Genome-wide alterations of uracil distribution patterns in human DNA upon chemotherapeutic treatments
- Genome Metabolism Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Hungary
- Department of Applied Biotechnology and Food Sciences, Budapest University of Technology and Economics, Hungary
- Doctoral School of Multidisciplinary Medical Science, University of Szeged, Hungary
- Department of Biochemistry and Molecular Pharmacology, New York University School of Medicine, United States
- Perlmutter Cancer Center, New York University School of Medicine, United States
- Howard Hughes Medical Institute, New York University School of Medicine, United States
- Cancer Biomarker Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Hungary
- Department of Bioinformatics and 2nd Department of Pediatrics, Semmelweis University, Hungary
- Genome Stability Research Group, Institute of Enzymology, Research Centre for Natural Sciences, Hungary
- Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital Campus, United Kingdom
Figures
Figure 1 with 2 supplements
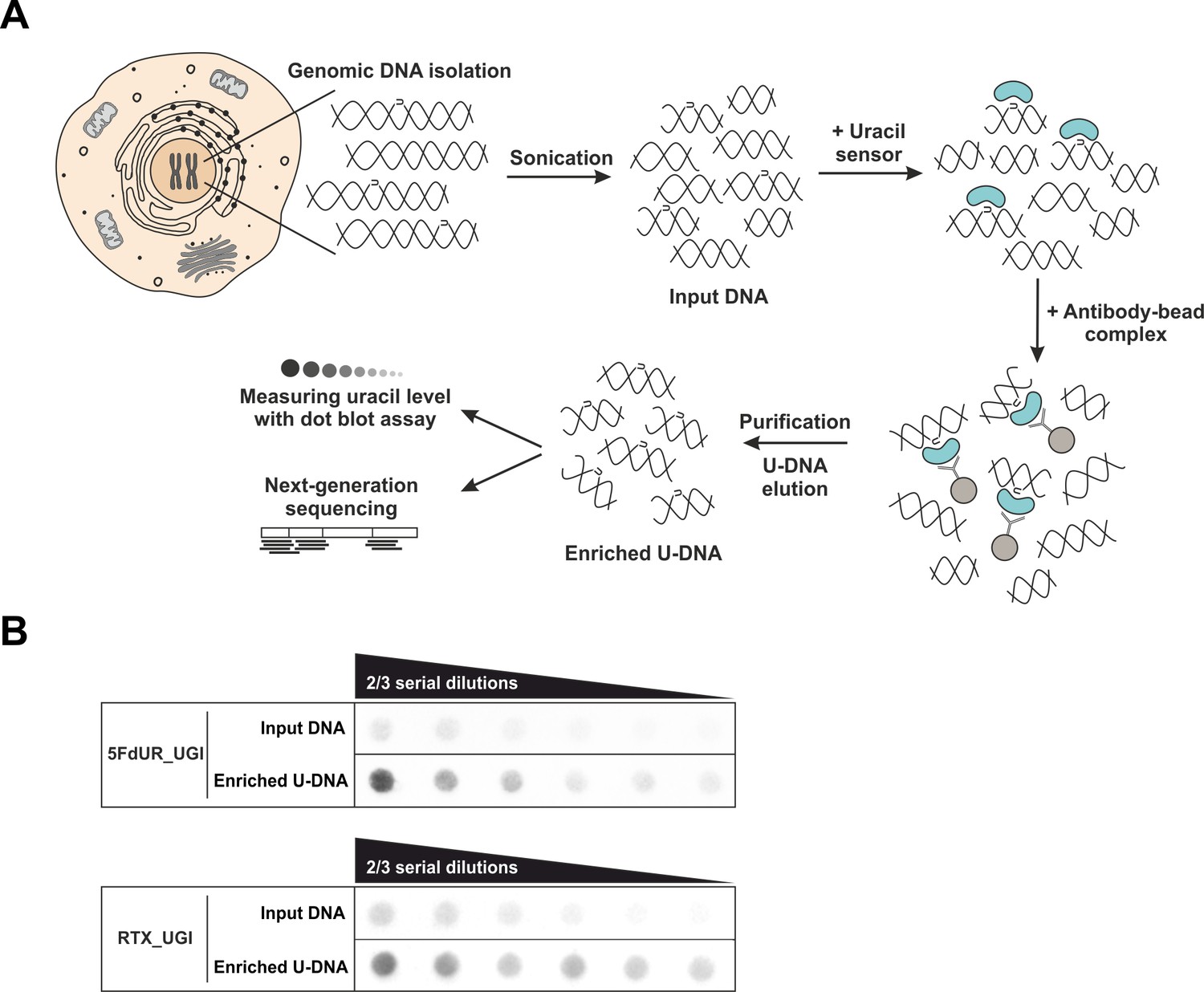
U-DNA-Seq provides genome-wide mapping of uracil-DNA distribution.
(A) Schematic image of the novel U-DNA immunoprecipitation and sequencing method (U-DNA-Seq). After sonication, enrichment of the fragmented U-DNA was carried out by the 1xFLAG-ΔUNG sensor construct followed by pull-down with anti-FLAG agarose beads. U-DNA enrichment compared to input DNA was confirmed by dot blot assay before samples were subjected to NGS. (B) Immunoprecipitation led to elevated uracil levels in enriched U-DNA samples compared to input DNA in case of both 5FdUR (5FdUR_UGI) and RTX (RTX_UGI) treated, UGI-expressing HCT116 samples. For each treatment, the same amount of DNA was loaded as input and enriched U-DNA samples providing a correct visual comparison. Two-third serial dilutions were applied.
Figure 1—figure supplement 1
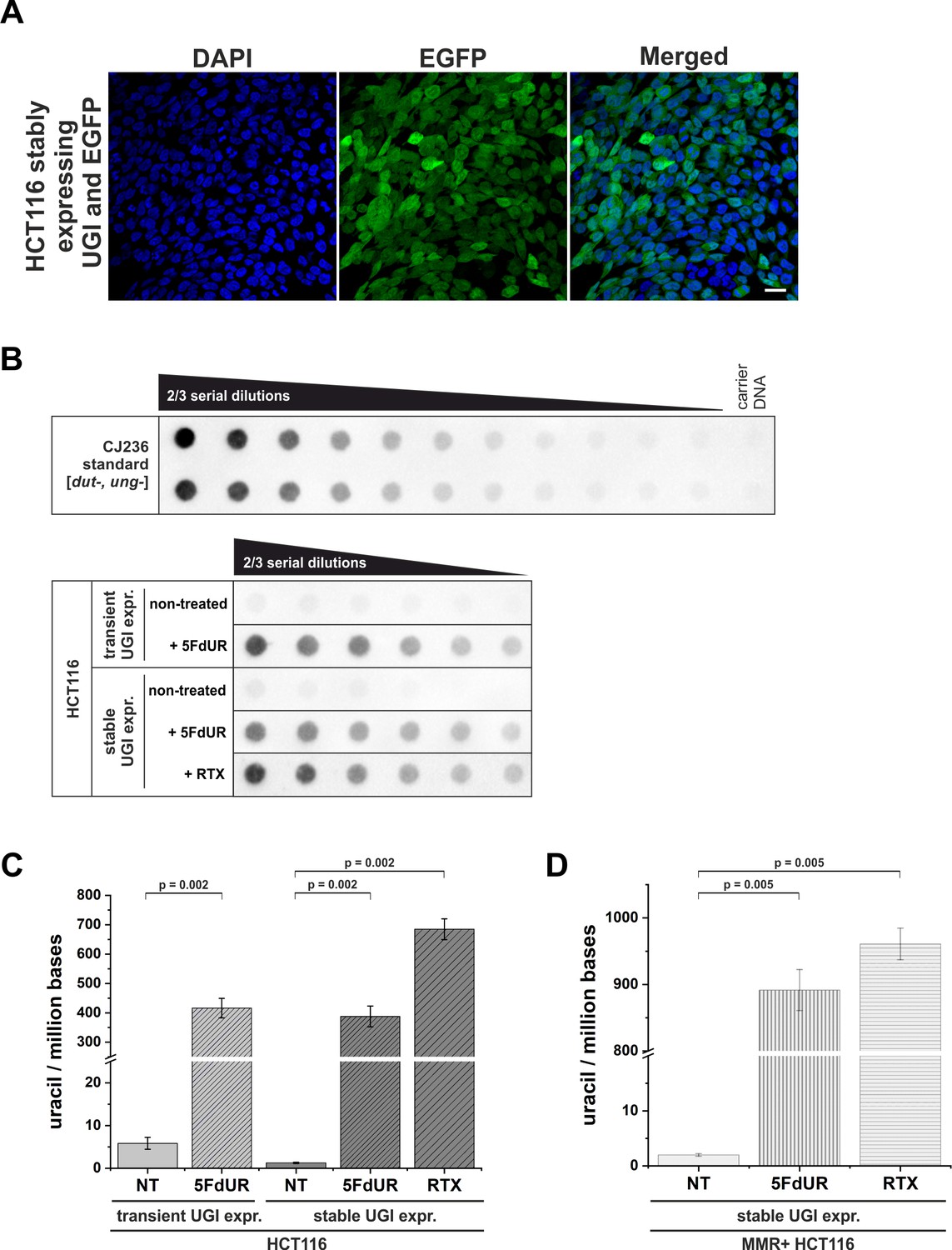
Elevation of genomic uracil content upon stable UGI expression and drug treatments.
