidic lysosomes and autolysosomes. The LysoTracker Green staining was faint and diffuse in well fed control animals, whereas nutrient deprivation resulted in a strong punctate LysoTracker Green staining (Figure 1D). Notably, loss of Atg9 dramatically blocked the starvation-induced punctate staining, compared to the controls (Figure 1E). Because inhibiting autophagic activity often results in the accumulation of autophagic substrate p62/SQSTM1 and ubiquitinated protein aggregates (Komatsu and Ichimura, 2010), we investigated the effect of Atg9 on protein ubiquitination and autophagic degradation of Ref(2)P, the Drosophila homolog of p62. Compared to the control, Atg9 mutants had a dramatic increase in Ref(2)p levels and ubiquitin aggregate formation (Figure 1F and G). Moreover, Atg9 null mutants displayed impaired developmental autophagy in the larval fat body and larval midgut (Figure 1—figure supplement 1A and B). These results together demonstrate the essential role of Atg9 in autophagy during development and in response to starvation.
Atg9 mutants exhibit shortened lifespan, locomotor defects, and increased susceptibility to stresses
Many Drosophila autophagy mutants display reduced lifespan and decreased climbing activity (Juhász et al., 2007; Kim et al., 2013). To gain more insight in the physiological function of Atg9, we analyzed the effect of Atg9 gene ablation on the lifespan of Drosophila. As shown in Figure 2A, we found that the lifespan of Atg9 mutants was greatly reduced compared with that of Atg9 heterozygous control flies (48% decrease in male mean lifespan and 53% in female, p<0.001) or Atg7 mutants (38% decrease in male mean lifespan and 40% in female, p<0.001) under normal conditions. The negative geotaxis assay also revealed that Atg9 mutants exhibited a significantly lowered climbing activity than that of the control flies (Figure 2B). The locomotion defects of Atg9 mutants were substantially suppressed by expressing the Atg9 genomic rescue construct, suggesting that the mobility defects were indeed caused by interruption of Atg9 gene expression. Moreover, loss of Atg9 leads to dramatically decreased viability under starvation and oxidative stress conditions (Figure 2C,D). We thus conclude that, like other autophagy mutants, Atg9 also regulates Drosophila lifespan, mobility, and susceptibility to stresses.
Figure 2
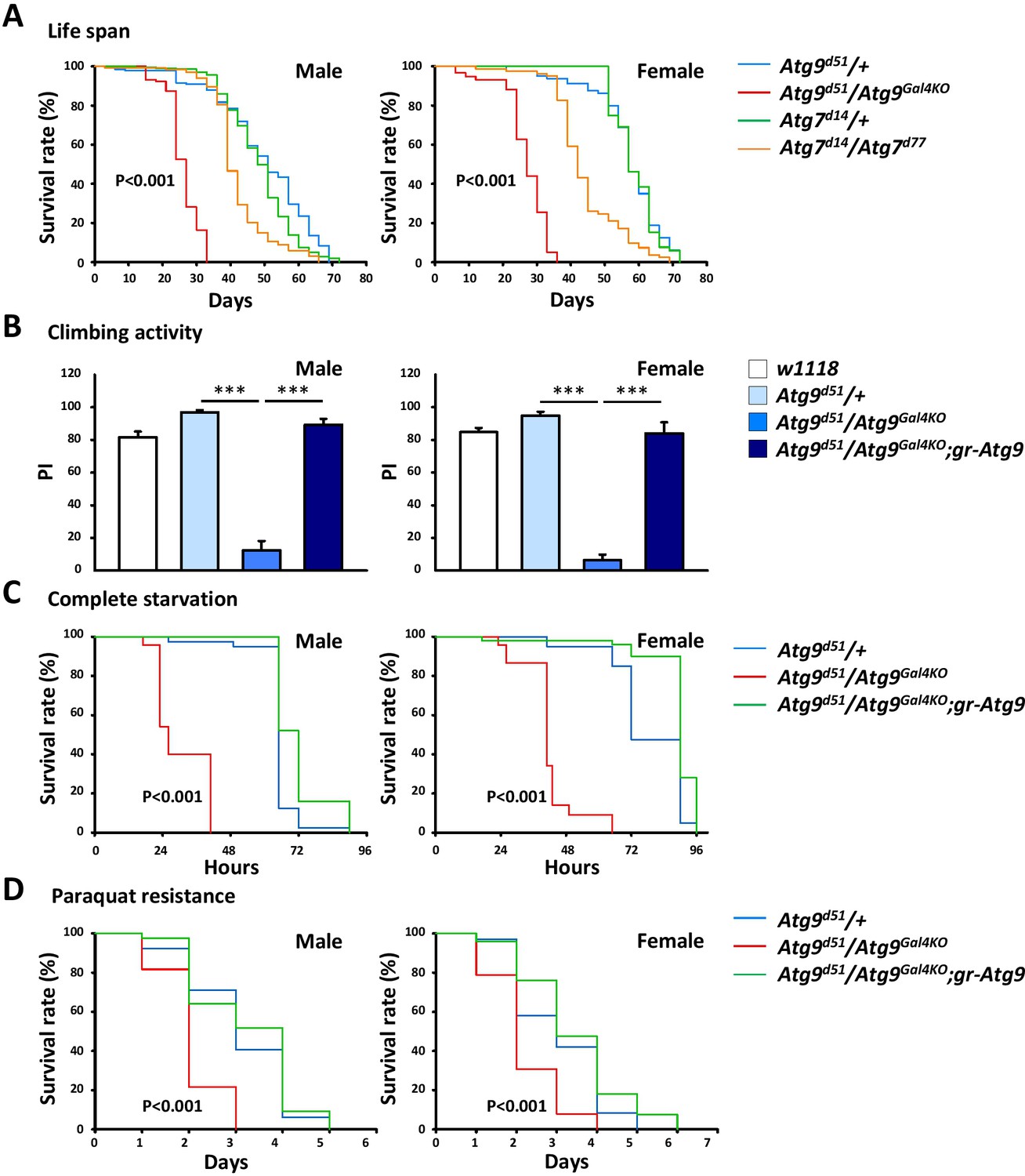
The Atg9 mutant flies display shortened lifespan, locomotor defects and decreased stress tolerance.
(A) Both Atg7 and Atg9 mutant flies showed shortened lifespan compared with control (Log-rank test, p<0.001). Comparing with Atg7 mutant, Atg9 mutant flies exhibited dramatically shortened lifespan. (B) Climbing analysis showed that Atg9 mutant exhibit decreased climbing activity in both male and female flies, compared with controls (***p<0.001). The locomotion defects can be rescued by expressing the Atg9 genomic rescue construct. (C–D) Both male and female Atg9 mutant flies die faster than control flies under complete starvation conditions (Log-rank test, p<0.001) or on paraquat treatment (Log-rank test, p<0.001).
-
Figure 2—source data 1
Survival rate and climbing activity of control and Atg9 mutants.
- https://doi.org/10.7554/eLife.29338.007
Atg9 is required for proper adult midgut morphogenesis
The Drosophila adult midgut shares many similarities with the mammalian intestine and has emerged as an attractive model system to study stem cell proliferation and differentiation (Jiang and Edgar, 2012). Our previous study showed that Drosophila Atg9 is involved in regulating adult midgut homeostasis upon ROS stimulation and bacterial infection (Tang et al., 2013). To examine whether Atg9 has a function in maintaining intestinal homeostasis under normal conditions, we examined the adult midgut morphology of the Atg9 mutant. The Drosophila adult midgut consists of a tubular, monolayered epithelium surrounded by visceral muscles (Micchelli and Perrimon, 2006). In striking contrast, the Atg9 mutant adult midgut is markedly shortened and significantly thickened in the posterior region, compared to control flies (Figure 3A). Moreover, phalloidin staining of actin filaments in the visceral muscles revealed a severe disruption in the continuity of the visceral mesoderm layer surrounding the gut (Figure 3B). To assess whether the loss of Atg9 would affect intestinal barrier function, we examined the intestinal integrity by feeding flies of different ages with a nonabsorbable blue food dye (Rera et al., 2011). As expected, the dye was restricted to the digestive tract in young control flies (10 days, Smurf- fly), whereas the dye was seen throughout the body in approximately 7% of the aged control flies (30 days, Smurf + fly) due to a loss of intestinal integrity (Figure 3—figure supplement 1A and B). Although we found no significant intestinal barrier dysfunction in young Atg9 mutants, there was a dramatic increase of Smurf + flies in aged Atg9 mutant animals, compared to controls (Figure 3—figure supplement 1B).
