Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy
- University of Geneva, Switzerland
Figures
Figure 1 with 1 supplement
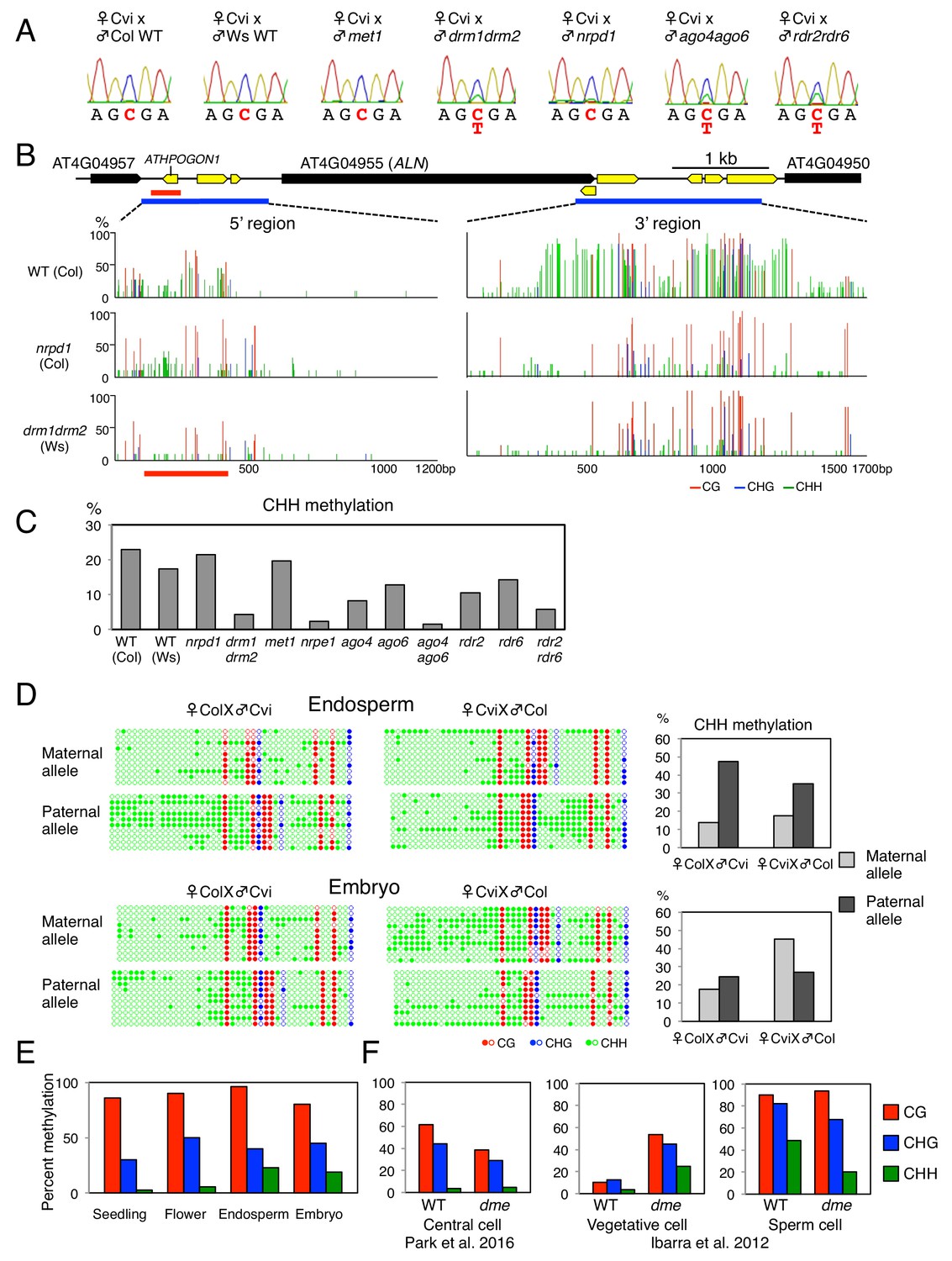
ALN imprinting necessitates non-canonical RdDM.
(A) Sanger sequencing chromatograms at SNPs of ALN. WT plants (Cvi) were pollinated with WT (Col and Ws) and mutant pollen as indicated. RNA was extracted from endosperm of F1 seeds and subjected to RT-PCR followed by Sanger sequencing. Nucleotides at SNP sites are highlighted in red: ‘C’ originates from Cvi and ‘T’ originates from Ws and Col. Representative results are shown from experiments repeated at least 5 times for each genotype. (B) DNA methylation on the ALN 5’ and 3’ flanking regions in endosperm of mature seed. Black box arrows and yellow box arrows show genes and TEs respectively. Blue lines show regions where DNA methylation was studied. Red line (300 bp) shows ALN’s highly methylated 5’ upstream region (referred as ‘POGO region’). All remaining methylation data shown in this figure only correspond to the 300 bp POGO region. Red, blue and green vertical lines represent CG, CHG, and CHH methylation levels, respectively. (C) Percentages of CHH methylation levels corresponding to the POGO region in different RdDM mutants (Materials and methods). drm1drm2 is in Ws background; ago4ago6 is generated after crossing ago4 with ago6 in Ler and C24 background, respectively; all other mutants are in Col-0 background. Percentages of DNA methylation at CG and CHG sites are shown in Figure 1—figure supplement 1. (D) DNA methylation levels in maternal and paternal alleles. DNA extracted from endosperm and embryo of F1 seeds obtained after reciprocally crossing Cvi and Col WT plants was analyzed by sodium bisulfite sequencing. SNPs were used to distinguish maternal and paternal alleles. Filled and open circles represent methylated and unmethylated cytosines, respectively. (E) Percentage of DNA methylation in the POGO region in different tissues. (F) Percentage of DNA methylation in the POGO region in female and male gametes. The data were extracted from published whole genomic DNA methylation data of female (Park et al., 2016) and male (Ibarra et al., 2012) gametes.