(A) HCT116 cells stably expressing UGI along with EGFP following retroviral transduction. GFP positive cells were selected by fluorescence-activated cell sorting and cultured for further analysis. DAPI was used for DNA staining. Scale bar represents 20 µm. (B) Dot blot assay for measuring genomic uracil levels (Róna et al., 2016) of non-treated and drug (5FdUR or RTX) treated HCT116 cells either transiently or stably expressing UGI. Genomic DNA (8 ng) isolated from log-phase growing CJ236 [dut-, ung-] Escherichia coli strain was applied as uracil standard in a ½ dilution series (upper panel). Two-third dilution series from HCT116 samples started with 600 ng DNA for non-treated or 5 ng DNA for 5FdUR or RTX treated samples (lower panel). The dot blot image presented is a representative of six independent biological experiments. (C) Bar graph shows the uracil moieties/million bases of each sample. Drug treatment led to significantly elevated uracil levels in HCT116 cells either transiently or stably expressing UGI (~400 uracil moieties/million for 5FdUR and ~700 uracil moieties/million for RTX) as compared to non-treated (NT) cells (~2–5 uracil moieties/million). Error bars indicate standard errors of the mean (SEM). Calculations were based on six independent datasets (n = 6). p=0.002. Source data are available in Figure 1—figure supplement 1—source data 1. (D) Bar graph shows the uracil moieties/million bases in MMR proficient HCT116 cells stably expressing UGI. Drug treatment resulted in even higher U-DNA content (~900 uracil moieties/million for 5FdUR and ~950 uracil moieties/million for RTX) as compared to MMR deficient cells. Error bars indicate standard errors of the mean (SEM). Calculations were based on six independent datasets (n = 6). p=0.005. Source data are available in Figure 1—figure supplement 1—source data 1.
-
Figure 1—figure supplement 1—source data 1
Quantification of genomic uracil content based on densitometry of dot blot measurements.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig1-figsupp1-data1-v2.xlsx
Figure 1—figure supplement 2
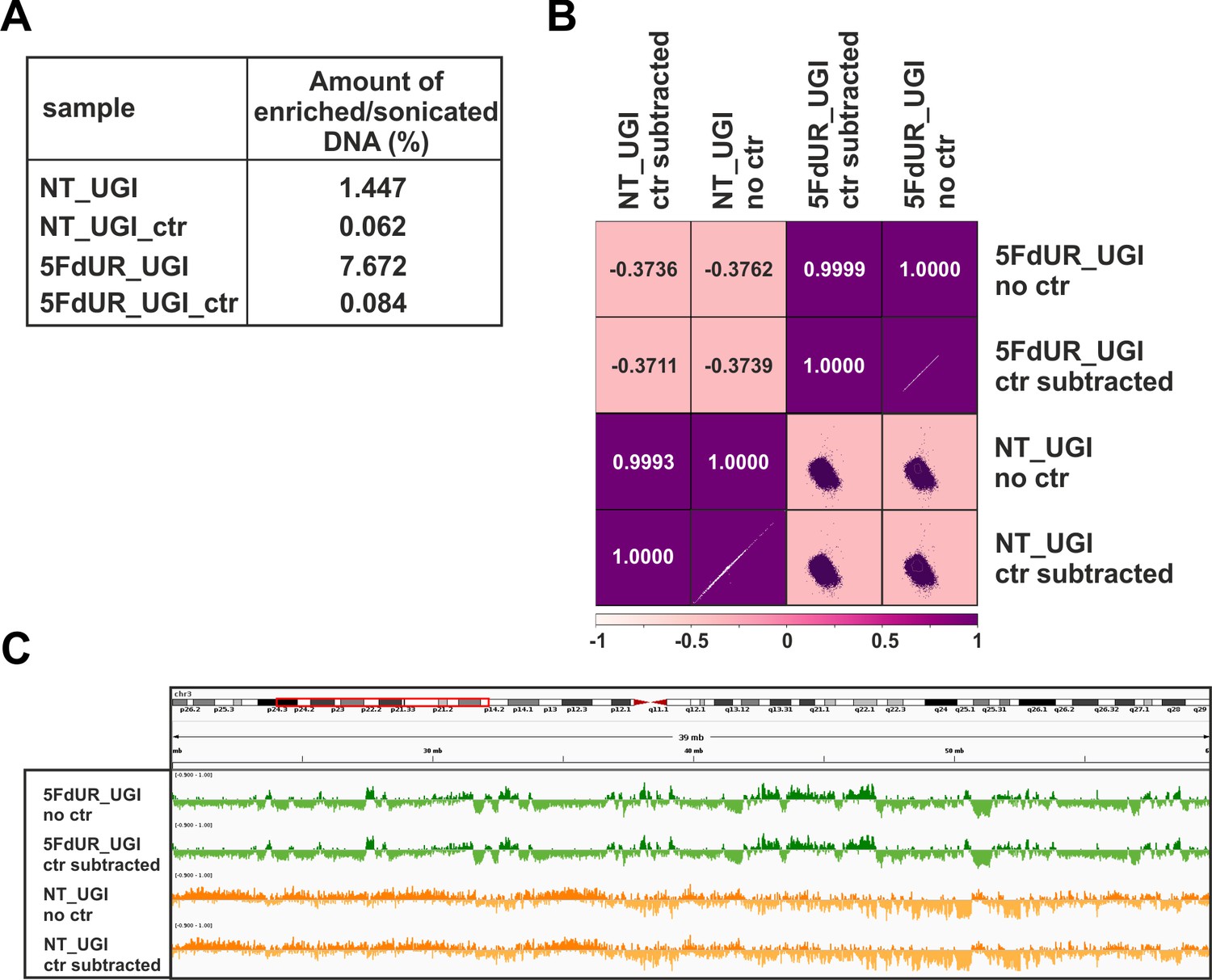
Negative control of U-DNA-IP using ΔUNG sensor free (empty) anti-FLAG beads.
(A) Relative amount of the pulled down DNA as compared to the input DNA amount. Samples were the following: U-DNA-IP from UGI-expressing, non-treated (NT_UGI) and 5FdUR treated (5FdUR_UGI) samples, and the corresponding mock pulled down controls (NT_UGI_ctr and 5FdUR_UGI_ctr) where ΔUNG sensor free, empty anti-FLAG beads were used. (B) Pearson correlation among log2 ratio coverage tracks calculated from the genome scaled input coverage track and either the original (NT_UGI_no_ctr and 5FdUR_UGI_no_ctr) or the control-subtracted (NT_UGI_ctr_subtracted and 5FdUR_UGI_ctr_subtracted), enriched coverage tracks (Materials and methods, Supplementary file 1). (C) A representative IGV view on original (NT_UGI_no_ctr and 5FdUR_UGI_no_ctr) and the control-subtracted (NT_UGI_ctr_subtracted and 5FdUR_UGI_ctr_subtracted) log2 enrichment (on chr3: 26,000,000–62,000,000).
Figure 2 with 3 supplements
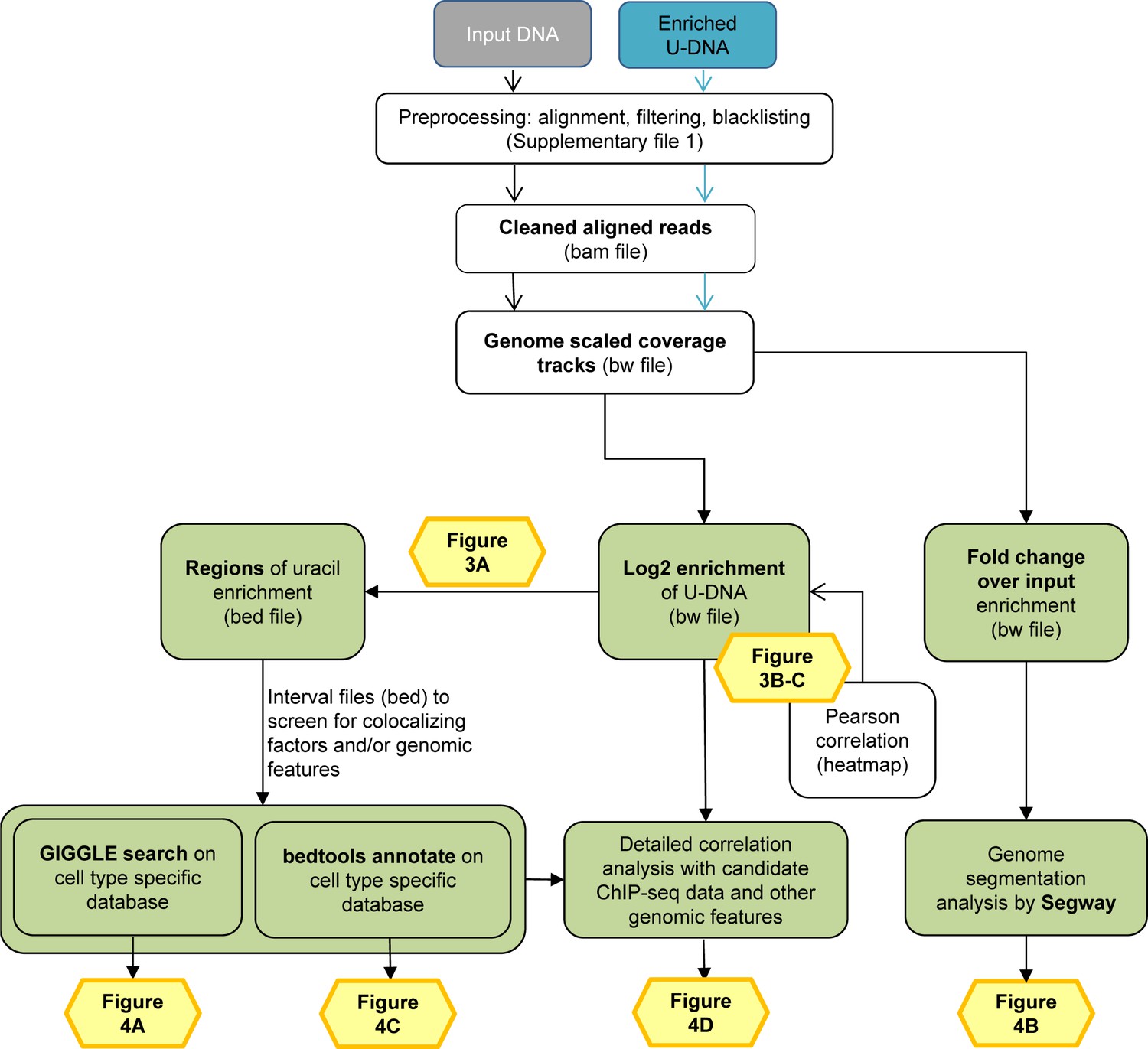
Data analysis pipeline.