Figure 3 with 1 supplement see all
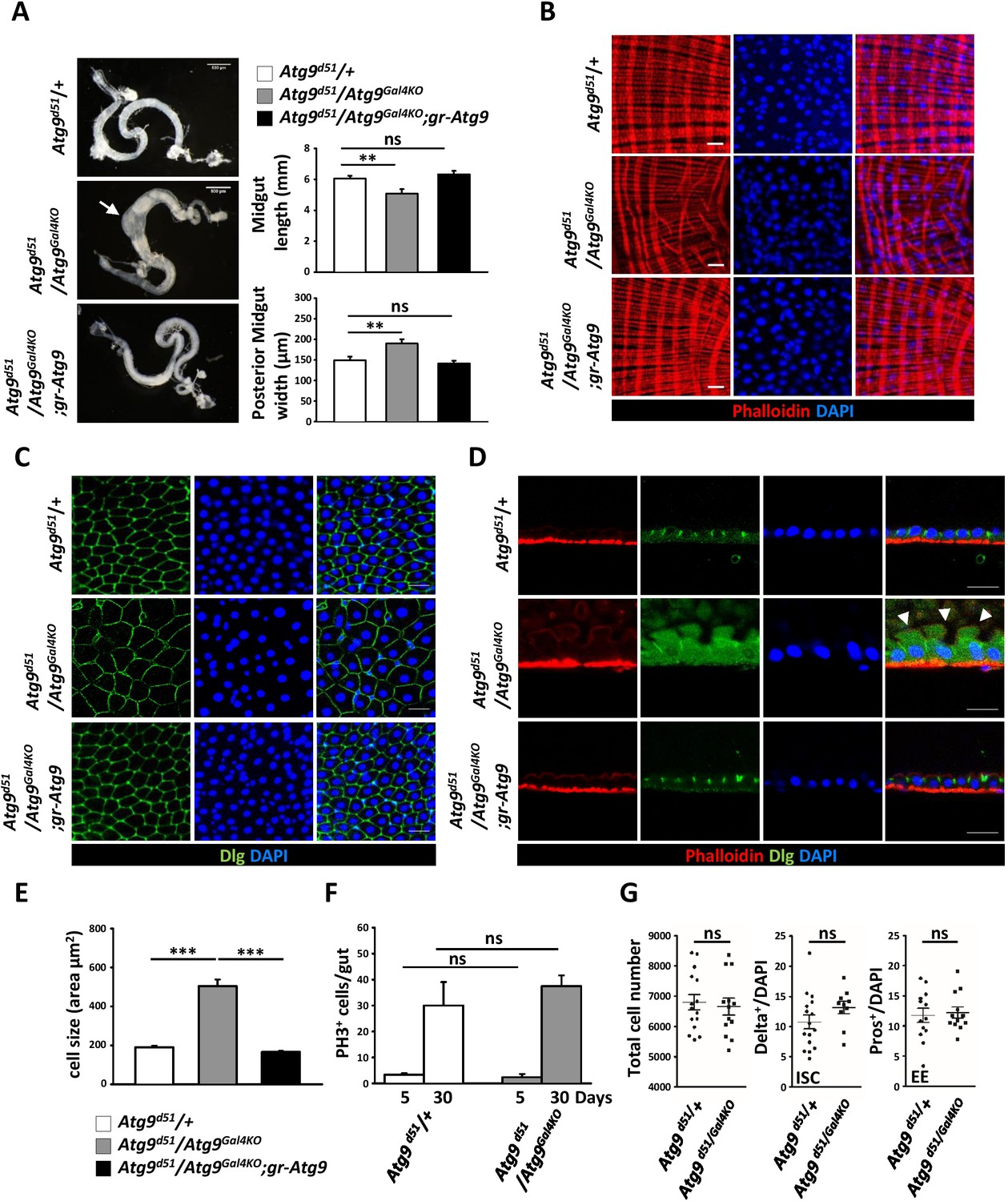
Atg9 is required for adult Drosophila midgut morphogenesis.
(A) Atg9 mutant midguts are shortened and display enlargement in the posterior region (arrow). Scale bar: 500 μm. Quantification of adult midgut length and posterior midgut width of control and Atg9 mutant flies. n = 10, **p<0.01. (B) Phalloidin staining of midgut visceral muscles revealed that loss of Atg9 leads to disruption of actin filaments. Scale bar: 20 μm. (C–D) Optical sections of control and the Atg9 mutant midgut epithelium layer stained with anti-Dlg showing that Atg9 mutants display abnormally enlarged cells with apical protrusions (arrowheads) into the lumen. Scale bar: 20 μm. (E) Quantification of cell size (shown in panel C) in control and Atg9 mutant posterior midgut. n ≥ 25, *p<0.05, **p<0.01. (F) Quantification of phospho-Histone3 positive (PH3+) cells per midgut of control and Atg9 mutant flies at 5 days and 30 days of age. n ≥ 8. (G) Quantification of total midgut cell numbers, posterior midgut ISC (Delta+) and EE (Pros+) cell numbers of 5-day-old control and Atg9 mutant adults. n ≥ 10. Data are mean ±s.e.m. ns, not statistically significant.
-
Figure 3—source data 1
Quantification of midgut length and width, cell size, total cell number, and PH3+, Delta+, Pros+ cells per gut.
- https://doi.org/10.7554/eLife.29338.010
The Drosophila adult midgut epithelium homeostasis is maintained by self-renewing intestinal stem cells (ISC) that divide asymmetrically to generate renewed ISCs and enteroblasts (EB) (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). The EB further differentiate into either absorptive enterocytes (EC) or secretory enteroendocrine cells (EE). In wild-type or Atg9 heterozygous mutant flies, the intestinal epithelium consistently showed a tight, polarized monolayer (Figure 3C,D). In Atg9 mutant flies, the epithelium was also a monolayer, but composed of dramatically enlarged cells and abnormal apical membrane protrusions that often expanded into the midgut lumen (Figure 3C–E). Notably, the aberrant midgut defects in Atg9 mutants can be fully rescued by the Atg9 genomic transgene (Figure 3C–E), further demonstrating that the intestinal defects were a direct consequence of the Atg9 mutation.
To investigate whether the aberrant posterior midgut enlargement in Atg9 mutants was due to an increase in cell growth and proliferation, we measured the ISC mitotic index in the Atg9 mutant adult midgut. Immunostaining with an antibody for the mitotic marker phospho-histone 3 (PH3) in the whole midgut revealed no statistical difference in the number of PH3-positive cells between Atg9 null and control flies at both 5 days and 30 days of age (Figure 3F). Moreover, the Atg9 mutant midgut showed a similar number of total intestinal cells, Delta-positive (ISC-specific marker) cells and Pros-positive (EE marker) cells, compared with controls (Figure 3G). Together, these results indicate that Atg9 is not required for the regulation of ISC proliferation under normal conditions.
Atg9 acts in ECs to regulate cell growth
To gain insights on the cell type requirement of Atg9 function, we expressed Atg9RNAi with Dl-Gal4 driver in ISCs, Su(H)GBE-Gal4 in EBs, and Myo1A-Gal4 (NP1-Gal4) in ECs. While Atg9 depletion in ISCs and EBs did not cause any observable defects in the midgut, Atg9 depletion in ECs resulted in pronounced defects in the midgut epithelium, with a markedly increased cell size and aberrant cell morphology (Figure 4A). We further utilized the TARGET system (McGuire et al., 2004) to specifically knockdown Atg9 in adulthood. The negative control was flies maintained at 18°C, and the positive control was flies shifted to 29°C within 24 hr after eclosion to inactivate Gal80ts and enable expression of the RNAi targeting Atg9 gene. Because the midgut defects was only observed when Atg9RNAi was induced by temperature shift in adult flies with Tub-Gal4; Tub-Gal80ts as driver, the aberrant midgut enlargement is likely not caused by defects during development (Figure 4B). Moreover, although the visceral muscle defects were observed in Atg9 mutants (Figure 3B), we found that, like Atg9 depletion in ISCs and EBs, ablation of Atg9 with muscle-specific How-Gal4ts did not cause any observable defects in the adult midgut (Figure 4—figure supplement 1). To further explore the role of Atg9 in adult midguts, we generated Atg9 mitotic clones using the heat shock-inducible Flp-FRT system. Mosaic analysis of Atg9 mutant clones revealed a dramatic increase in cell size of Atg9 mutant cells (Figure 4C, GFP-negative, encircled). Next, we utilized the MARCM (mosaic analysis with a repressible cell marker) technique to generate clones of cells homozygous for the Atg9 mutation (Figure 4D, marked by GFP). Immunostaining with Pdm1 (an EC specific marker) (Lee et al., 2009) revealed that many of the enlarged GFP-positive cells in Atg9 mutant clone were stained positive for Pdm1 (Figure 4D), suggesting that Atg9 acts largely in ECs to regulate cell growth.