-
Figure 1—source data 1
Figure 1B and 2B: DNA methylation in the 5’ and 3’ flanking regions of ALN in the endosperm of mature seeds.
- https://doi.org/10.7554/eLife.37434.004
-
Figure 1—source data 2
Figure 1C: Percentages of DNA methylation levels in the POGO region of different RdDM mutants.
- https://doi.org/10.7554/eLife.37434.005
Figure 1—figure supplement 1
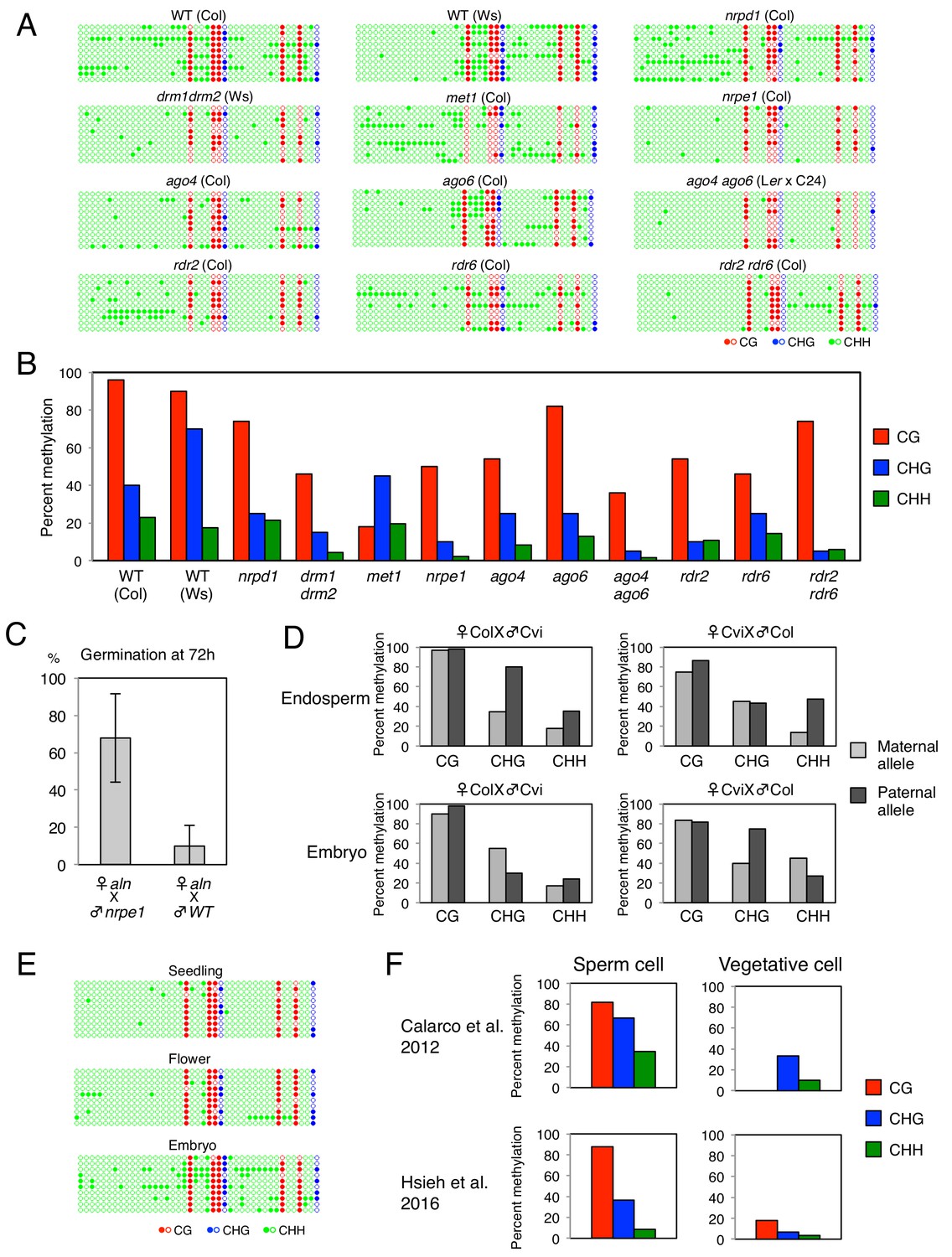
DNA methylation in the POGO region.