Both input and enriched U-DNA samples were pre-processed the same way: initial trimming and alignment were followed by filtering for uniquely mapped reads and blacklisting of regions suffering from alignment artefacts, resulting in cleaned read alignments in the format of bam files. The key steps of our proposed data processing are (1) calculation of genome scaled coverage tracks (bigwig/bw files), (2) calculation of log2 (enriched coverage/input coverage) ratio tracks (bigwig/bw files), (3) extraction of interval (bed) files of uracil enriched regions from the corresponding log2 ratio tracks. To correlate the uracil enrichment profiles with other published data, first quick screens using interval files were done, and then detailed correlation analysis with a promising candidate of colocalizing genomic features was performed using coverage track files. GIGGLE search (Layer et al., 2018) and bedtools annotate (Quinlan and Hall, 2010) were used for scoring the similarities between query uracil-DNA and the database interval files. Genome segmentation analysis was performed on fold change over input bigwig files either from the ENCODE database, or our own ChIP-seq data and U-DNA profiles using Segway package (Chan et al., 2018; Hoffman et al., 2012). Figures corresponding to the different analysis steps are also indicated. A more detailed pipeline is shown in Figure 2—figure supplement 1, and the full methodology is described in the Supplementary file 1, 3–5.
Figure 2—figure supplement 1
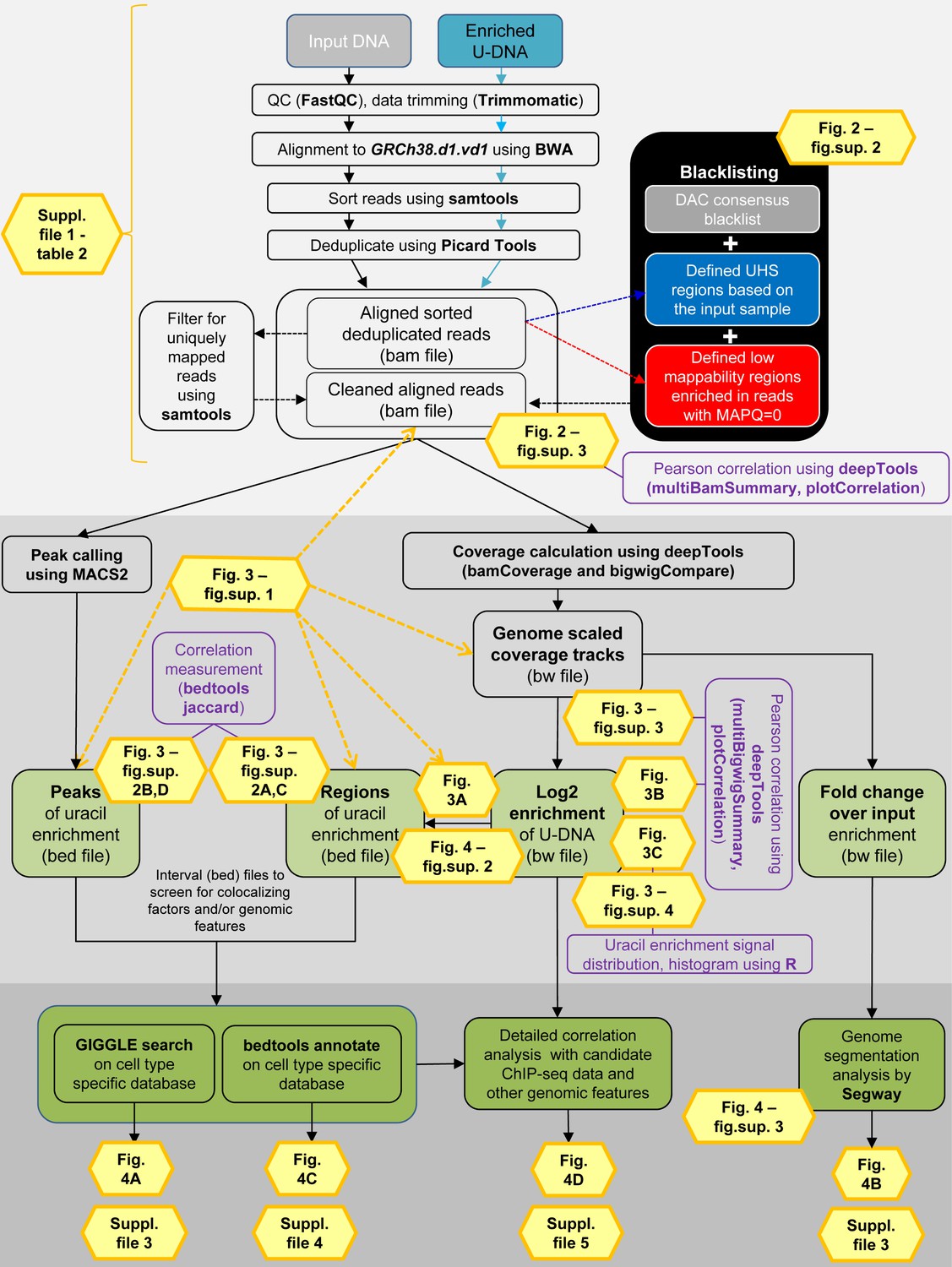
Detailed analysis pipeline.
Input DNA and enriched U-DNA mean genomic DNA fragmented by sonication and uracil-containing fragments pulled-down by FLAG-ΔUNG sensor, respectively. Pre-processing steps (light grey field) include quality check, data trimming, read alignment, sorting, deduplication, filtering out ambiguously mapped reads (mapping quality, MAPQ = 0) and blacklisting. The cell line specific blacklists are defined based on consensus DAC blacklist recommended by ENCODE Consortium (Amemiya et al., 2019), the ultra-high signal (UHS) and low mappability (more than 50% of the reads are with MAPQ = 0) regions. Pearson correlation among samples at the level of aligned reads in bam files is calculated using deepTools package (Ramírez et al., 2016). Data processing (darker grey field) can follow two main streams: (1) peak calling originally designed for ChIP-seq (using MACS2 [Feng et al., 2011; Zhang et al., 2008], resulting in peaks’ interval files in bed format); and (2) coverage track calculation and analysis (using deepTools, resulting in coverage track files in bigwig/bw format). Interval (bed) files are derived not only from peak calling but also from the log2 (enriched coverage/input coverage) ratio bigwig files (log2 enrichment). Ratio of enriched coverage/input coverage is also calculated from the genome scaled coverage tracks, that provides the same type of enrichment profiles as ‘fold change over control’ bigwig tracks used in the ENCODE (fold change over input). Additional calculations for sample characterization are indicated in purple boxes. In the third part (darkest grey field), uracil enrichment profiles were further analyzed by comparing to available data. First, a quick screen using interval (bed) files was performed, and then a detailed correlation analysis was carried out with the promising candidates using coverage track (bw) files. For the initial screen, we collected a set of HCT116 specific ChIP-seq and DNA-IP-seq data, as well as other genomic features in the format of interval (bed) files. GIGGLE search (Layer et al., 2018) and bedtools annotate (Quinlan and Hall, 2010) were used for scoring the similarities between the query uracil-DNA and the database intervals. Moreover, the ENCODE set of these HCT116 specific ChIP-seq data was selected together with our U-DNA fold change over input profiles to perform a genome segmentation analysis using the Segway software package (Chan et al., 2018).
Figure 2—figure supplement 2

Creation of cell line specific blacklist.
(A) Concept of blacklisting. United cell line specific blacklist was composed from (1) universal DAC blacklist (https://www.encodeproject.org/files/ENCFF419RSJ, grey), (2) Ultra High Signal (UHS, blue) regions derived from input samples, and (3) low mappability regions where more than 50% of the reads are ambiguously mapped (MAPQ = 0, red). (B) Representative IGV view of genomic region (chr2:32,863,802–32,872,901) where both UHS and low mappability regions are present. Tracks from the top are as follows: DAC blacklist (no intervals in the presented region); coverage track (bw file, blue), and the derived UHS regions (bed file, blue); log2 ratio (original/filtered) track (bw file, red), and the derived low mappability regions (bed file, red); intervals after combination of DAC, UHS, and low mappability regions (bed file, purple); and the same after merging intervals separated by less than 500 bases (bed file, the final blacklist, black). The bw files and the derived bed files are shown for one input sample, while the blacklists presented here are calculated from all the input data corresponding to HCT116 cell line. Raw alignments (bam files) of the corresponding enriched and input samples are also shown. Reads in white are characterized with MAPQ = 0. Alterations in reads comparing to the reference genome are indicated with different colors according to the IGV standard. In regions suffering from alignment artefacts, reads often appear with multiple alterations, as it is visible here for the UHS region. (C) Histogram of coverage signal of a representative input sample (the same as above). Both full data range and a zoomed version (inset) with better resolution are shown. In the zoomed histogram, the defined UHS threshold (blue) is also indicated. Source data are available in Figure 2—figure supplement 2—source data 1. (D) Histogram of log2 ratio (original/filtered) signal of a representative input sample (the same as above). Both full data range and a zoomed version (inset) with better resolution are shown. In the zoomed histogram, the defined low mappability threshold (red) is also indicated. Source data are available in Figure 2—figure supplement 2—source data 1.