Figure 4 with 2 supplements see all
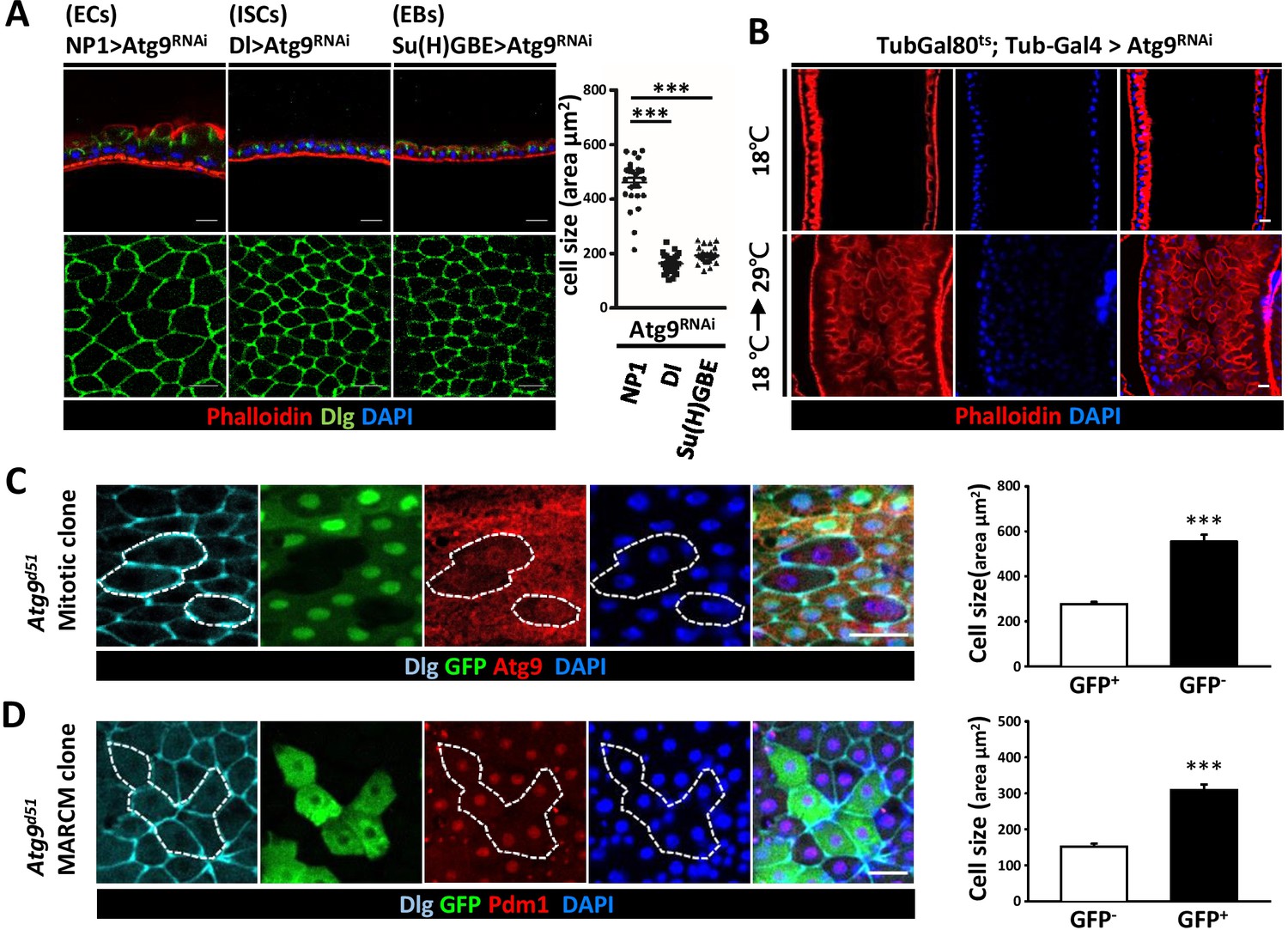
Loss of Atg9 leads to enlarged enterocytes.
(A) Expression Atg9RNAi in ISCs, EBs and ECs with Dl-Gal4, Su(H)GBE-Gal4, or NP1-Gal4, respectively. Ablation of Atg9 in ECs, but not ISCs or EBs, caused enlarged cell size. n ≥ 25, ***p<0.001. (B) Temporal control of Atg9RNAi expression using the Gal80ts; Tub-Gal4 inducible system. The flies were either maintained at 18°C throughout development or shifted to 29°C after eclosion for 5 days to inactivate Gal80ts and enable expression of the RNAi targeting Atg9. (C) Clonal analysis in adult midgut using Flp-FRT-mediated recombination revealed that Atg9d51 mutant cells (marked by lack of GFP and Atg9 expression) are larger than the controls (GFP-positive cells). n ≥ 17, ***p<0.001. (D) MARCM analysis showed that the enlarged Atg9d51 mutant cells (marked by GFP) are Pdm1 positive EC cells. n ≥ 21, ***p<0.001. Scale bar: 20 μm. Genotypes: (C) hsFLP; FRT42D Ubi-GFP/FRT42D Atg9d51 (D) hsFLP; FRT42D tubGal80/FRT42D Atg9d51; Tub-Gal4/UAS-mCD8GFP.
-
Figure 4—source data 1
Quantification of cell size.
- https://doi.org/10.7554/eLife.29338.015
To investigate the involvement of other Drosophila Atg genes in midgut cell growth and homeostasis, we systematically depleted Atg1, Atg13, Atg17/FIP200, Atg9, Atg7, Atg12, Atg16, Atg18, and Vps34 with NP1-Gal4; tubulin-Gal80ts as driver. Surprisingly, only RNAi targeting Atg9 and components of the Atg1 kinase complex, but not other Atg genes, caused prominent defects in the midgut epithelium (Figure 5A,B). Similar to the Atg9 mutant, depletion of Atg1, Atg13, and Atg17 showed enlarged and disorganized midgut epithelial cells. Moreover, we found that knockdown of Atg1, Atg13, and Atg17 in the adult fly also caused intestinal barrier dysfunction and shortened lifespan (Figure 5—figure supplement 1A and B). We further confirmed that autophagy activity is efficiently blocked by temporal knockdown of these Atg genes (Figure 5—figure supplement 2A and B). Studies in yeast have shown that Atg9 is a direct target of Atg1 kinase during early autophagsome formation (Papinski et al., 2014). Activation of the Atg1 complex recruits and tethers Atg9-containing vesicles for the initiation of autophagy (Rao et al., 2016). In Drosophila, we have previously shown that ablation of Atg9 suppresses Atg1-induced eye roughness and wing vein defects (Tang et al., 2013). We thus investigated whether Atg1 also genetically interacts with Atg9 in the adult midgut. Indeed, we found that overexpression of Atg1 can rescue the midgut defects caused by depletion of Atg9, whereas Atg1 depletion enhanced the phenotype (Figure 5C). These results highlight the critical role of Atg9 and the Atg1 kinase complex in maintaining adult midgut epithelium homeostasis.
Figure 5 with 2 supplements see all
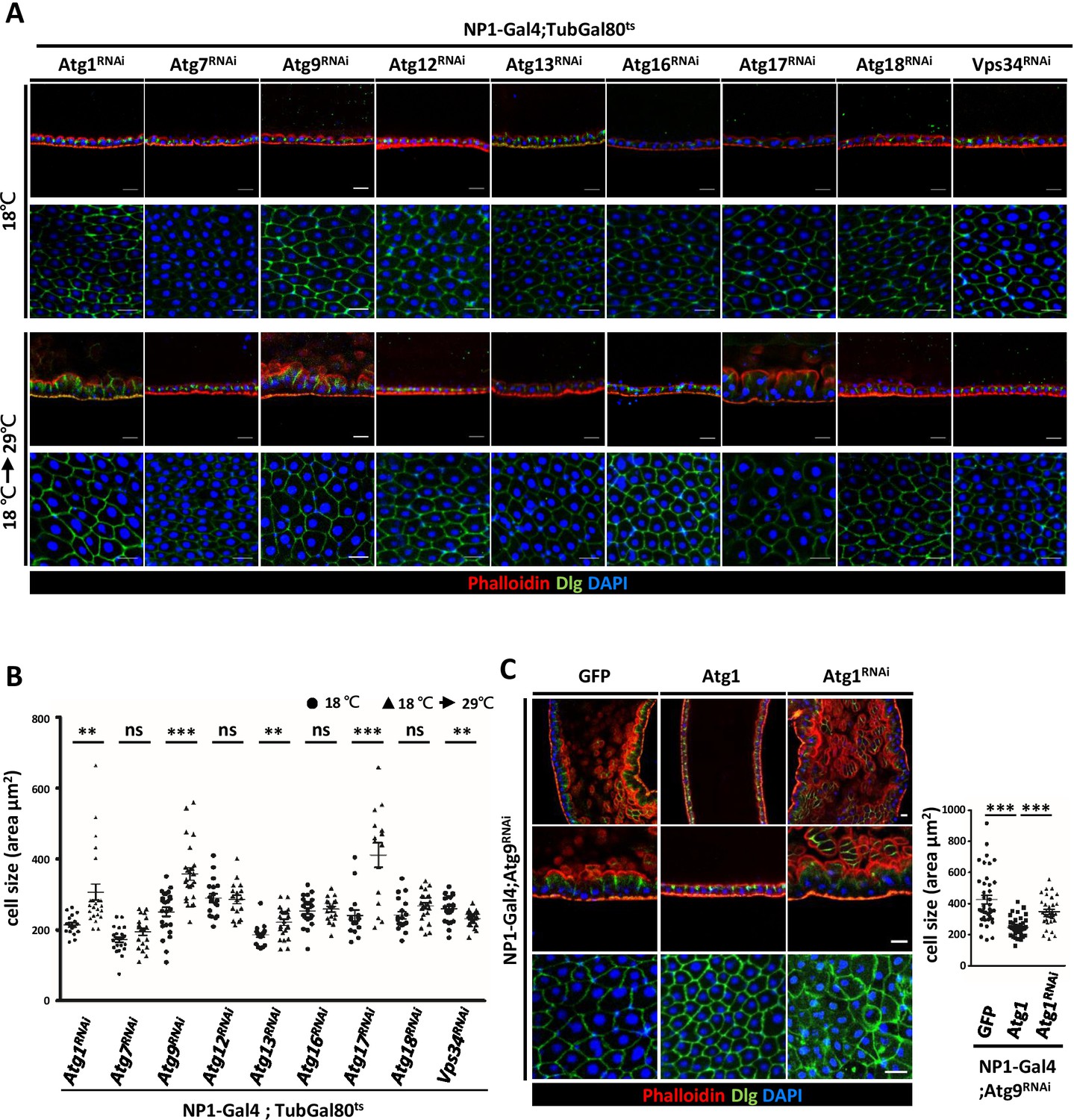
Components of Atg1 kinase complex are required for adult midgut epithelium homeostasis.