(A) DNA methylation in the POGO region upstream of ALN in the endosperm of WT seeds and RdDM mutant seeds. The studied region is depicted by a red line in Figure 1B. Filled and open circles represent methylated and unmethylated cytosines respectively. (B) Histograms show percentages of CG and CHG, and CHH methylation in the POGO region of WT and different RdDM mutants as indicated. (C) aln x nrpe1 and aln x WT F1 seeds were after-ripened for 7 days. Germination was scored 72 hr upon seed imbibition (eight replicates, n = 50). (D) DNA methylation levels in the POGO region of maternal and paternal alleles. DNA from endosperm and embryo of F1 seeds obtained after reciprocally crossing Cvi and Col WT plants was analyzed by sodium bisulfite sequencing. SNPs were used to distinguish maternal and paternal alleles. (E) DNA methylation in the POGO region in different tissues of WT (Col) plants. (F) Histograms show percentage of DNA methylation in the POGO region in sperm cells and vegetative cell from two independent published whole genomic DNA methylation data of male gamete (Calarco et al., 2012 and Hsieh et al., 2016).
Figure 2 with 1 supplement
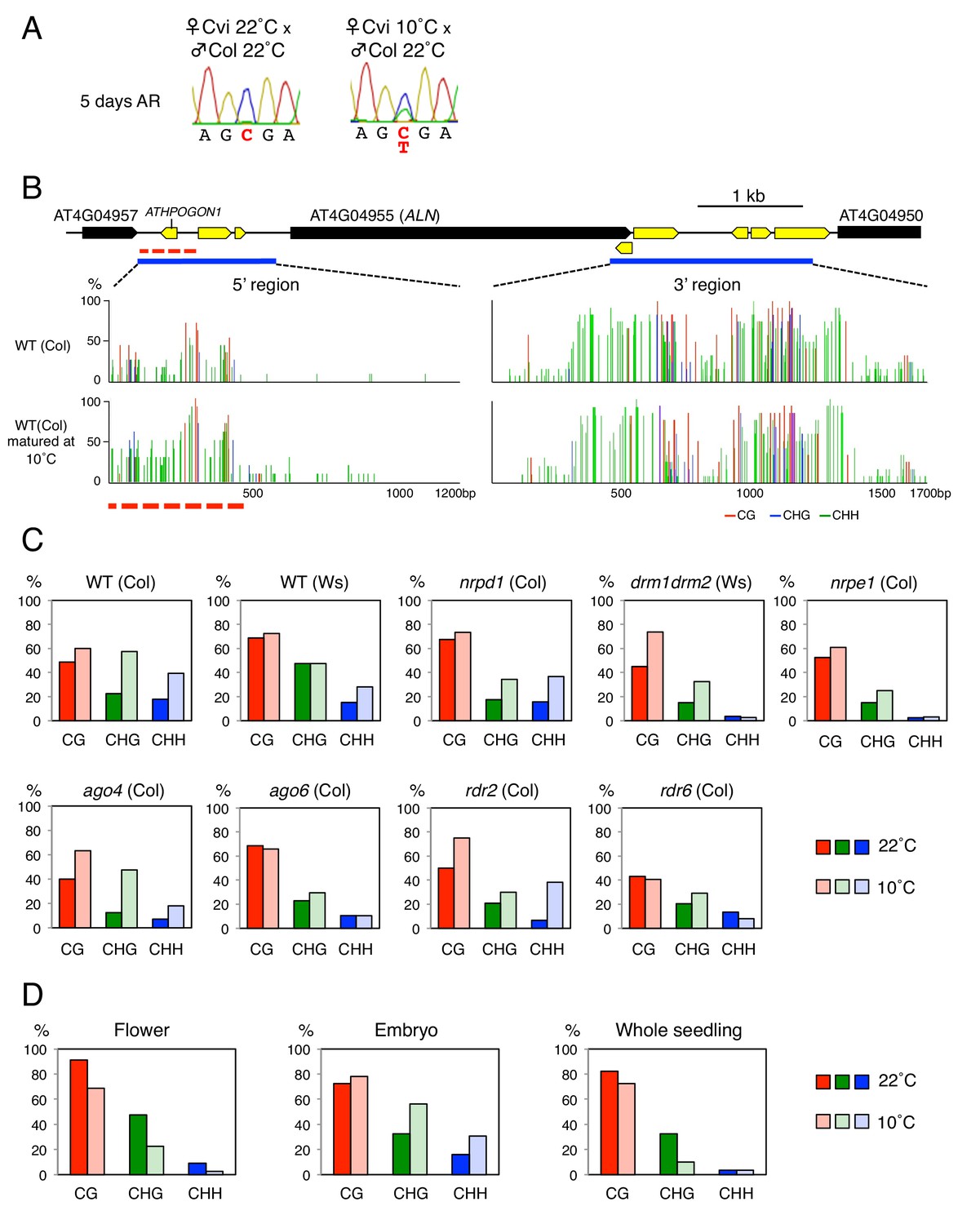
Cold temperatures increase methylation levels and expand the length of the ATHPOGON1-containing region subjected to methylation through RDR6-RdDM.