-
Figure 2—figure supplement 2—source data 1
Histograms to determine the UHS and the low mappability regions for cell line specific blacklists.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig2-figsupp2-data1-v2.xlsx
Figure 2—figure supplement 3
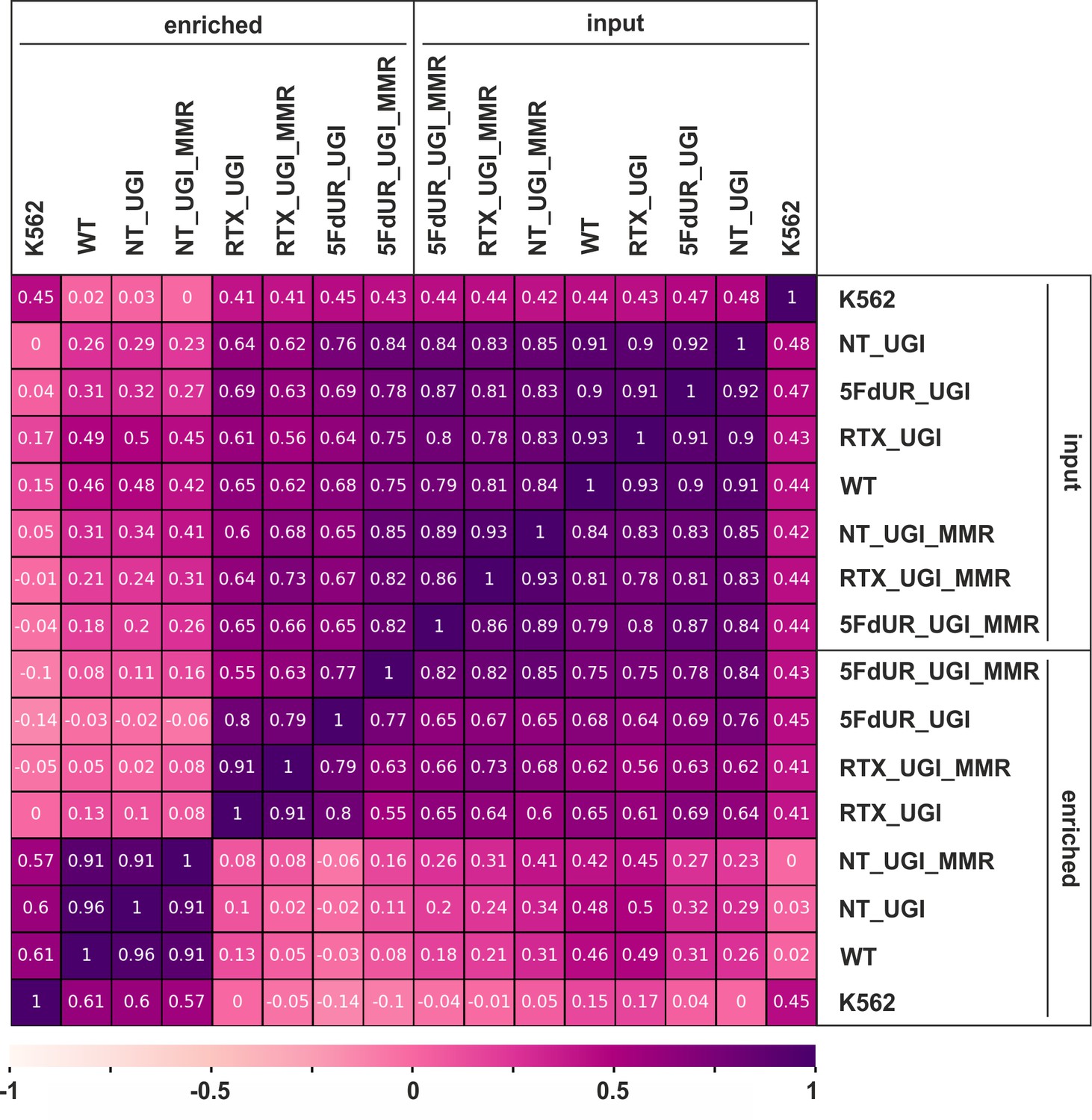
Pearson correlation among processed BAM files.
Calculation was done on uniquely mapped reads after blacklisting and merging replicates, using deepTools multiBamSummary and plotCorrelation (Ramírez et al., 2016) as described in the Supplementary file 1. Heatmap was plotted using python seaborn/matplotlib (Hunter, 2007). The input and the corresponding enriched samples are as follows: non-treated wild type (WT), non-treated UGI-expressing (NT_UGI, NT_UGI_MMR), 5FdUR treated UGI-expressing (5FdUR_UGI, 5FdUR_UGI_MMR), RTX treated UGI-expressing (RTX_UGI, RTX_UGI_MMR) HCT116 cells which are MMR deficient (no distinction) or proficient (labeled with MMR), and non-treated wild type K562 cells (K562).
Figure 3 with 4 supplements
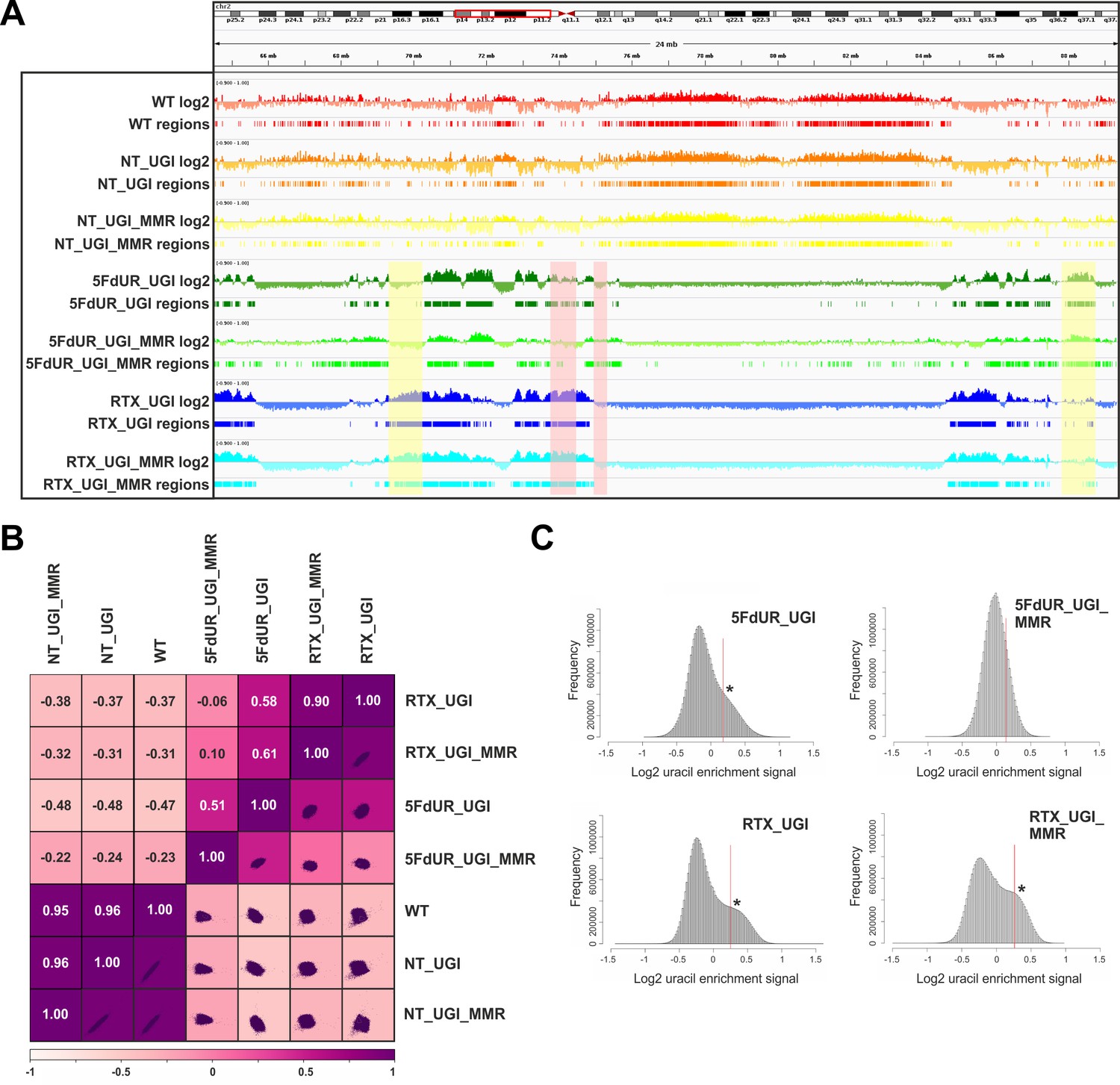
Comparison of processed U-DNA-Seq data among samples.
(A) Representative IGV view in genomic segment (chr2:64,500,000–89,500,001) shows log2 ratio signal tracks of enriched versus input coverage (log2, upper tracks) and derived regions of uracil enrichment (regions, bottom tracks) for non-treated: wild type (WT, red), UGI-expressing (NT_UGI, orange), and MMR proficient UGI-expressing (NT_UGI_MMR, yellow); and for treated: with 5FdUR (5FdUR_UGI, green; 5FdUR_UGI_MMR, light green) or raltitrexed (RTX_UGI, blue; RTX_UGI_MMR, cyan) HCT116 samples. Two replicates for each sample were merged before coverage calculation. Differences between treated and non-treated samples are clearly visible. Furthermore, 5FdUR and RTX treatments caused similar but not identical uracil enrichment profiles (differences are highlighted with yellow shade). The impact of the MMR status in case of the 5FdUR treated samples is highlighted with pink shade. (B) Comparison of log2 uracil enrichment profiles among samples was performed using multiBigwigSummary (deepTools) and Pearson correlation was plotted using plotCorrelation (deepTools). A heatmap combined with scatter plots is shown for the seven samples. Two replicates for each sample were merged before coverage calculation, and the same analysis for individual replicates are shown in Figure 3—figure supplement 3. (C) Histograms of log2 ratio profiles were calculated and plotted using R for the drug-treated samples. A sub-population of data bins with elevated log2 uracil enrichment signal is clearly visible (indicated with asterisk) in most cases, where high uracil incorporation was detected (Figure 1—figure supplement 1B–D). Thresholds applied in determination of uracil enriched regions are indicated with red line and also provided in Figure 3—source data 1 together with the histogram data.