(A) Systematic knock-down of Drosophila Atg1, Atg7, Atg9, Atg12, Atg13, Atg16, Atg17, Atg18 and Vps34 in adult midgut with the EC-specific driver NP1-Gal4; Gal80ts. The flies were either kept at 18°C throughout development or shifted to 29°C after eclosion for 5 days to inactivate Gal80ts and enable expression of the RNAi targeting Atg genes. Ablation of Atg1, Atg13, and Atg17, but not other Atg genes, caused increased cell size. (B) Quantification of posterior midgut cell size (shown in A) of Atg depleted flies. n ≥ 15, data are mean ±s.e.m. **p<0.01, ***p<0.001. ns, not statistically significant. (C) Overexpression of Atg1 suppressed Atg9RNAi-induced adult midgut defects. n ≥ 30, ***p<0.001. Scale bar: 20 μm.
-
Figure 5—source data 1
Quantification of cell size.
- https://doi.org/10.7554/eLife.29338.019
Functional interaction between Atg9 and the TOR pathway
The target of rapamycin (TOR) signaling pathway has been shown to regulate cell growth and proliferation (Miron and Sonenberg, 2001; Zhang et al., 2000). TOR is activated by the phosphatidylinositol 3-kinase (PI3K)/AKT pathway in response to nutrients or growth factors such as insulin stimulation (Dibble and Cantley, 2015; Oldham and Hafen, 2003). The activated TOR kinase phosphorylates ribosomal protein S6 kinase (S6K) and the eukaryotic translation initiation factor 4E-binding protein (4EBP) to regulate protein translation and cell size (Katewa and Kapahi, 2011). Studies in Drosophila and mammals have shown that loss of Atg1 and Atg17 can provoke TOR/S6K-dependent cell growth and development (Kim et al., 2013; Lee et al., 2007; Scott et al., 2007). This prompted us to investigate whether TOR signaling is activated in Atg9 mutants. Intriguingly, Western blot analysis showed a marked increase in S6K and 4EBP phosphorylation levels in the midguts of Atg9 mutants, compared to controls (Figure 6A). Inhibition of TOR activity by feeding Atg9 mutant flies with rapamycin effectively suppressed the enlarged midgut cell size and aberrant epithelial morphology of the Atg9 mutants (Figure 6B and C). Moreover, we found that rapamycin treatment significantly rescued the intestinal barrier dysfunction (Figure 6—figure supplement 1A), but not the lifespan defects of Atg9 mutants (Figure 6—figure supplement 1B). We next checked whether modulation of the components of TOR signaling could rescue the Atg9 midgut defects. Overexpression of the TOR negative regulator TSC1-TSC2 (the tuberous sclerosis complex 1 and 2), the dominant-negative TOR (TORTED), the dominant-negative S6K (S6KKQ), or knockdown of TOR activator Rheb (RhebRNAi) strongly suppressed Atg9RNAi-induced midgut defects (Figure 6D–H,L). Whereas overexpression of TOR activator Rheb or depletion of TSC1 or TSC2 enhanced Atg9RNAi-induced midgut defects (Figure 6I–K,L). Moreover, ablation of components of the insulin receptor (InR)-PI3K-AKT pathway, which is upstream of TOR, also rescues Atg9RNAi-induced midgut defects (Figure 6—figure supplement 2). Taken together, these results suggest that Atg9 antagonizes TOR signaling in regulating cell growth and tissue homeostasis in the adult midgut.
Figure 6 with 2 supplements see all
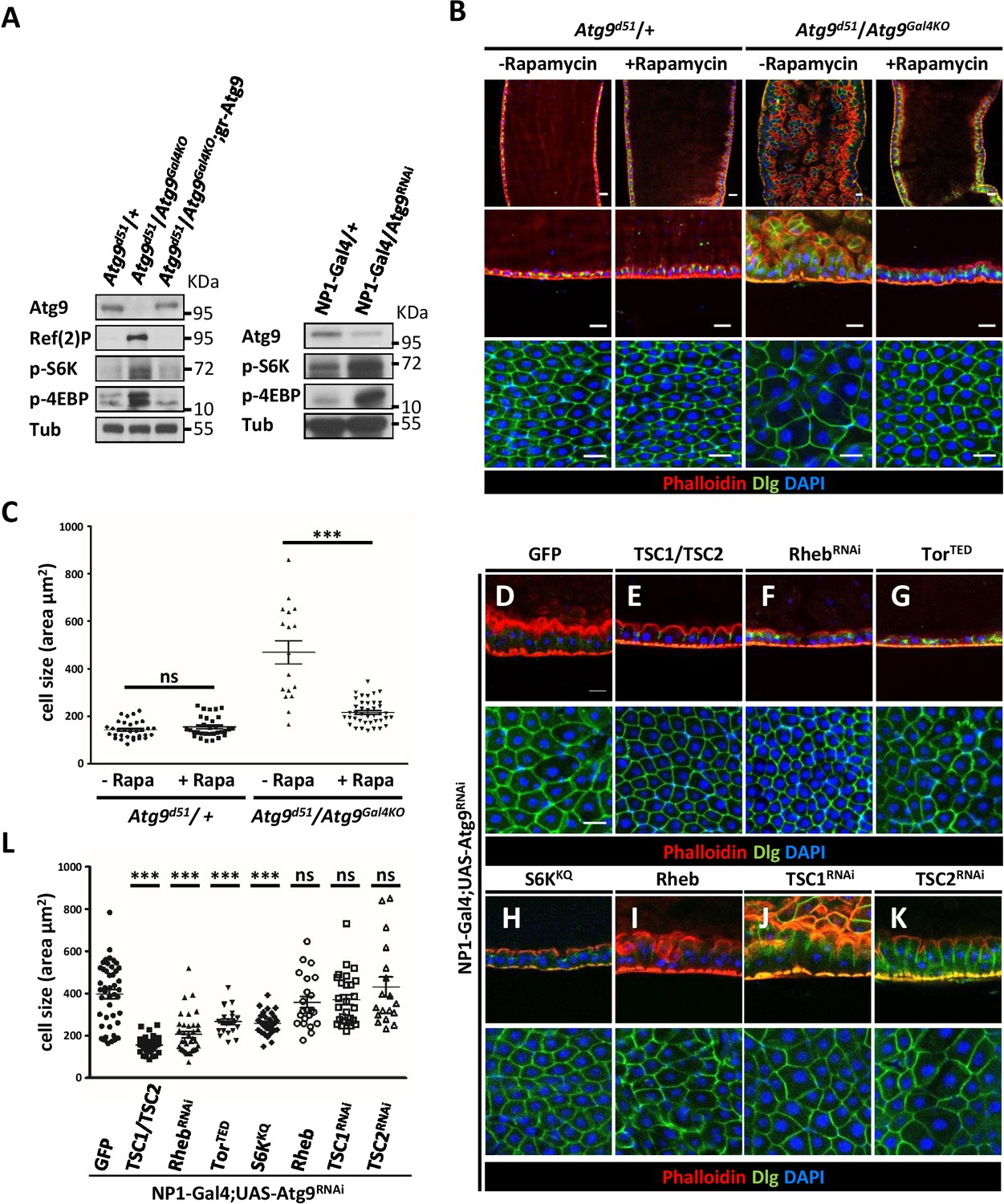
Loss of Atg9 enhances TOR activity in Drosophila adult midgut.
(A) The adult midguts of denoted genotypes were dissected, lysed, and subjected to Western blot analysis with anti-Atg9, anti-Ref(2)P, anti-p-S6K, anti-p-4EBP and anti-tubulin antibodies. (B) Inhibition of TOR activity by feeding flies with rapamycin rescued Atg9 mutant midgut defects. (C) Quantification of posterior midgut cell size shown in (B). n ≥ 17, data are mean ±s.e.m. ***p<0.001. (D–K) Atg9 genetically interacts with components of the TOR signaling pathway. The Atg9RNAi-induced midgut defects (D) could be suppressed by the coexpression of TSC1-TSC2 (E), RhebRNAi (F), dominant-negative TOR (TORTED) (G), or dominant-negative S6K (S6KKQ) (H), whereas coexpression of TOR activator Rheb (I) or knock-down of TSC1 (J) or TSC2 (K) could not rescue the Atg9RNAi-induced midgut defects. Genetic analyses were performed for three times with 100% penetrance of the phenotype. (L) Quantification of posterior midgut cell size shown in (D–K). n ≥ 18, data are mean ±s.e.m. ***p<0.001. ns, not statistically significant. Scale bar: 20 μm.
-
Figure 6—source data 1
Quantification of cell size.