(A) Sanger sequencing chromatograms at SNPs of ALN. WT (Cvi) plants cultivated at 22 ˚C or 10 ˚C were pollinated with WT (Col) pollen. RNA was extracted from endosperm of F1 seeds and subjected to RT-PCR followed by Sanger sequencing. Nucleotides at SNP sites are highlighted in red: ‘C’ originates from Cvi and ‘T’ originates from Col. Representative results are shown from experiments repeated at least five times for each genotype. (B) DNA methylation in the ALN 5’ and 3’ flanking regions of the endosperm of mature seeds. Blues lines show regions where DNA methylation was studied. Dashed red line (500 bp) shows the region analyzed for DNA methylation levels. All remaining methylation data shown in this figure only correspond to this 500 bp region. Red, blue and green vertical lines represent CG, CHG, and CHH methylation levels, respectively. (C) Histograms show percentage of DNA methylation in the 500 bp region. DNA extracted from endosperm of WT seeds and RdDM mutant seeds produced at 22 ˚C or 10 ˚C was analyzed by sodium bisulfite sequencing. (D) Cold-induced and seed-specific DNA methylation. Seeds produced at 22 ˚C or 10 ˚C were sown and the resulting seedlings cultivated at 22 ˚C for three weeks prior to DNA extraction and sodium bisulfite sequencing analysis (Whole seedling).
-
Figure 2—source data 1
Figure 2C: Percentages of DNA methylation levels in the POGO region.
- https://doi.org/10.7554/eLife.37434.008
Figure 2—figure supplement 1

DNA methylation in the POGO region.
The region analyzed for DNA methylation is indicated in Figure 2B. DNA was extracted from endosperm of mature seeds produced at 22 ˚C or 10 ˚C was analyzed by sodium bisulfite sequencing.
Figure 3
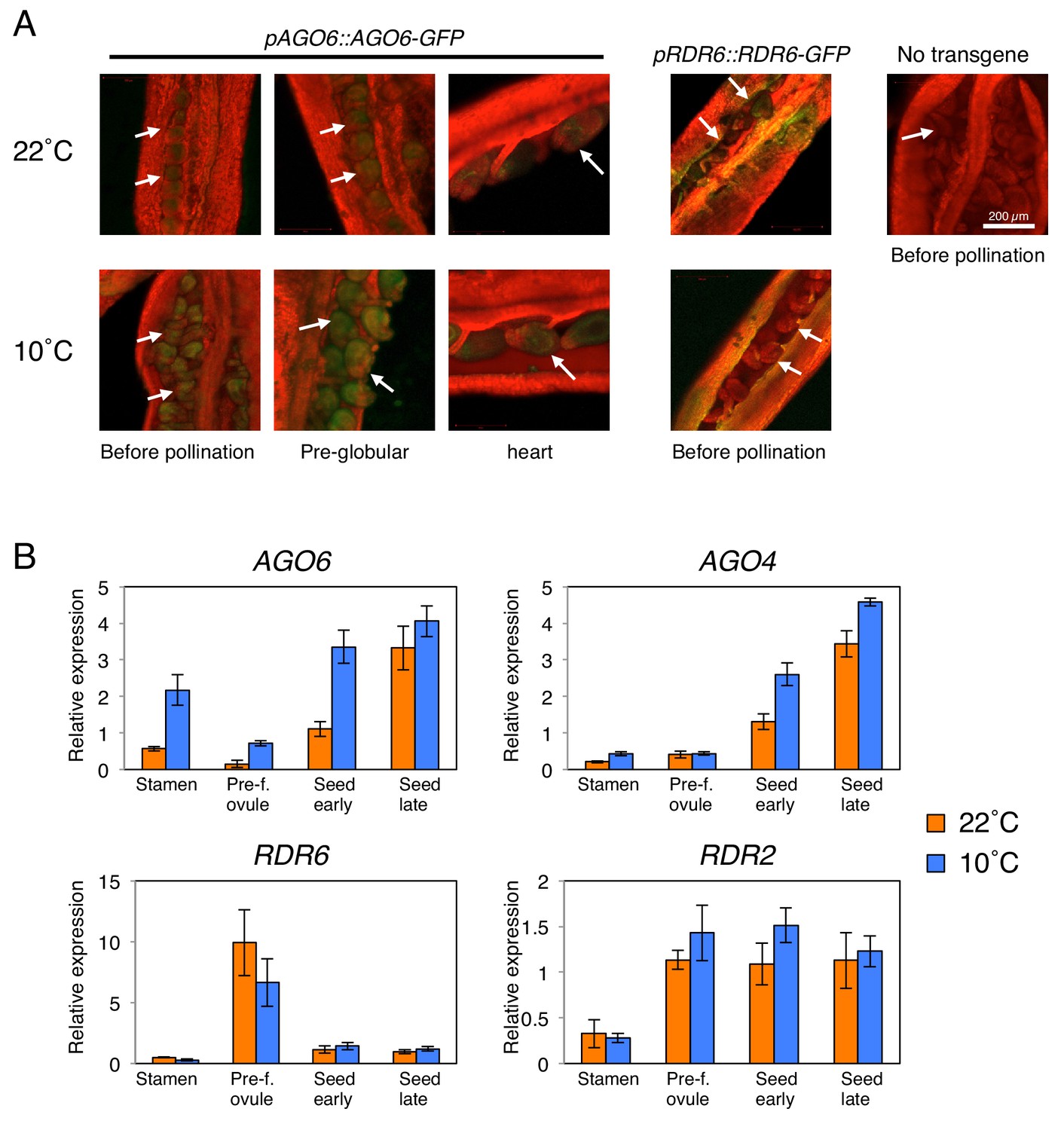
AGO6 expression is tissue-specific and is stimulated by cold.