-
Figure 3—source data 1
Histograms for the U-DNA signal distribution in drug-treated samples.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig3-data1-v2.xlsx
Figure 3—figure supplement 1

Representative IGV views from aligned reads to a 10 Mb cluster of uracil enriched regions compared to usual peak calling results.
Upper panel shows IGV view of 112.667.000–112.684.000 genomic segment to demonstrate the processing steps. The tracks from the top are as follows: cleaned aligned reads (bam files) for a pair of input (black title) and enriched (cyan title) samples; genome scaled coverage tracks for the input (black), and for the enriched (cyan) samples computed by deepTools/bamCoverage; log2 ratio track (enriched coverage/input coverage, positive values are shown in cyan, negative values are in black, computed by deepTools/bigwigCompare); regions of uracil enrichment derived from log2 ratio (cyan); peaks called by MACS2 with 0.05 and also with the more permissive 0.5 broad-cutoff (grey). In the presented 16 kb genomic segment a 6 kb region of uracil enrichment is detected, while peak calling either fails or detects only a shorter peak depending on the applied broad-cutoff value. The following panels are zooming out on the same region allowing visualization of its broader context. This 6 kb region of uracil enrichment (red arrows) is a part of a 250 kb super-region that is surrounded by similar super-regions separated by negative log2 value segments. On a larger scale, even these super-regions are clustered (to a 10 Mb cluster here, bottom panel). It is also visible that using more permissive broad-cutoff in peak calling allows higher coverage of the uracil enriched regions, however detects false positive peaks as well.
Figure 3—figure supplement 2
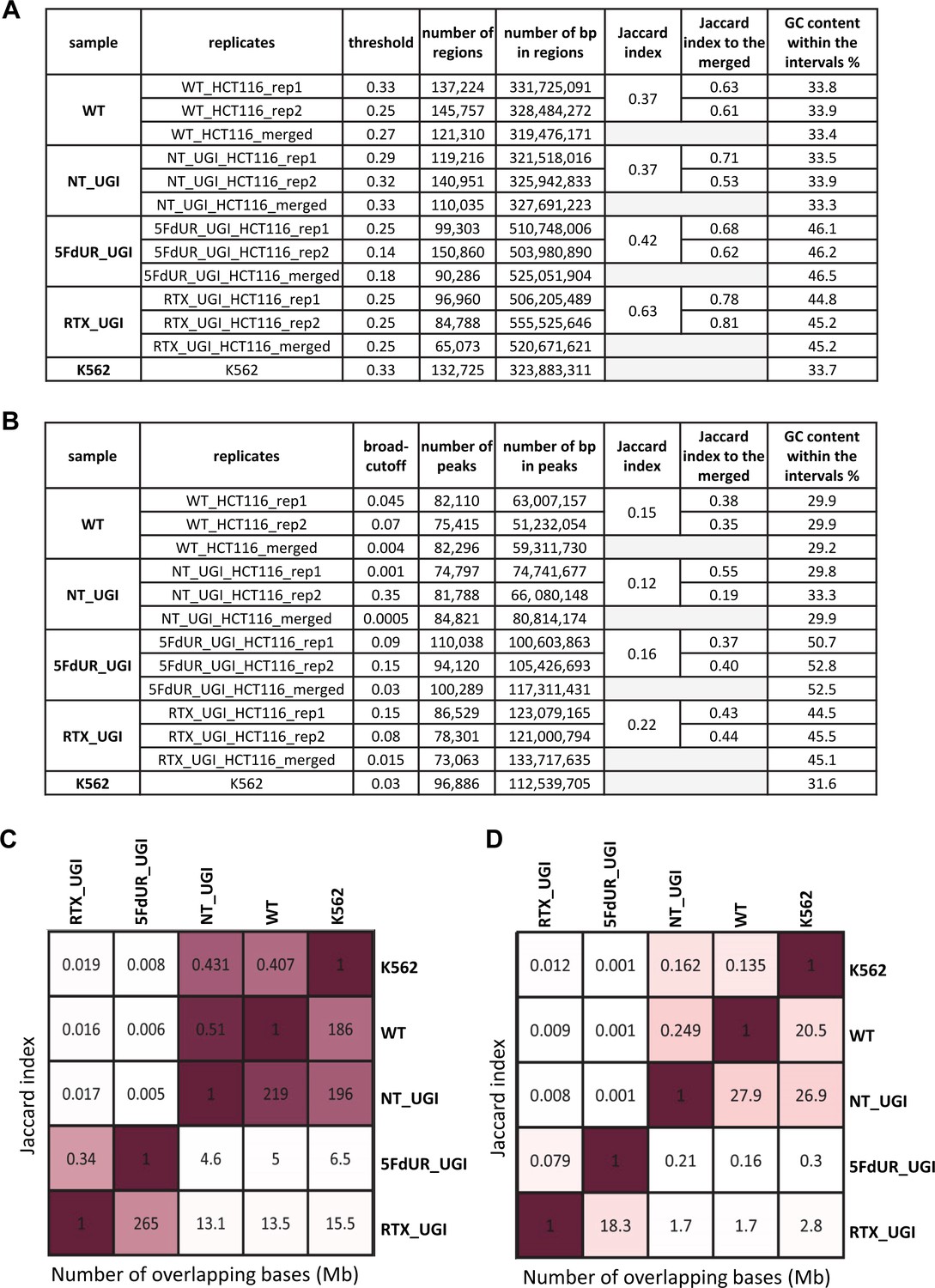
Comparison of the regions of uracil enrichment derived from log2 signal ratio and the peaks called by MACS2.
The samples are as follows: non-treated wild type (WT), non-treated UGI-expressing (NT_UGI), 5FdUR treated UGI-expressing (5FdUR_UGI), RTX treated UGI-expressing (RTX_UGI) HCT116 cells, non-treated wild type K562 cells (K562). The merged data are derived from the merged bam files in the same way as in case of the individual replicates (rep1, rep2). (A) Statistics on the log2 ratio derived regions of uracil enrichment. The applied threshold values were fine-tuned to have approximately the same length of intervals in the corresponding replicates and merged data. Bedtools Jaccard indices were calculated between replicates, as well as between individual replicates and the corresponding merge data. GC contents were also calculated for each interval file of regions. (B) Statistics on peaks called by MACS2. Broad-cutoff values were adjusted to have approximately the same length of intervals in the corresponding replicates and merged data. Bedtools Jaccard indices were calculated between replicates, as well as between individual replicates and the corresponding merged data. GC contents were also calculated for each interval file of peaks. (C) Comparison of samples based on log2 ratio derived regions (merged data). Jaccard indices and numbers of millions of intersecting bases are indicated above and below the diagonal, respectively. Drug-treated and non-treated samples are well separated. (D) Comparison of samples based on peaks (merged data). Jaccard indices and numbers of millions of intersecting bases are indicated above and below the diagonal, respectively. Dreated and non-treated samples are not that well separated anymore.
Figure 3—figure supplement 3

Pearson correlation among log2 ratio tracks of replicates.
The log2 ratio tracks were calculated from the genome scaled coverage tracks (enriched vs input) using deepTools bigwigCompare tool for the individual replicates. Pearson correlation was calculated by deepTools multiBigwigSummary and plotCorrelation (Ramírez et al., 2016) as described in the Supplementary file 1. The heatmap was plotted using python seaborn/matplotlib (Hunter, 2007). The samples are as follows: non-treated wild type (WT), non-treated UGI-expressing (NT_UGI, NT_UGI_MMR), 5FdUR treated UGI-expressing (5FdUR_UGI, 5FdUR_UGI_MMR), RTX treated UGI-expressing (RTX_UGI, RTX_UGI_MMR) HCT116 cells which are MMR deficient or proficient (labeled by MMR), and non-treated wild type K562 cells (K562). Replicates are indicated as rep1 and rep2.
Figure 3—figure supplement 4
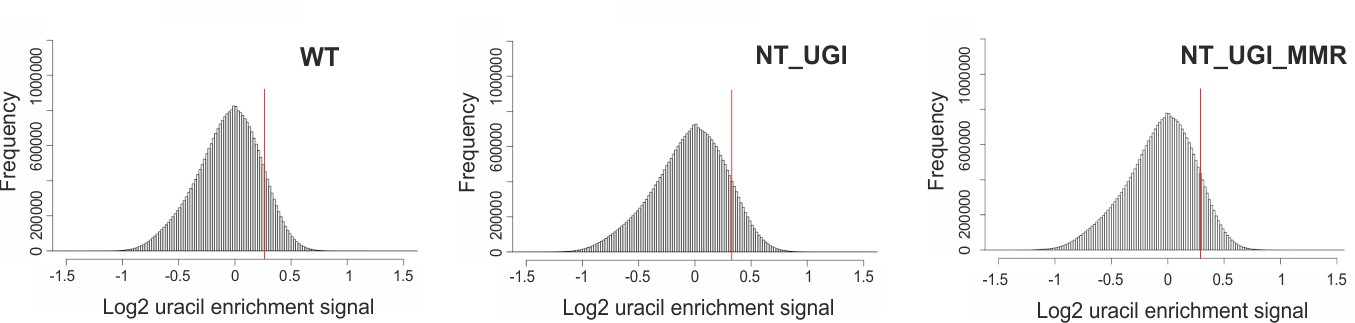
Histograms for the U-DNA signal distribution in non-treated samples.
Histograms of log2 ratio profiles were calculated and plotted using R for the non-treated samples: wild type (WT), UGI-expressing (NT_UGI) HCT116 cells and its MMR proficient variant (NT_UGI_MMR). Thresholds applied in determination of uracil enriched regions are indicated with red line and also provided in Figure 3—figure supplement 4—source data 1 together with the histogram data.