- https://doi.org/10.7554/eLife.29338.025
Atg9 interacts with Patj and TSC2 to regulate midgut cell growth
To understand the mechanism by which Atg9 negatively regulates TOR signaling and cell growth, we performed a pull down assay using GST-fused C-terminus (residues 668–845) of Atg9 and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identity Atg9-interacting proteins. Among the proteins that were identified to interact with Atg9, we focused on Pals1-associated tight junction protein (Patj). While the multi-PDZ domain containing protein Patj forms a complex with the apical polarity protein Crumbs (Crb) and Stardust (Sdt; Pals1), several studies have indicated that Patj is not essential for apical basal polarity in Drosophila (Pénalva and Mirouse, 2012; Sen et al., 2012; Zhou and Hong, 2012). In mammals, Patj binds to tight-junction associated proteins such as Pals1, Claudin 1, and ZO-3, and regulates tight junction formation and cell migration (Roh et al., 2002; Shin et al., 2007). Interestingly, recent findings have shown that Patj can interact directly with TSC2 and depletion of Patj leads to increased TOR activity in human intestinal epithelial cells (Massey-Harroche et al., 2007; Rosner et al., 2008), suggesting that Patj may regulate TOR signaling through its interaction with TSC2. We thus performed GST pull-down and co-immunoprecipitation assays to confirm the interaction between Atg9 and Patj. As shown in Figure 7A, GST-Atg9-C, but not GST, efficiently interacted with Patj. To determine whether full-length Atg9 interacts with Patj, HEK293 cells were transiently transfected with Flag-tagged Atg9 (Flag-Atg9) and Myc-tagged Patj (Myc-Patj). Immunoblotting of the anti-Flag immunoprecipitates from cell lysates showed that Patj co-precipitated with Atg9 (Figure 7B). Similarly, a reciprocal co-immunoprecipitation experiment with anti-Myc antibody revealed an interaction between Atg9 and Patj (Figure 7C). Moreover, co-immunoprecipitation assays showed that Patj can interact with TSC2 in Drosophila S2 cells (Figure 7D). We next determined whether Patj could genetically interact with Atg9 in maintaining intestinal homeostasis. Indeed, similar to the Atg9 mutant, we found that depletion of Patj with NP1-Gal4 results in aberrant intestinal epithelium (Figure 7E). Overexpression of Patj in the midgut largely rescued the Atg9 depletion-induced midgut defects (60%, n = 24) but not the midgut defects caused by Atg1 or Atg17 depletion (Figure 7F and Figure 7—figure supplement 1A and B), whereas Patj depletion exacerbated the midgut defects caused by Atg9 ablation (Figure 7F). We have previously shown that ablation of Atg9 in EC cells leads to increased lethality in response to paraquat ingestion (Tang et al., 2013). Strikingly, ectopic expression of Patj in EC cells significantly rescued the paraquat-induced lethality of Atg9 knockdown animals (Figure 7—figure supplement 1C).
Figure 7 with 1 supplement see all
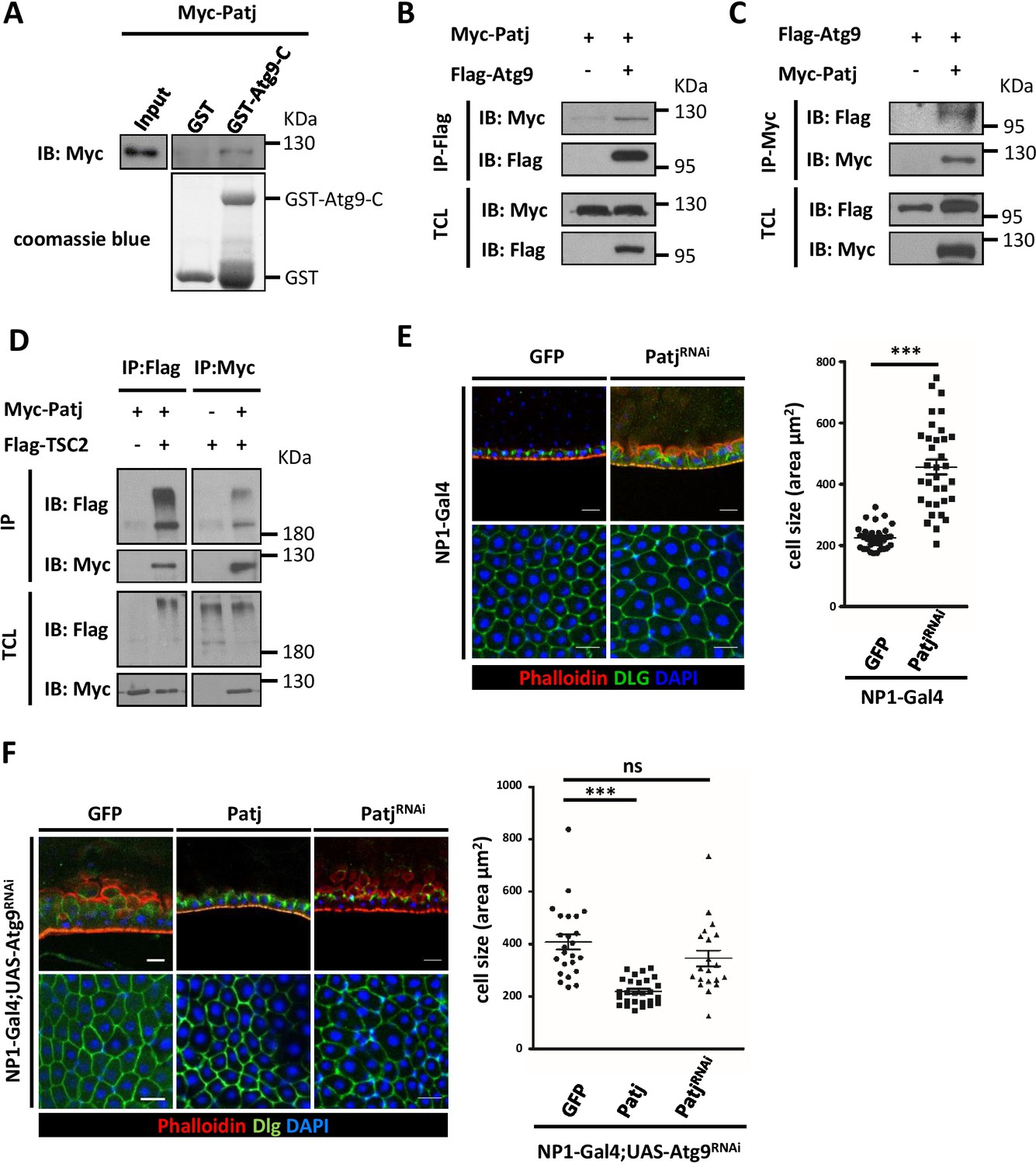
Atg9 interacts with Patj to regulate midgut cell growth.
(A) Lysates of cells expressing Myc-Patj were incubated with GST or GST–Atg9-C (amino acids 668–845) in GST pull-down assays. The pull-down products and input Myc-Patj were analyzed by Western blots with the anti-Myc antibody. (B–C) HEK293T cells transfected with Flag-Atg9 and Myc-Patj were subjected to immunoprecipitation with anti-Flag (B) or anti-Myc (C) antibody. The immunoprecipitates and total cell lysates (TCL) were analyzed by Western blot with antibodies as indicated. (D) S2 cells transfected with pWA-Gal4, pUAS-Flag-TSC2 and pUAS-Myc-Patj were subjected to immunoprecipitation with anti-Flag or anti-Myc antibody. The immunoprecipitates and total cell lysates were analyzed by Western blot with antibodies as indicated. (E) Depletion of Patj with NP1-Gal4 resulted in aberrant midgut epithelium and increased EC cell size. n ≥ 34, data are mean ±s.e.m. ***p<0.001. (F) Patj genetically interacts with Atg9. Overexpression of Patj suppressed the Atg9 depletion-induced midgut defects (60% penetrance, n = 24). The cell size of posterior midgut ECs of each genotype was quantified. n ≥ 20, data are mean ±s.e.m. ***p<0.001. Scale bar: 20 μm.
-
Figure 7—source data 1
Quantification of cell size.
- https://doi.org/10.7554/eLife.29338.030
Several studies have shown that TSC2 is a short-lived protein and is readily targeted for degradation (Chong-Kopera et al., 2006; Hu et al., 2008). We thus investigated whether Atg9 could interact with and regulate TSC2 stability. As shown in Figure 8A, Atg9 specifically interacted with TSC2 by immunoprecipitation experiments, and depletion of Atg1 does not affect the association between Atg9, Patj and TSC2 (Figure 8—figure supplement 1A). More strikingly, clonal depletion of Atg9 (RFP-positive cells) in the adult midgut caused a marked decrease in TSC2 levels (Figure 8B), whereas TSC2 levels were not affected in Atg1 or Atg17 knockdown cells (Figure 8—figure supplement 1B). Immunoblotting analysis showed that TSC2 level is markedly decreased in the midgut of Atg9 mutants, compared with controls (Figure 8C). Collectively, our results suggest that Atg9 antagonizes TOR signaling by interacting with Patj-TSC2 and regulates TSC2 stability (Figure 8D).