(A) pAGO6::AGO6-GFP and pRDR6::RDR6-GFP transgenic lines were used to image AGO6-GFP and RDR6-GFP fluorescence, respectively, at various stages of ovule and seed development using confocal laser-scanning microscopy. Plants lacking the transgene are shown as a negative control (No transgene). Chlorophyll auto-fluorescence appears in red. Arrows indicate pre- and after-fertilization ovules. (B) qRT-PCR analysis of AGO6, AGO4, RDR2, and RDR6 mRNA accumulation in plants cultivated at 22˚C and 10˚C. RNA was extracted from stamens (Stamen), pre-fertilization ovules (Pre-f. ovule), developing seeds at the heart stage (Seed early) and developing seeds at the mature green stage (Seed late). Expression levels were normalized to those of PP2A. Error bars indicate SD from three technical replicates. Experiments were repeated two times giving similar results.
-
Figure 3—source data 1
Figure 3B: qRT-PCR expression analysis of AGO6, AGO4, RDR2, and RDR6.
- https://doi.org/10.7554/eLife.37434.010
Figure 4 with 3 supplements
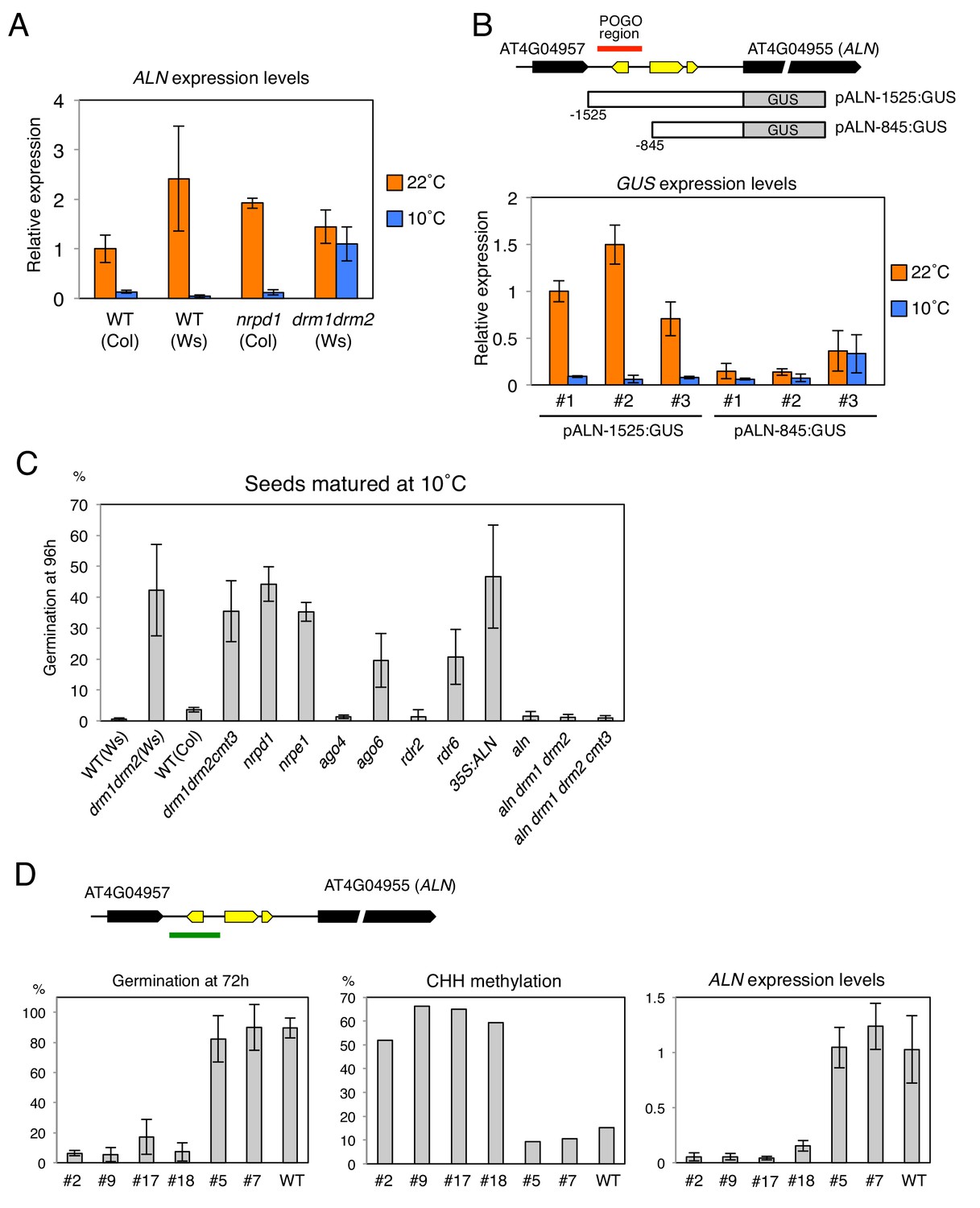
Changes in the POGO region methylation levels correlate with changes in ALN expression and dormancy levels.