-
Figure 3—figure supplement 4—source data 1
Histograms for the U-DNA signal distribution in non-treated samples.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig3-figsupp4-data1-v2.xlsx
Figure 4 with 3 supplements
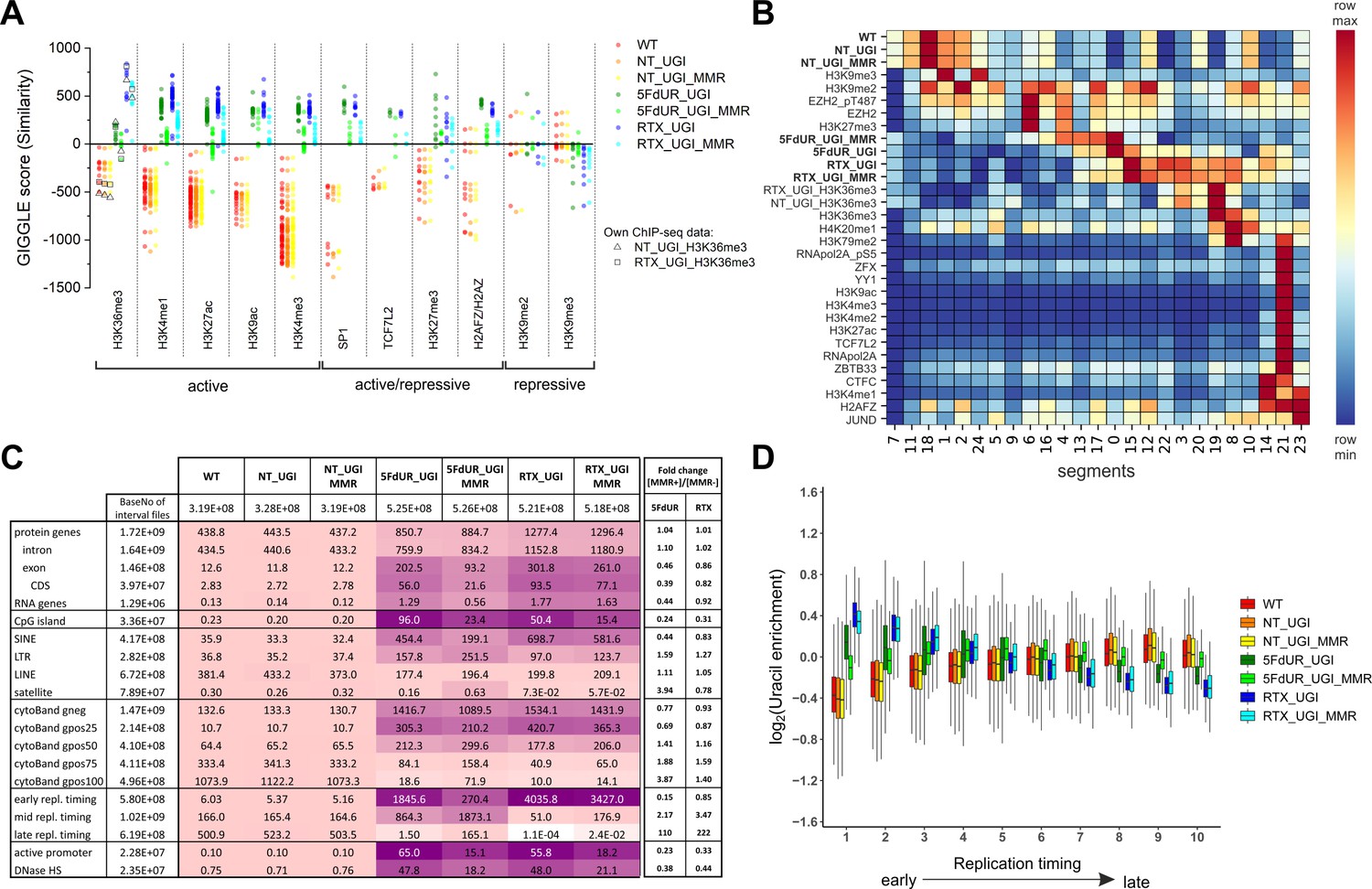
Characterization of U-DNA enrichment patterns.
(A) GIGGLE search was performed with interval (bed) files of uracil enriched regions on a set of HCT116 related ChIP-seq and DIP-seq experimental data (for details see Supplementary file 3). Factors corresponding to the top 10 hits for each sample were selected. GIGGLE scores between all seven samples and all experiments corresponding to these factors were plotted excluding those, where data were not informative (data are found in Supplementary file 3-table 1). Source data are available in Figure 4—source data 1. Histone marks and the transcription factors, SP1 and TCF7L2 are categorized depending on their occurrence in transcriptionally active or repressive regions. Notably, some of them have plastic behavior allowing either transcriptionally active or repressive function. U-DNA-Seq samples are as follows: non-treated wild type (WT, red), non-treated UGI-expressing (NT_UGI, orange), 5FdUR treated UGI-expressing (5FdUR_UGI, green) and RTX treated UGI-expressing (RTX_UGI, blue) HCT116 cells, and their MMR proficient counterparts (NT_UGI_MMR, yellow; 5FdUR_UGI_MMR, light green; RTX_UGI_MMR, light blue). GIGGLE scores are also indicated for our own H3K36me3 ChIP-seq experiments (RTX_UGI sample: empty squares, NT_UGI sample: empty triangles). The tendencies are even more pronounced if the RTX treated U-DNA-Seq is compared with the RTX treated ChIP-seq or if the non-treated U-DNA-Seq is compared with the non-treated ChIP-seq data. (B) Genome segmentation analysis was performed on signal tracks of 22 ChIP-seq data available for HCT116 cells in the ENCODE database, on our own ChIP-seq data for H3K36me3, and on the seven U-DNA enrichment profiles (bold). The Segway train was performed with 25 labels and the corresponding genomic segments were identified with Segway annotate (Chan et al., 2018). The signal distribution data were calculated using Segtools (Buske et al., 2011), and plotted using python seaborn/matplotlib modules (Hunter, 2007). Source data are available in Figure 4—source data 2. Details including the applied command lines are provided in Supplementary file 3. The color-code is applied for each factor (rows) independently, from the minimum to the maximum value as indicated. (C) Correlation with genomic features. Interval (bed) files of genomic features were obtained from UCSC, Ensembl, and ReplicationDomain databases (for details see Supplementary file 4-table 1), and correlation with interval files of uracil regions were analyzed using bedtools annotate software (details are provided in Supplementary file 4). Numbers of overlapping base pairs were summarized for each pair of interval files, and scores were calculated according the formula: (baseNo_overlap/baseNo_sample_file) * (baseNo_overlap/baseNo_feature_file) * 10000. Heatmap was created based on fold increase of the scores compared to the corresponding WT scores. Sizes of interval files in number of base pairs are also given in the second column and the second line. Upon drug treatments, a clear shift from non-coding/heterochromatic/late replicated segments towards more active/coding/euchromatic/early replicated segments can be seen. CDS, coding sequence; SINE, short interspersed element; LTR, long terminal repeat; LINE, long interspersed element; cytoBand, cytogenetic chromosome band negatively (gneg) or positively (gpos) stained by Giemsa; repl. timing, replication timing; DNaseHS, DNase hypersensitive site. (D) Correlation analysis with replication timing. Replication timing data (bigWig files with 5000 bp binsize) specific for HCT116 were downloaded from ReplicationDomain database (Weddington et al., 2008). Data bins were distributed to 10 equal size groups according to replication timing from early to late. Then log2 uracil enrichment signals for these data bin groups were plotted for each sample using R (Supplementary file 5). Source data are available in Figure 4—source data 3.
-
Figure 4—source data 1
GIGGLE similarity scores between U-DNA patterns and selected histone marks or transcription factors.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Signal distribution data from genome segmentation analysis by Segway.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig4-data2-v2.xlsx
-
Figure 4—source data 3
Correlation between U-DNA patterns and replication timing.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig4-data3-v2.xlsx
Figure 4—figure supplement 1

Comparison of our own H3K36me3 ChIP-seq data to each other and to the ENCODE data using Pearson correlation.
ChIP-seq experiments were performed for H3K36me3 in UGI-expressing non-treated (NT_UGI_H3K36me3) and RTX treated (RTX_UGI_H3K36me3) HCT116 cells (Materials and methods). Fold change over control tracks were calculated and compared to HCT116 specific H3K36me3 ChIP-seq data from the ENCODE database (ENCFF514ZYW (merged), ENCFF334KFI (rep1), ENCFF238GBP (rep2)) using Pearson correlation calculated by deepTools multiBigwigSummary and plotCorrelation (Ramírez et al., 2016) as described in Supplementary file 3. Notably, differences between our non-treated and RTX treated samples are not higher than difference between replicates of the ENCODE data.