Figure 8 with 1 supplement see all
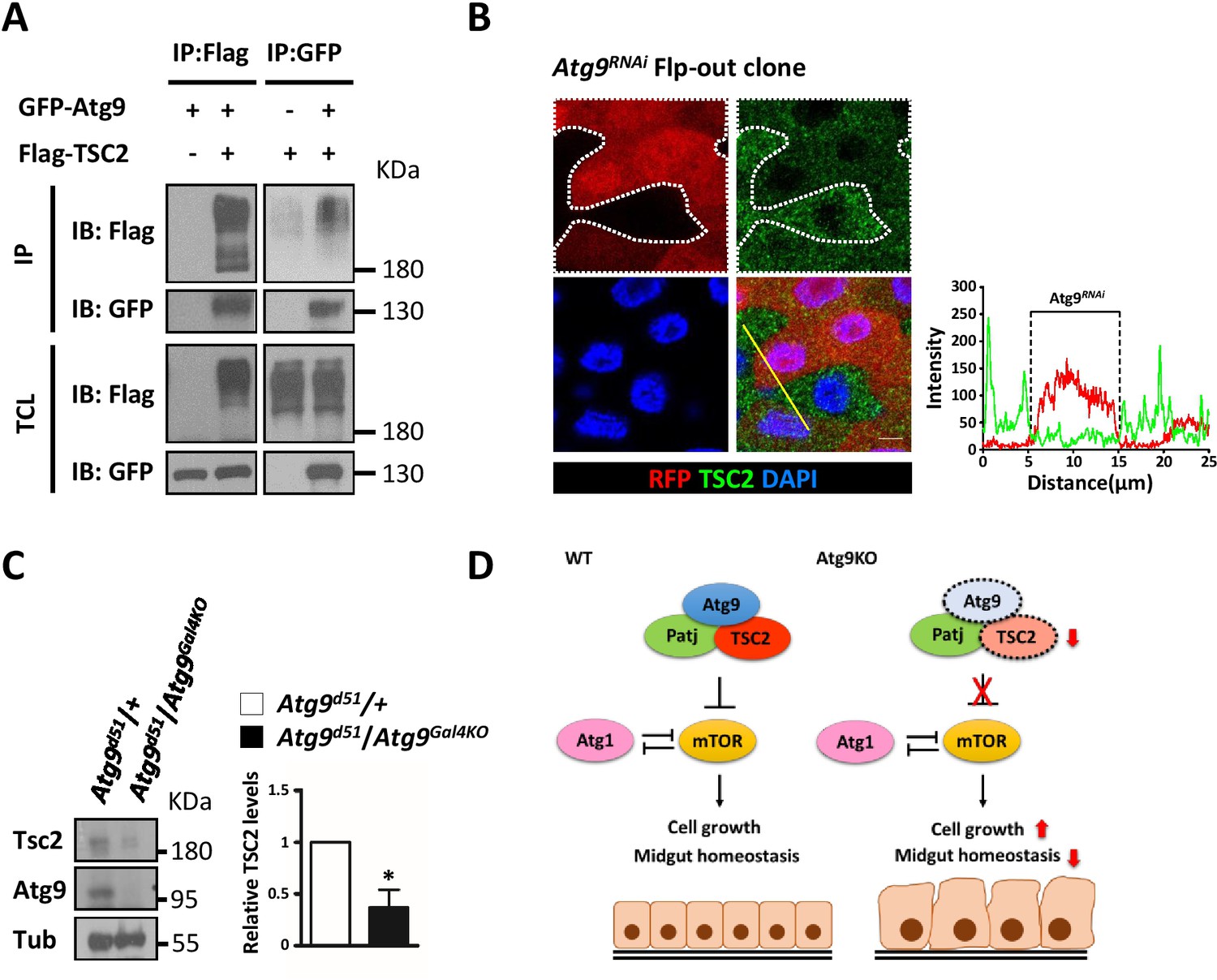
Atg9 interacts with TSC2 to regulate midgut cell growth.
(A) S2 cells transfected with pWA-Gal4, pUAS-Flag-TSC2 and pUAS-GFP-Atg9 were subjected to immunoprecipitation with anti-Flag or anti-GFP antibody. The immunoprecipitates and total cell lysates were analyzed by Western blot with antibodies as indicated. (B) Clonal expression of Atg9RNAi (marked in Red) in the adult midgut resulted in marked decrease in TSC2 levels (green). Line scan across the Atg9RNAi clone to show the relative fluorescent intensities of TSC2 in control (dsRed-negative) and Atg9 depletion (dsRed-positive) cells. Scale bar: 5 μm. (C) The adult midguts of denoted genotypes were dissected, lysed, and subjected to Western blot analysis with anti-Atg9, anti-TSC2 and anti-tubulin antibodies. N = 3, data are mean ±s.e.m. *p<0.05. (D) Model for the antagonistic effect of Atg9 on TOR signaling in the regulation of intestinal cell growth and midgut homeostasis in Drosophila. Genotypes: (G) hsflp; UAS-Atg9RNAi/+; Act-CD2-Gal4-UAS-dsRed..
-
Figure 8—source data 1
Quantification of fluorescent intensity and Western blots.
- https://doi.org/10.7554/eLife.29338.034
Discussion
In this study, we generated Drosophila Atg9 null mutants to determine the developmental and physiological function of Atg9. Similar to other autophagy mutants such as Atg7 and Atg17/Fip200 null flies (Juhász et al., 2007; Kim et al., 2013), Atg9 mutants exhibit severe defects in autophagy, shortened lifespan, impaired motility, and hypersensitivity to stresses. Atg9 loss-of-function also leads to aberrant midgut enlargement and intestinal barrier dysfunction. Interestingly, we found that, unlike Atg9 mutant, depletion of Atg7 in adult flies did not cause aberrant midgut enlargement or intestinal barrier dysfunction (Figure 5 and Figure 5—figure supplement 1A). It is possible that the adult midgut defects may contribute to the much shorter lifespan of Atg9 mutants. Our findings indicate that Atg9 not only acts as a key regulator in autophagy but also functions in maintaining adult Drosophila midgut homeostasis.
The Drosophila adult midgut is composed of a monolayer of epithelial cells including nutrient absorbing enterocytes (ECs), secretory enteroendocrine (EE) cells, and multipotent intestinal stem cells (ISCs) (Lemaitre and Miguel-Aliaga, 2013). A number of conserved signaling pathways, including insulin, Notch, EGFR, Wingless (Wg)/Wnt, Hippo, TOR, and JAK-STAT pathways, have been shown to be involved in the regulation of ISC proliferation and in the maintenance of tissue homeostasis of the Drosophila midgut (Guo et al., 2016; Jiang et al., 2016). The enlarged adult midguts observed in Atg9 mutants may be due to dysregulation of cell proliferation and cell growth. ISCs are the only dividing cells in the Drosophila adult midgut and play an essential role in maintaining tissue homeostasis. However, we found that the total number of intestinal cells and Delta+ ISC population were not affected in Atg9 mutants. PH3 staining of control or Atg9 mutant midguts at young (5 day) or old (30 day) stages showed similar numbers of mitotic ISCs in both animals, thus indicating that loss of Atg9 does not affect ISC proliferation.
Insulin and TOR signaling are conserved nutrient-sensing pathways involved in regulating cell growth, metabolism and tissue homeostasis (Oldham and Hafen, 2003). Recent studies have reported that these pathways regulate enterocyte growth and endoreduplication in the adult Drosophila midgut (Amcheslavsky et al., 2011; Kapuria et al., 2012; Xiang et al., 2017). As the loss of Atg9 leads to a marked increase in TOR activity, while inhibition of insulin/TOR signaling rescues Atg9 midgut defects, Atg9 may act as a negative regulator of TOR-mediated cell growth. It is also interesting to note that although Atg9 depletion caused a dramatic enlargement in adult midgut ECs, we did not find an increase in larval disc cell size (Figure 4—figure supplement 2). It has been shown that the normal development and tissue homeostasis is maintained by a delicate coordination between cell growth and division (Jorgensen and Tyers, 2004; Zeng et al., 2013). One possible explanation for this difference is that ECs in adult midgut are non-dividing differentiated cells, whereas larval disc cells are active dividing cells and therefore maintain a moderate cell size.
Besides Atg9, we found that depletion of components of the Atg1 kinase complex also resulted in enlarged adult midgut ECs. While TOR inhibits Atg1/Ulk1-mediated autophagy induction under nutrient-rich conditions, recent reports have shown that Atg1 and Ulk1 negatively regulate TOR signaling in Drosophila and mammalian cells, respectively (Lee et al., 2007; Scott et al., 2007). Although Atg9 was found to be a downstream target of Atg1 during autophagy, several lines of evidence suggest that Atg1 inhibits TOR independent of Atg9. First, our genetic results showed that overexpression of Atg1 rescued Atg9RNAi-induced midgut defects; in contrast, overexpression of Atg9 or Patj could not suppress the midgut defects caused by Atg1 depletion (Figure 7—figure supplement 1A). Second, co-immunoprecipitation assays showed that Atg9 can still interact with Patj and TSC2 in Atg1 knockdown S2 cells (Figure 8—figure supplement 1A). Thirdly, clonal analysis showed that although the TSC2 level was markedly decreased in Atg9 knockdown cells, TSC2 levels were not affected in Atg1 or Atg17 knockdown cells, compared with controls (Figure 8—figure supplement 1B). In mammals, it has been shown that Atg1/Ulk1 inhibits TOR signaling by phosphorylating Raptor and impairs substrate binding to Raptor (Dunlop et al., 2011; Jung et al., 2011), and the inhibitory effect of Ulk1 on TOR signaling occurred independently of TSC2 (Jung et al., 2011). Our results and findings from mammalian cells together strongly indicate that Atg9 inhibits TOR activity independent of Atg1.