(A) qRT-PCR analysis of ALN mRNA accumulation in WT, nrpd1 and drm1drm2 endosperm. RNA was extracted from endosperm of seeds produced at 22˚C and 10˚C and imbibed for 48 hr. ALN expression levels were normalized to those of PP2A. Error bars indicate SD of results from three technical replicates. Experiments were repeated three times giving similar results. (B) Analysis of reporter expression driven by ALN upstream sequences. Two different lengths of ALN upstream sequences (open box) were fused to GUS gene (filled box) and transformed into WT plants (Ws background). RNA was extracted from endosperm of seeds produced at 22˚C and 10˚C and imbibed for 48 hr. GUS expression levels were normalized to those of PP2A. Analysis was performed on three independent transgenic lines. Error bars indicate SD of results from three technical replicates. (C) Histograms show percent germination in populations of seeds produced 10 ˚C. Germination was scored 96 hr after seed imbibition (three biological replicates, n = 80–120). drm1drm2 is in Ws background; all other mutants are in Col-0 background. (D) Inducing DNA methylation of the AthPOGON1-containing region upstream of ALN. A DNA fragment covering AthPOGON1 (indicated by a green line) was cloned into a RNAi vector to produce double stranded inverted repeats (Materials and methods) and transformed into WT plants (Ws background). Dormancy levels, CHH methylation levels of endogenous ALN upstream sequences (Materials and methods) and ALN expression levels were analyzed for independent transgenic lines as shown. For the analysis of dormancy levels, seeds were after-ripened for 10 days, and germination was scored 72 hr upon seed imbibition (three replicates, n = 100). RNA was extracted from endosperm 48 hr after imbibition. ALN expression levels were normalized to those of PP2A. Error bars indicate SD of results from three technical replicates.