Figure 4—figure supplement 2
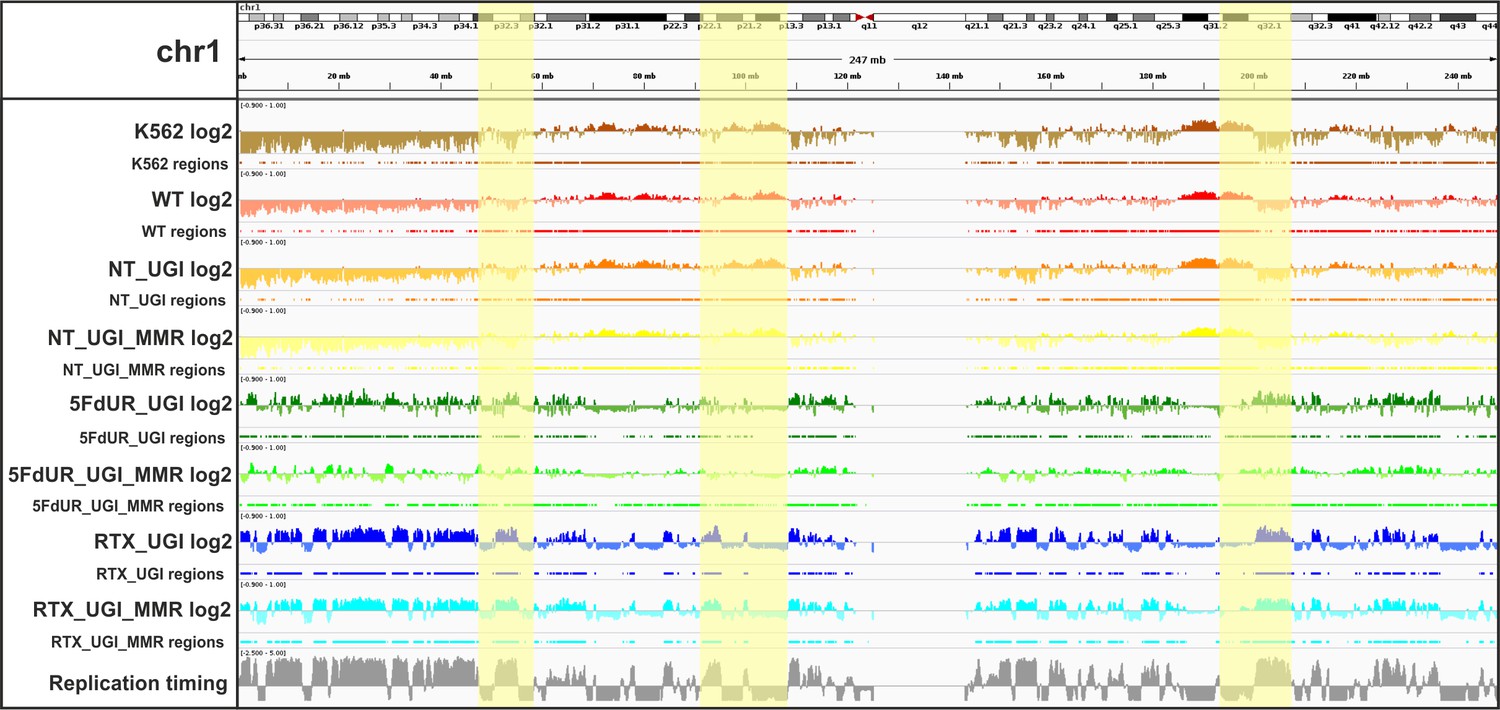
IGV view of log2 ratio and regions of uracil enrichment on chromosome 1 (for all chromosomes see Supplementary file 2).
At the upper part cytogenetic bands by Giemsa staining are visible, the staining intensity (from white to black) correlates to the chromatin structure. Log2 ratio tracks (enriched coverage/input coverage, computed by deepTools/bigwigCompare) are colored by samples (non-treated wild type K562 cells (K562, brown), non-treated wild type (WT, red), non-treated UGI-expressing (NT_UGI, orange), 5FdUR treated UGI-expressing (5FdUR_UGI, green), RTX treated UGI-expressing (RTX_UGI, blue) HCT116 cells, and their MMR proficient variants (NT_UGI_MMR, yellow; 5FdUR_UGI_MMR, light green; RTX_UGI_MMR, light blue)). The negative values of log2 tracks are shown with a bit lighter color than the positives. For all the log2 ratio tracks, the same range was applied from −0.9 to 1. Regions of uracil enrichment derived from log2 ratio tracks are shown with the same color directly below the corresponding log2 tracks. The bottom track shows replication timing data (grey) for HCT116 downloaded from Replication Domain database (Weddington et al., 2008). Replication timing scores are derived from E/L Repli-seq experiments, where cycling cells are pulse-labeled with BrdU, then sorted to early and late S-phase fractions by flow cytometry, and BrdU labeled genomic DNA fragments are pulled down and subjected to NGS. Signal tracks are computed from the read coverages in early over late S-phase samples, therefore the higher score means earlier replication (Marchal et al., 2018). The scale goes from −2.5 to 5. Correlation among samples, replication timing and the cytogenetic bands are well visible (highlighted regions, Figure 4C–D, and Supplementary file 4).
Figure 4—figure supplement 3

Replication timing scores and AT content calculated on the genomic segments that were defined by the Segway analysis.
25 genomic segments from the Segway were analyzed. Average replication timing scores (gray bars) were calculated from two replicates available in Replication Domain database (Weddington et al., 2008), the standard deviation is also indicated. Replication timing scores are derived from E/L Repli-seq experiments, the higher score means earlier replication (Marchal et al., 2018). AT content of the segments (%, black square) were calculated using bedtools nuc (Quinlan and Hall, 2010) and the GRCh38 reference genome. Details of the calculations are provided in Supplementary file 4. For better comparison, signal distribution patterns of U-DNA-Seq samples derived from the Segway analysis (Figure 4B) are repeated at the bottom.
-
Figure 4—figure supplement 3—source data 1
Replication timing scores and AT content calculated on genomic segments that were determined by the Segway analysis.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig4-figsupp3-data1-v2.xlsx
Figure 5 with 1 supplement

Cell cycle analysis showing the impact of UGI expression with or without drug treatments in MMR deficient and proficient HCT116 cells.
Scatter plots represent the flow cytometric measurements of BrdU incorporation and propidium iodide (PI) DNA-staining. (A) Cell cycle distribution in non-treated, MMR deficient (WT), and UGI-expressing (NT_UGI); or in MMR proficient (NT_MMR), and UGI-expressing (NT_UGI_MMR) HCT116 cells. (B) Cell cycle differences caused by 5FdUR or RTX drug treatments in MMR deficient, UGI-expressing (5FdUR_UGI and RTX_UGI); or in MMR proficient, UGI-expressing (5FdUR_UGI_MMR and RTX_UGI_MMR) HCT116 cells.
Figure 5—figure supplement 1
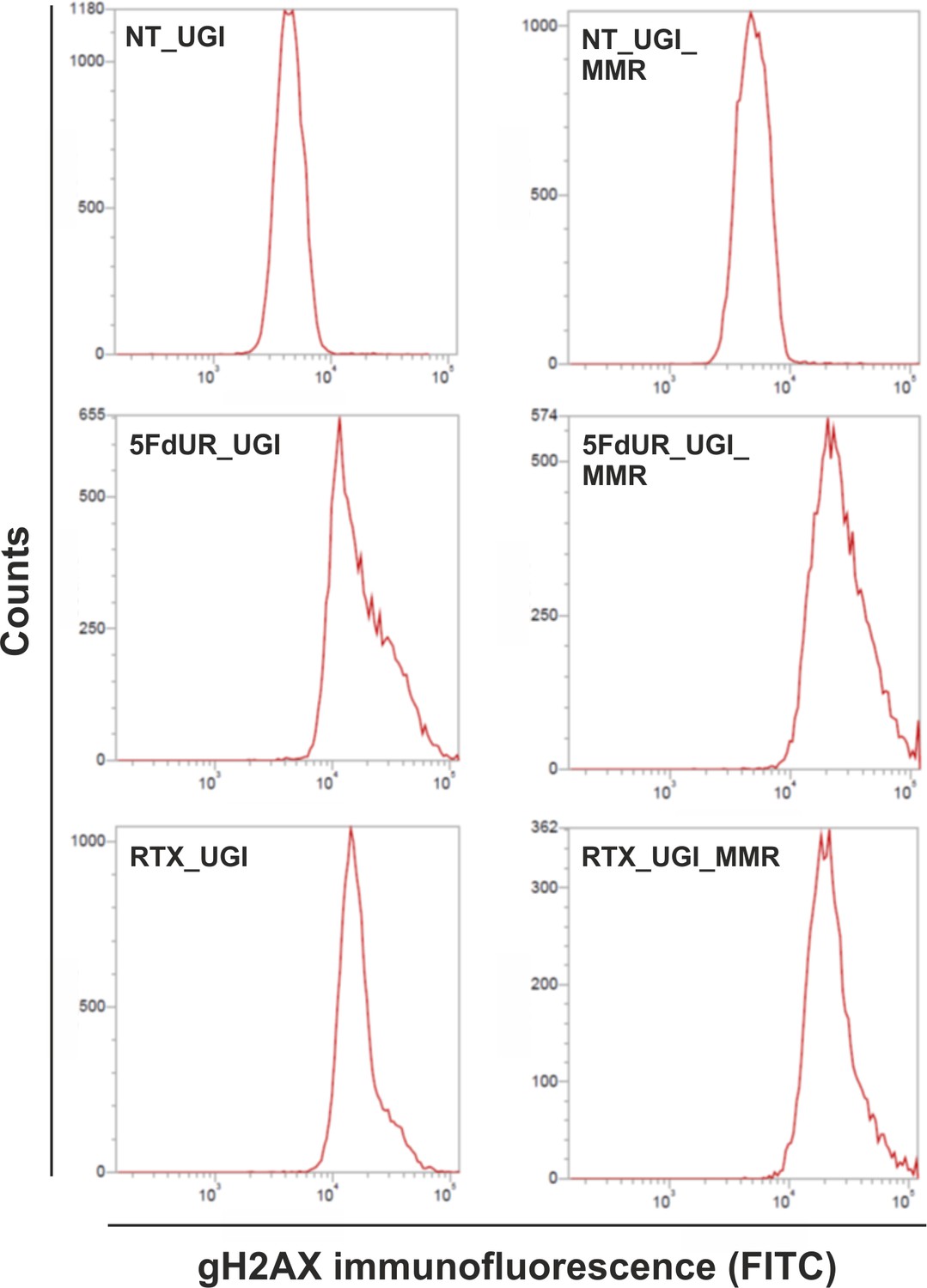
Flow cytometry-based detection of γH2AX, indicative to DSBs involved in DNA damage response.
Representative histograms show increased γH2AX immunofluorescent signal upon drug (5FdUR or RTX) treatments both in MMR deficient, UGI-expressing (5FdUR_UGI, RTX_UGI); and MMR proficient, UGI-expressing (5FdUR_UGI_MMR, RTX_UGI_MMR) HCT116 cells, compared to the corresponding non-treated ones (NT_UGI, NT_UGI_MMR).