The formation of the midgut epithelium in Drosophila depends on the establishment of cell polarity and adhesion (Müller, 2000). One striking phenotype observed in Atg9 mutant midgut epithelium is the appearance of enlarged epithelial cells with aberrant apical membrane expansion. The Drosophila epithelial polarity is regulated by evolutionarily conserved polarity protein complexes including the Crumbs (Crb)/Stardust (Sdt)/Patj complex, the Bazooka (Baz)/Par6/aPKC complex and the Scribble/Discs large (Dlg)/Lethal giant larva (Lgl) complex (Tepass, 2012). While Crumbs and Par complexes are localized apically in epithelial cells for apical domain maintenance, the Scribble complex is localized in the basolateral region for the maintenance of the basolateral membrane. It has been reported that the Par proteins Par3/Baz, Par6, and aPKC are involved in regulating asymmetric cell division and the differentiation of adult Drosophila ISCs (Goulas et al., 2012). Here, we identified Patj as a novel interactor of Atg9. The scaffolding protein Patj interacts with both Crb-Std and Baz-Std apical polarity protein complexes, and has been shown to play a non-essential role in apical-basal polarity during Drosophila development (Pénalva and Mirouse, 2012; Sen et al., 2015; Zhou and Hong, 2012). However, the role of Patj in midgut epithelium formation and the molecular mechanisms underlying Atg9-Patj-TSC2-mediated intestinal cell growth remain unknown. Interestingly, like Atg9, Patj has been shown to cycle between plasma membrane and endosomal compartments in mammalian cells (Heller et al., 2010; Wells et al., 2006). Ablation of Patj results in accumulation of Crumbs3 in EEA-1 positive early endosomes (Michel et al., 2005). It will be exciting to determine how Atg9 coordinates with Patj to regulate TSC2 stability and intestinal epithelial homeostasis. Further research should aid our understanding in the regulation of TOR signaling by Atg9 and its role in human diseases.
Materials and methods
Drosophila strains and genetics
Request a detailed protocolFlies were raised at 25°C following standard procedures unless otherwise noted. The following Drosophila strains were used: Atg7d14, Atg7d77 (Juhász et al., 2007), UAS-Atg1 (Scott et al., 2007), Dl-Gal4, Su(H)GBE-Gal4, (Zeng et al., 2010), How-Gal4ts (gift from Bruno Lemaitre, EPFL), NP1-Gal4, UAS-TSC1, UAS-TSC2 (Tang et al., 2013), UAS-Rheb (RRID:BDSC_9688), UAS-S6KKQ (RRID:BDSC_6911), UAS-TorTED (RRID:BDSC_7013), UAS-Atg1RNAi (RRID:BDSC_44034), UAS-RhebRNAi (RRID:BDSC_33966), UAS-Atg7RNAi (RRID:BDSC_34369), UAS-Atg9RNAi (RRID:BDSC_34901), UAS-Atg12RNAi (RRID:BDSC_34675), and Df(2R)Exel7142 (RRID:BDSC_7886) flies were obtained from the Bloomington Stock Center; UAS-PatjRNAi (RRID:FlyBase_FBst0473750), UAS-Atg13RNAi (RRID:FlyBase_FBst0475239), UAS-Atg16RNAi (RRID:FlyBase_FBst0477819), UAS-Atg17RNAi (RRID:FlyBase_FBst0476692), UAS-Atg18RNAi (RRID:FlyBase_FBst0477193), UAS-Vps34RNAi (RRID:FlyBase_FBst0472170), UAS-TSC1RNAi (RRID:FlyBase_FBst0454493), and UAS-TSC2RNAi (RRID:FlyBase_FBst0470276) were obtained from the Vienna Drosophila Resource Center.
Generation of Atg9 mutants
Request a detailed protocolThe Atg9Gal4KO mutant was generated by knock-in replacement of the Atg9 genomic region with the Gal4 cassette as described previously (Chan et al., 2011). In brief, we first generated two 500 bp homology arms flanking the 20 kb Atg9-containing genomic fragment for first-round recombineering. The following primers were used in a SOEing PCR reaction:
LA500-fwd: 5’-ACAAGTTTGTACAAAAAAGCAGGCT TTGGCAGGCACACGACATTT −3’
LA500-rev: 5’-CACGCAGGATCCCTTCAATCCAGAGCAACAGG-3’
RA500-fwd: 5’-TTGAAGGGATCCTGCGTGGAACCCATCTTTGG-3’
RA500-rev: 5’-ACCACTTTGTACAAGAAAGCTGGGT TTGCATTTTGTTTGCTAAGT −3’
The 1 kb PCR product was then cloned into P[acman]-KO to generate P[acman]-KO-Atg9-1kb, and the resultant plasmid was digested with BamHI and transformed into DY380 cells containing the Atg9 genomic DNA. To replace the Atg9 open-reading frame with the Gal4-RFP-Kan cassette, the following primers were used in second-round recombineering:
Atg9-fwd:5’-TCTCTTAGGAGAGTCAGCTGTTTGCTGAGAAGGTTCAGCAGAATCAAACAACAAAGAATTTTCCAACTTATACTATACAGCGATATAAATAGTCAGAACGGTTGACCTTGACGTTGGGCG-3’
Atg9-rev:5’-GTTTAGCTTAGTTTCAGATTAGTTTAGCTACGCACTAGACGACGTCGTTCGTTCGTTTACACTTTAAAATTTAGGTTAATCACTAATAGCAGAATGGGTGGTCCTTAGCTCTACAGGTGG-3’
The PCR product was transformed into DY380 cells containing P[acman]-KO-Atg9-20kb. The transformants containing P[acman]-Atg9-Gal4 were PCR-verified followed by sequencing. The confirmed P[acman]-Atg9-Gal4 was then injected into the embryo following standard transgenesis protocol. The transgenic Atg9Gal4 allele was then excised in vivo and targeted to the endogenous atg9 locus to generate Atg9Gal4KO flies.
The Atg9d51 mutant was generated using CRISPR/cas9-based genome editing (Kondo and Ueda, 2013). In brief, gRNA sequence GAGGGATGGTGCTCCAGGAA[CGG] was cloned into pBFv-U6.3. Cassette attPX-3-frame Stop-floxed 3xP3-RFP flanked by two Atg9 homology arms (500 bp upstream and downstream of CRISPR targeting site) was cloned into pCR2-TOPO. Atg9-targeting gRNA plasmid and donor template for repair were co-injected into embryos of nanos-Cas9 expressing flies. Progeny flies carrying the selection marker of 3xP3-RFP were further validated by genomic PCR and sequencing. The CRISPR-mediated mutagenesis was performed with the help of WellGenetics, Inc. To induce mitotic clones, hsFLP; FRT42D Ubi-GFP/FRT42D Atg9d51 flies were heat shocked in a 37°C water bath for 1 hr twice a day. MARCM clones were generated by placing hsFLP; FRT42D tubGal80/FRT42D Atg9d51; Tub-Gal4/UAS-mCD8GFP flies in a 37°C water bath for 1 hr twice a day. The flies were kept at 25°C for seven additional days before dissection.
RT-PCR
Request a detailed protocolTotal RNA was isolated from adult female flies using TRIzol (Invitrogen). cDNA was synthesized from 1 ug RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer’s instructions. The following pairs of specific primers were used: 5’-TTGTCCAGATCCGAATCCTC-3’ (Atg9-L); 5’-TCGTCTGGCTACTTGCCTTT-3’ (Atg9-R); 5’-TTGTCTGGGCAAGAGGATCAG-3’ (Actin5C-L); 5’-ACCACTCGCACTTGCACTTTC-3’ (Actin5C-R).
Plasmids and antibodies
Request a detailed protocolThe pUAS-GFP-Atg9 transgene was generated by PCR amplification Atg9 from RE14003 into the pUAST vector. The Myc-tagged Patj expression plasmid was generated by PCR amplification of Patj from LD22238, then cloned into the pUAST or pcDNA3.1 vector. Drosophila pUAS-Flag-TSC2 was kindly provided by Jun Hee Lee (University of Michigan). For the Atg9 genomic rescue construct, the Atg9 genomic locus flanked by 1 kb each of upstream and downstream genomic sequence was cloned into pCaSpeR4. Antibodies used for the study were: anti-Dlg (1:100, DSHB, University of Iowa), anti-Delta (1:100, RRID:AB_2056641), and anti-Prospero (1:100, RRID:AB_528440), anti-Atg9 (1:100) (Tang et al., 2013), anti-TSC2 (1:100, gift from Aurelio Teleman, German Cancer Research Center) (Tsokanos et al., 2016), anti-Pdm1 (1:100, gift from Xiaohang Yang, Zhejiang University), anti-Atg8 (1:100, RRID:AB_297935), anti-Ref(2)p (1:500, Abcam, Cat# ab178440), anti-Ub (1:100, RRID:AB_10691572), anti-phospho-S6K (1:1000, Cell Signaling, Cat# 9209), and anti-phospho-4EBP (1:1000, Cell Signaling, Cat# 2855), anti-GFP (1:100, RRID:AB_1563142), anti-Myc (1:500, RRID:AB_2298152), anti-PH3 (1:1000, RRID:AB_477043), anti-Flag (1:1000, RRID:AB_2687448), and anti-tubulin (1:5000, RRID:AB_1844090).
Cell culture, transfection and immunoprecipitation
Request a detailed protocolDrosophila S2 cells were cultured at 25°C in Schneider’s Drosophila medium (Thermo Fisher) containing 10% fetal bovine serum (FBS) and antibiotics. HEK293T cells were cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) medium (Thermo Fisher) supplemented with 10% FBS and antibiotics. S2 cells were transfected with Lipofectamine 2000 (Invitrogen), whereas HEK293T were transfected with PolyJet (SignaGen) according to the manufacturer's protocol. The dsRNAs were generated using the T7 RiboMAX Express RNAi System (Promaga). For immunoprecipitations, cells transiently transfected with the indicated plasmids were scraped from dishes in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton, 10% glycerol, 1 mM EDTA, 10 mM NaF, 1 mM PMSF, and protease inhibitor cocktail (Roche)). Cell lysates were immunoprecipitated with Myc or Flag antibody at 4°C overnight and protein G-Sepharose beads (GE Healthcare) at 4°C for 1 hr. These beads were washed three times with the lysis buffer.