-
Figure 4—source data 1
Figure 4A, 4B, 4C and 4D: qRT-PCR expression analysis of ALN, GUS. Germination percentages of populations of seeds of different genotypes and produced at 10˚C.
POGO region DNA methylation levels and germination percentages of different transgenic lines
- https://doi.org/10.7554/eLife.37434.015
Figure 4—figure supplement 1
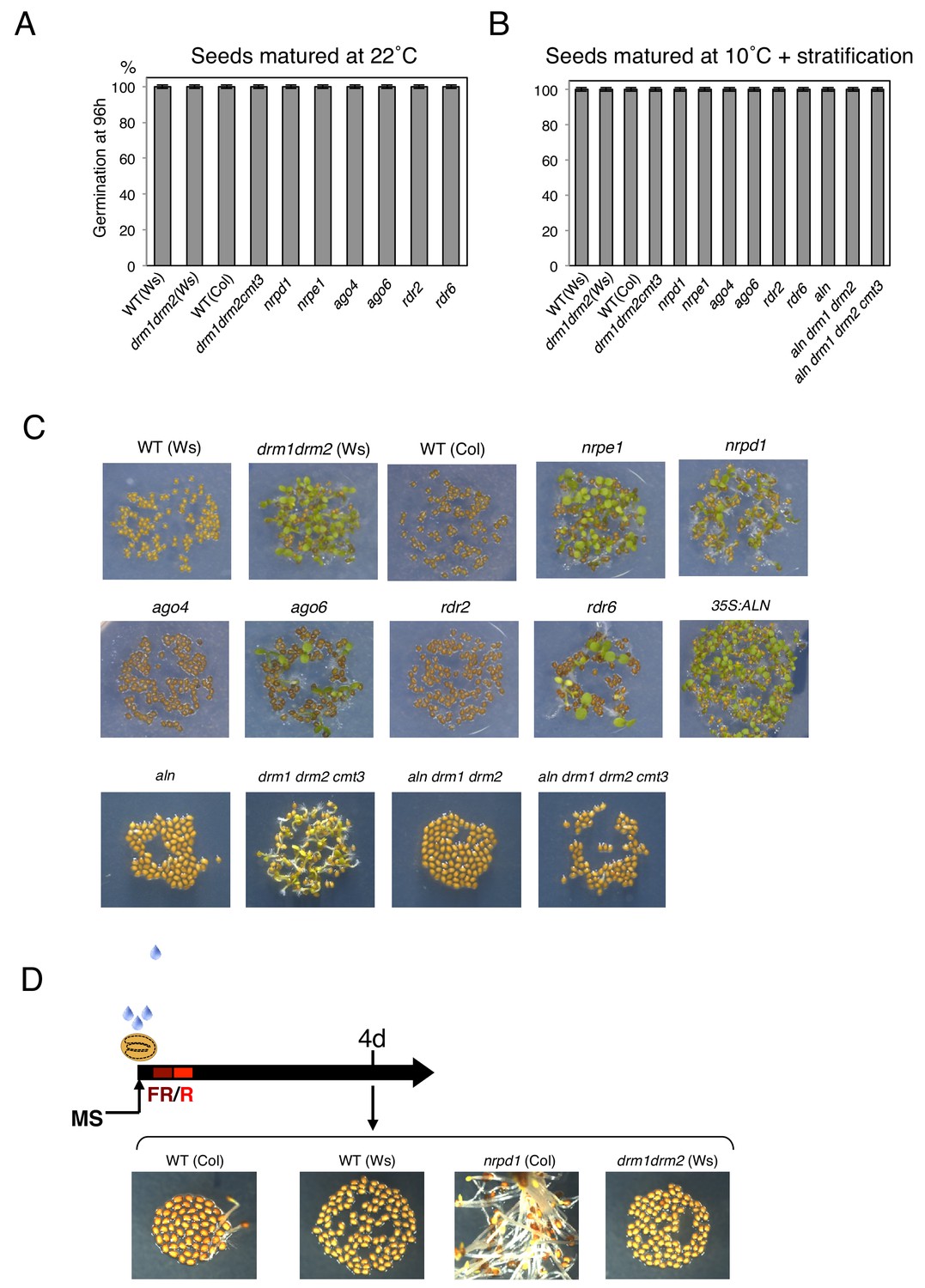
Germination percentages of populations of seeds of different genotypes and produced under different temperatures
(A) Histograms show percent germination in populations of seeds produced 22 ˚C. Germination was scored 96 hr after seed imbibition (three biological replicates, n = 80–120). (B) Histograms show percent germination after four days stratification in populations of seeds produced at 10 ˚C. Germination was scored 96 hr after seed imbibition (two biological replicates, n = 80–120). (C) Pictures show plants from seeds produced at 10 ˚C and imbibed for 96 hr. (D) nrpd1 mutant seeds produced at 22 ˚C are less dormant. Germination assay was performed under suboptimal conditions (De Giorgi et al., 2015): seeds were sown in the dark and exposed to a far red (FR) light pulse for 5 min; germination was induced by exposing seeds to a red light pulse (R) for 5 min. Germination assay was performed using WT (Col and Ws), nrpd1, drm (drm1drm2) seeds produced at 22 ˚C and dry after-ripened for one week. Pictures show plants 4 days after the red pulse.
Figure 4—figure supplement 2
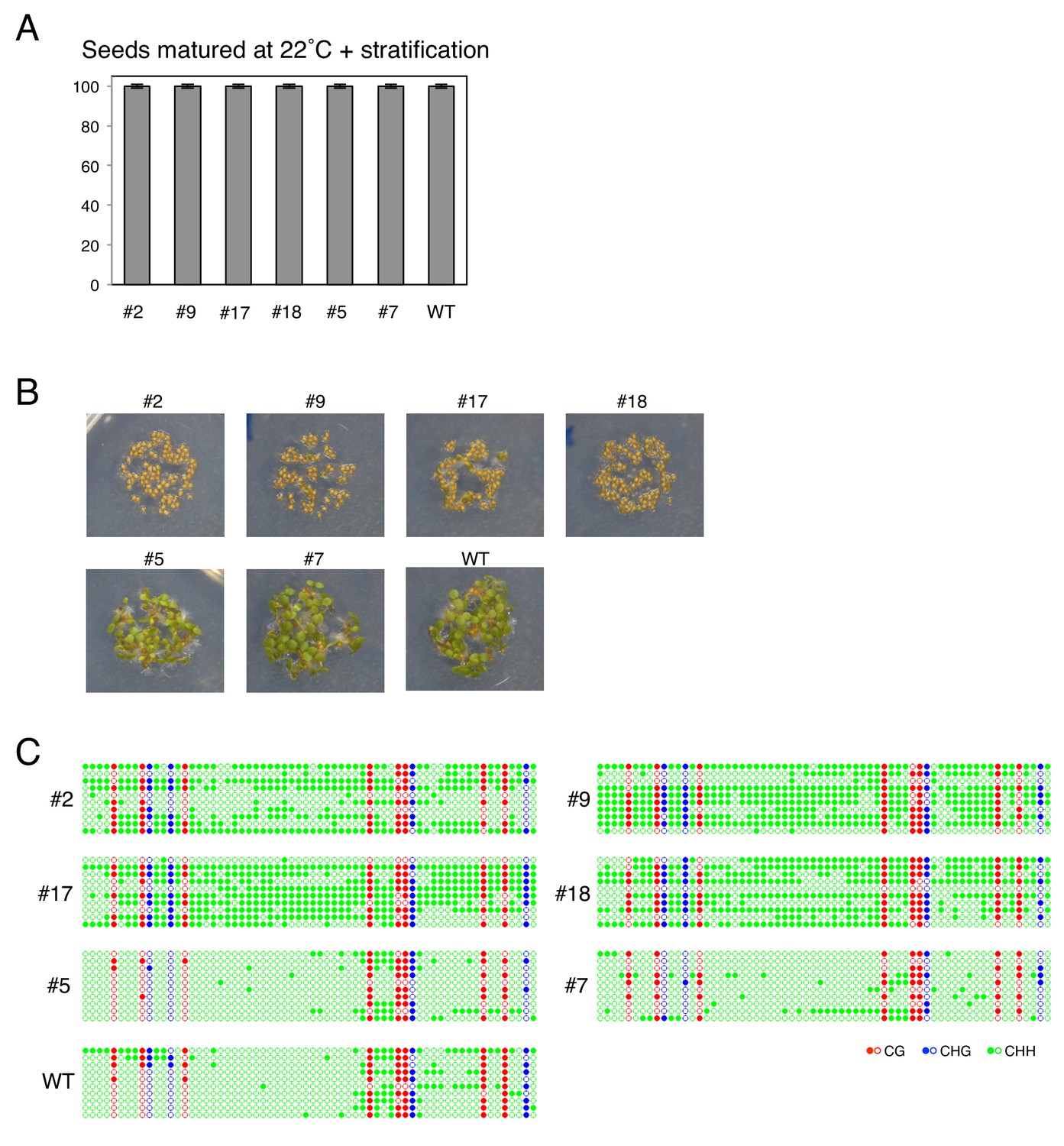
Germination percentages and POGO region DNA methylation levels of different transgenic lines
(A) Histograms show percent germination after stratification in populations of transgenic lines shown in Figure 4D produced at 22 ˚C. Germination was scored 48 hr after seed imbibition (two biological replicates, n = 80–120). (B) Pictures show transgenic lines shown in Figure 4D. Seeds from each line were after-ripened for 10 days and imbibed for 72 hr. (D) DNA methylation in the POGO region. The region analyzed for DNA methylation is indicated in Figure 2B. DNA was extracted from endosperm transgenic lines shown in Figure 4D was analyzed by sodium bisulfite sequencing.
Figure 4—figure supplement 3

Model for parental and environmental control of seed dormancy through epigenetic control of ALN expression.
(A) Under warm temperatures (22 ˚C), CHH methylation levels in the ALN promoter are lower in vegetative and flower tissues. During gametogenesis, CHH methylation is established in the sperm cells as a result of tissue-specific expression of AGO6, thus promoting non-canonical RdDM, which involves RDR2, RDR6, AGO6 and AGO4 components but not Pol IV. The paternal allele CHH methylation is maintained during seed development, which is likely due to pollen- and seed-specific non-canonical RdDM activity. (B) Under cold temperatures (10 ˚C) AGO6 accumulation is stimulated in pre-fertilization ovular tissues, which can increase RDR6-RdDM-dependent maternal allele methylation of the POGO region upstream of ALN. Cold also enhances AGO6 accumulation in stamens and post-fertilization ovular tissues, which can further increase methylation of the POGO region in mature seeds. Suppression of ALN expression promotes seed dormancy.
Author response image 1

Author response image 2

Author response image 3
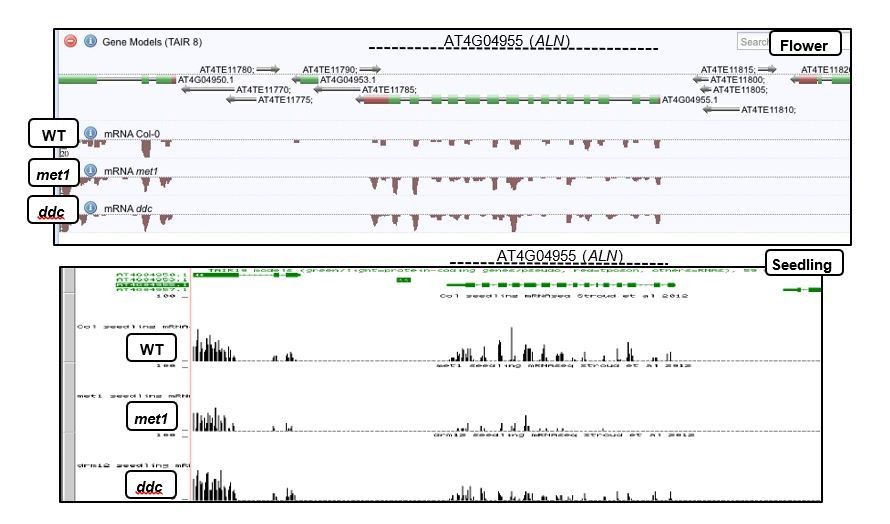
Author response image 4

Additional files
-
Supplementary file 1
Primers used in this study.
- https://doi.org/10.7554/eLife.37434.016
-
Transparent reporting form
- https://doi.org/10.7554/eLife.37434.017
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy
eLife 8:e37434.
https://doi.org/10.7554/eLife.37434




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}