Immunofluorescence
Request a detailed protocolFor Drosophila midgut immunohistochemistry, female adult midguts were dissected in PBS and immediately fixed in 4% paraformaldehyde for 40 min at room temperature. After fixation, the samples were permeabilized in PBST (PBS containing 0.5% Triton X-100) for 10 min, then blocked in PBST containing 5% Normal Goat Serum (NGS) for 1 hr. The samples were incubated with primary antibodies in PBST-NGS at 4°C for 16 hr. On the following day, samples were washed with PBST and incubated with fluorescent-labeled secondary antibodies at 4°C overnight. Nuclei were stained using DAPI (1 μg/ml). Images were acquired using the Zeiss LSM510 or Olympus FV3000RS confocal laser scanning microscope. For lysotracker staining, Drosophila larval fat bodies dissected under fed or starved conditions were incubated for 45 s in 100 μM LysoTracker Green DND-26 (Thermo) with DAPI in PBS, and immediately photographed live on an Olympus BX61 fluorescence microscope.
Lifespan, climbing, and stress resistance analyses
Request a detailed protocolThe lifespan assays were performed as described previously (Chen et al., 2012). Female or male flies were housed in groups of 20 and the flies were transferred to fresh food every 2–3 days until all were dead. Climbing assays (Martinez et al., 2007) and stress resistance experiments were performed as preciously described (Juhász et al., 2007; Tang et al., 2013). At least three independent measurements were performed for each experiment.
Rapamycin feeding
Request a detailed protocolRapamycin feeding was performed as previously described (Amcheslavsky et al., 2011), with flies being incubated on the food medium containing 50 μM rapamycin (Merck) for 7 days.
Statistical analyses
Request a detailed protocolStatistical analysis was performed by Student’s t test. Log-rank test was used for lifespan statistical analysis. Differences were considered significant if p values were less than 0.05 (*), 0.01 (**), or 0.001 (***).
References
-
Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacksMolecular and Cellular Biology 32:2–11.https://doi.org/10.1128/MCB.06159-11
-
Tuberous sclerosis complex and Myc coordinate the growth and division of Drosophila intestinal stem cellsThe Journal of Cell Biology 193:695–710.https://doi.org/10.1083/jcb.201103018
-
TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligaseJournal of Biological Chemistry 281:8313–8316.https://doi.org/10.1074/jbc.C500451200
-
The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagyThe Journal of Cell Biology 191:155–168.https://doi.org/10.1083/jcb.201002100
-
Regulation of mTORC1 by PI3K signalingTrends in Cell Biology 25:545–555.https://doi.org/10.1016/j.tcb.2015.06.002
-
Maintenance of the adult Drosophila intestine: all roads lead to homeostasisCurrent Opinion in Genetics & Development 40:81–86.https://doi.org/10.1016/j.gde.2016.06.009
-
Amot recognizes a juxtanuclear endocytic recycling compartment via a novel lipid binding domainJournal of Biological Chemistry 285:12308–12320.https://doi.org/10.1074/jbc.M109.096230
-
Atg9A trafficking through the recycling endosomes is required for autophagosome formationJournal of Cell Science 129:3781–3791.https://doi.org/10.1242/jcs.196196
-
Intestinal stem cell function in Drosophila and miceCurrent Opinion in Genetics & Development 22:354–360.https://doi.org/10.1016/j.gde.2012.04.002
-
Intestinal stem cell response to injury: lessons from DrosophilaCellular and Molecular Life Sciences 73:3337–3349.https://doi.org/10.1007/s00018-016-2235-9
-
How cells coordinate growth and divisionCurrent Biology 14:R1014–R1027.https://doi.org/10.1016/j.cub.2004.11.027
-
Role of TOR signaling in aging and related biological processes in Drosophila melanogasterExperimental Gerontology 46:382–390.https://doi.org/10.1016/j.exger.2010.11.036
-
TBC1D14 regulates autophagy via the TRAPP complex and ATG9 trafficThe EMBO Journal 35:281–301.https://doi.org/10.15252/embj.201592695
-
The digestive tract of Drosophila melanogasterAnnual Review of Genetics 47:377–404.https://doi.org/10.1146/annurev-genet-111212-133343
-
An Atg9-containing compartment that functions in the early steps of autophagosome biogenesisThe Journal of Cell Biology 190:1005–1022.https://doi.org/10.1083/jcb.200912089
-
Age-related changes in climbing behavior and neural circuit physiology in DrosophilaDevelopmental Neurobiology 67:778–791.https://doi.org/10.1002/dneu.20388
-
Evidence for a molecular link between the tuberous sclerosis complex and the Crumbs complexHuman Molecular Genetics 16:529–536.https://doi.org/10.1093/hmg/ddl485
-
PATJ connects and stabilizes apical and lateral components of tight junctions in human intestinal cellsJournal of Cell Science 118:4049–4057.https://doi.org/10.1242/jcs.02528
-
Regulation of translation via TOR signaling: insights from Drosophila melanogasterThe Journal of Nutrition 131:2988S–2993.
-
Autophagy in mammalian development and differentiationNature Cell Biology 12:823–830.https://doi.org/10.1038/ncb0910-823
-
Insulin/IGF and target of rapamycin signaling: a TOR de force in growth controlTrends in Cell Biology 13:79–85.https://doi.org/10.1016/S0962-8924(02)00042-9
-
Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagyMolecular Biology of the Cell 23:1860–1873.https://doi.org/10.1091/mbc.E11-09-0746
-
The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagyNature Communications 7:10338.https://doi.org/10.1038/ncomms10338
-
The carboxyl terminus of zona occludens-3 binds and recruits a mammalian homologue of discs lost to tight junctionsJournal of Biological Chemistry 277:27501–27509.https://doi.org/10.1074/jbc.M201177200
-
The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partnersMutation Research/Reviews in Mutation Research 658:234–246.https://doi.org/10.1016/j.mrrev.2008.01.001
-
ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinaseNature Cell Biology 15:741–750.https://doi.org/10.1038/ncb2757
-
Autophagy regulation by nutrient signalingCell Research 24:42–57.https://doi.org/10.1038/cr.2013.166
-
Drosophila PATJ supports adherens junction stability by modulating Myosin light chain activityThe Journal of Cell Biology 199:685–698.https://doi.org/10.1083/jcb.201206064
-
Localization and function of pals1-associated tight junction protein in Drosophila is regulated by two distinct apical complexesJournal of Biological Chemistry 290:13224–13233.https://doi.org/10.1074/jbc.M114.629014
-
The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survivalAnnual Review of Cell and Developmental Biology 28:655–685.https://doi.org/10.1146/annurev-cellbio-092910-154033
-
The role of membrane proteins in mammalian autophagySeminars in Cell & Developmental Biology 21:677–682.https://doi.org/10.1016/j.semcdb.2010.03.007
-
eIF4A inactivates TORC1 in response to amino acid starvationThe EMBO Journal 35:1058–1076.https://doi.org/10.15252/embj.201593118
-
Atg9 vesicles are an important membrane source during early steps of autophagosome formationThe Journal of Cell Biology 198:219–233.https://doi.org/10.1083/jcb.201202061
-
Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomesJournal of Cell Science 119:3888–3900.https://doi.org/10.1242/jcs.03172
-
Stem cells in the Drosophila digestive systemAdvances in Experimental Medicine and Biology 786:63–78.https://doi.org/10.1007/978-94-007-6621-1_5
-
Regulation of cellular growth by the Drosophila target of rapamycin dTORGenes & Development 14:2712–2724.https://doi.org/10.1101/gad.835000
Article and author information
Author details
Funding
Ministry of Science and Technology, Taiwan (MOST104-2311-B-002-017-MY3)
- Chih-Chiang Chan
Ministry of Science and Technology, Taiwan (MOST105-2311-B-001-062-MY3)
- Guang-Chao Chen
Academia Sinica (101CDA-Lo4)
- Guang-Chao Chen
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Chun-Hong Chen, Gábor Juhász, Elisabeth Knust, Bruno Lemaitre, Jun Hee Lee, Thomas Neufeld, Henry Sun, Aurelio Teleman, Xiaohang Yang, Xiankun Zeng, the Bloomington Stock Center, Vienna Drosophila RNAi Center, Developmental Studies Hybridoma Bank, and Fly Core Taiwan for reagents and fly stocks. We thank Horng-Dar Wang for technical advice and Chiou-Yang Tang and WellGenetics for Drosophila embryo injection. We are grateful to Chi-Kuang Yao for helpful comments on the manuscript and Cindy Lee for English editing. This work was supported in part by the Ministry of Science and Technology of Taiwan (MOST105-2311-B-001–062-MY3 to GCC and MOST104-2311-B-002–017-MY3 to CCC) and the Academia Sinica Career Development Award (101CDA-L04 to GCC).
Copyright
© 2017, Wen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,544
- views
-
- 709
- downloads
-
- 51
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 51
- citations for umbrella DOI https://doi.org/10.7554/eLife.29338
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Atg9 antagonizes TOR signaling to regulate intestinal cell growth and epithelial homeostasis in Drosophila
eLife 6:e29338.
https://doi.org/10.7554/eLife.29338




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}