Figure 6 with 1 supplement
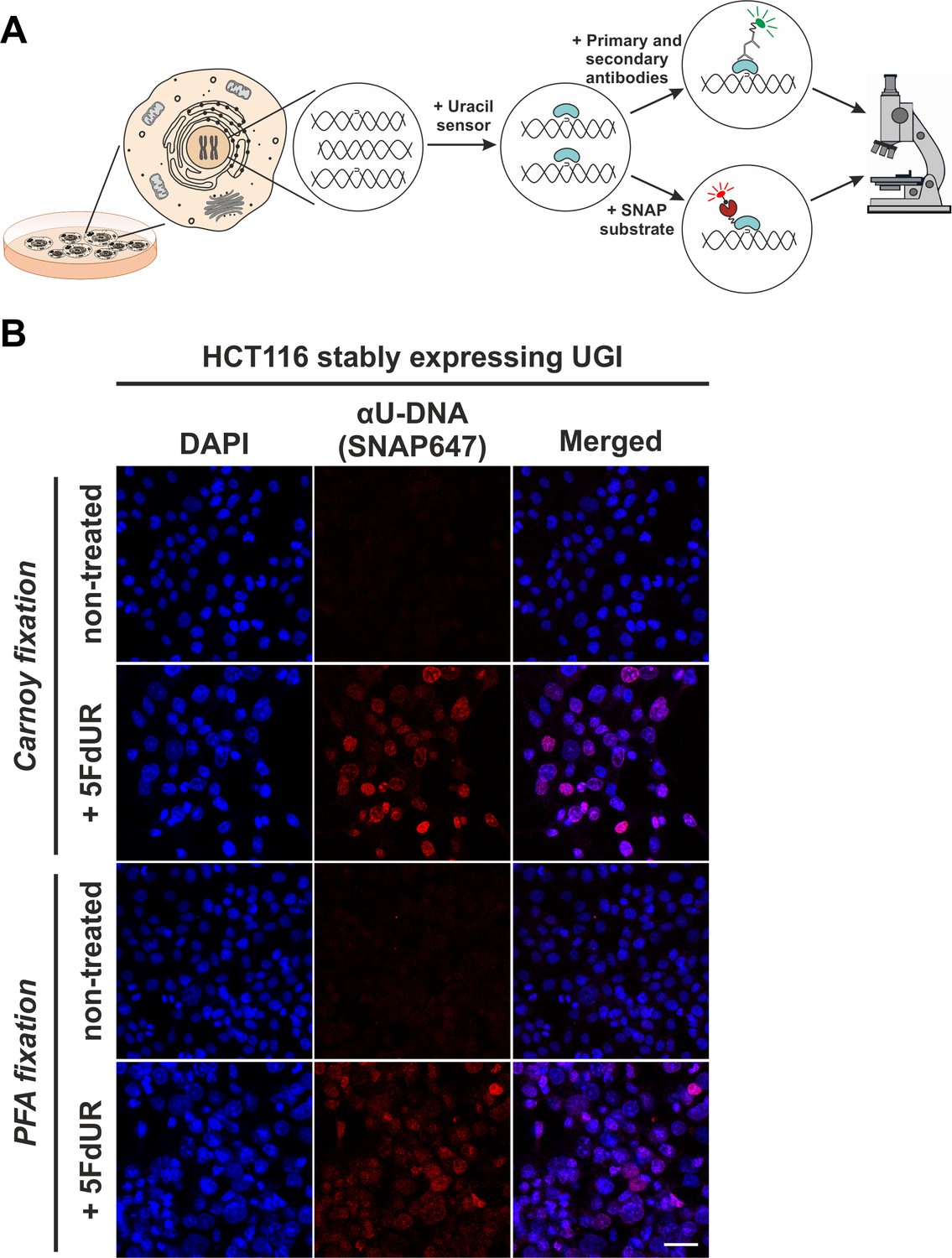
In situ detection of the cellular endogenous U-DNA content.
(A). Scheme represents that genomic uracil residues can be visualized in situ using our further developed U-DNA sensor construct via immunocytochemistry (through FLAG-tag) or directly via SNAP-tag chemistry. (B) HCT116 cells expressing UGI and treated with 5FdUR show efficient staining with the uracil sensor compared to non-treated cells, detected by confocal microscopy. Uracil residues are labelled by our FLAG-ΔUNG-SNAP sensor protein visualized by the SNAP647 substrate. DAPI was used for DNA counterstaining. Our optimized staining method is capable of comparable, specific uracil detection in HCT116 cells even with paraformaldehyde (PFA) fixation compared to the Carnoy fixation applied previously (Róna et al., 2016). Scale bar represents 40 µm. Note that the nuclei of the treated cells (5FdUR_UGI) are enlarged as compared to the non-treated ones (NT_UGI) presumably due to cell cycle arrest (Huehls et al., 2016; Yan et al., 2016).
Figure 6—figure supplement 1
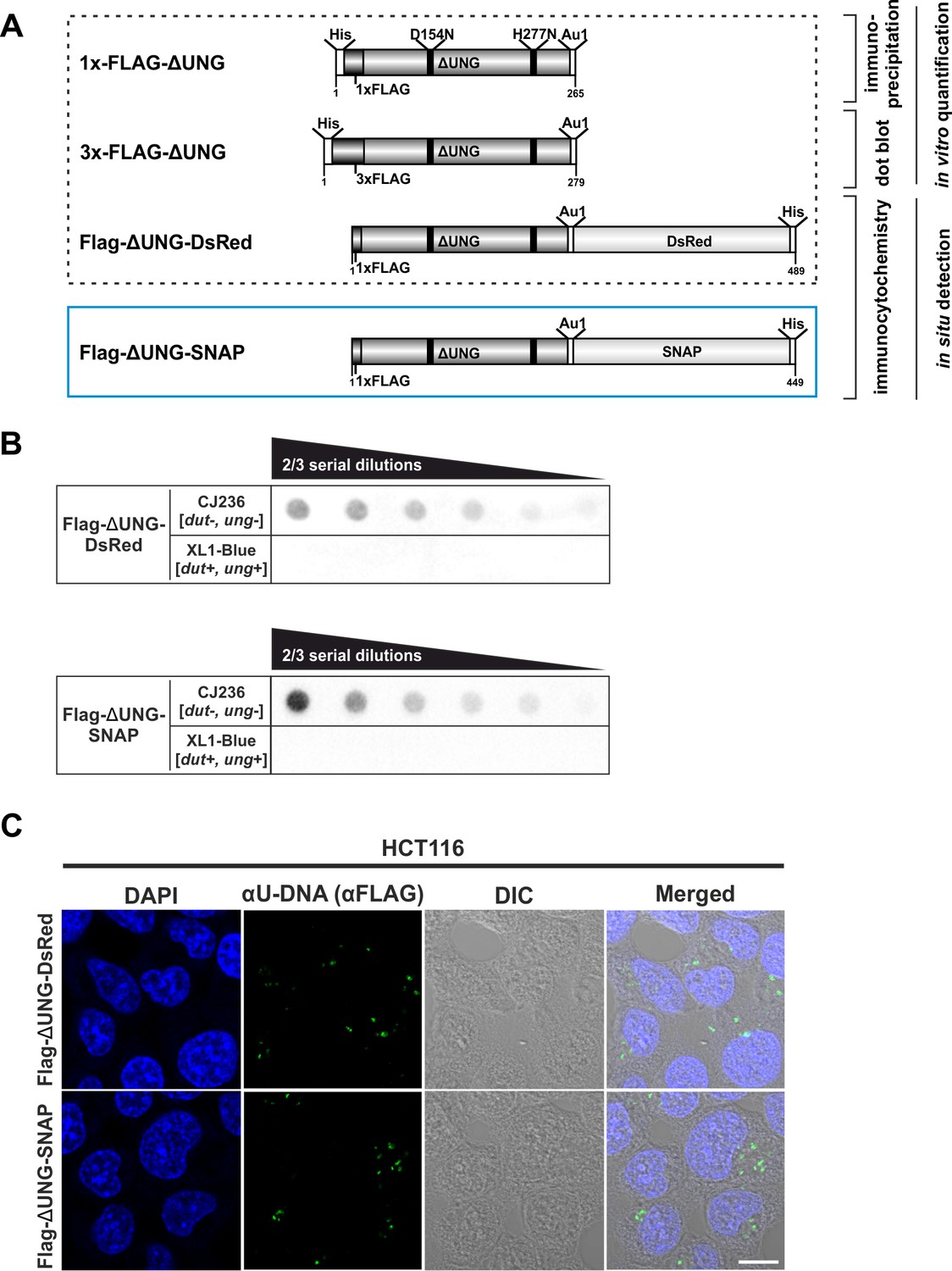
Design and validation of the new FLAG-ΔUNG-SNAP uracil-sensor.
(A) Scheme of the used constructs in this study, with their applications highlighted. The catalytically inactive (D154N and H277N, mutated sites indicated with black lines) and truncated ΔUNG (the N-terminal 84 residues, responsible for the binding to RPA and PCNA, were also removed) was created from human UNG2. The ΔUNG uracil-recognizing core was fused to different epitope tags: His-tag for affinity purification, 1x/3xFLAG and Au1 for antibody-based detection, DsRed-monomer or SNAP-tag for direct fluorescent detection. Our newly developed FLAG-ΔUNG-SNAP sensor construct is indicated with blue frame, separately from the constructs characterized previously (Róna et al., 2016). (B) Dot blot assay was used to compare uracil binding capability of FLAG-ΔUNG-DsRed and FLAG-ΔUNG-SNAP sensor constructs. CJ236 [dut−, ung−] (positive control) and XL1-Blue [dut+, ung+] (negative control) E. coli genomic DNA samples (8 ng) were measured in two-third dilution series. (C) Comparison of the FLAG-ΔUNG-DsRed and the FLAG-ΔUNG-SNAP constructs in detecting uracil-rich plasmid DNA aggregates in HCT116 in an immunocytochemistry assay. The sensors were visualized through the FLAG epitope tag. DAPI was used to counterstain DNA. Scale bar represents 10 μm.
Figure 7 with 5 supplements
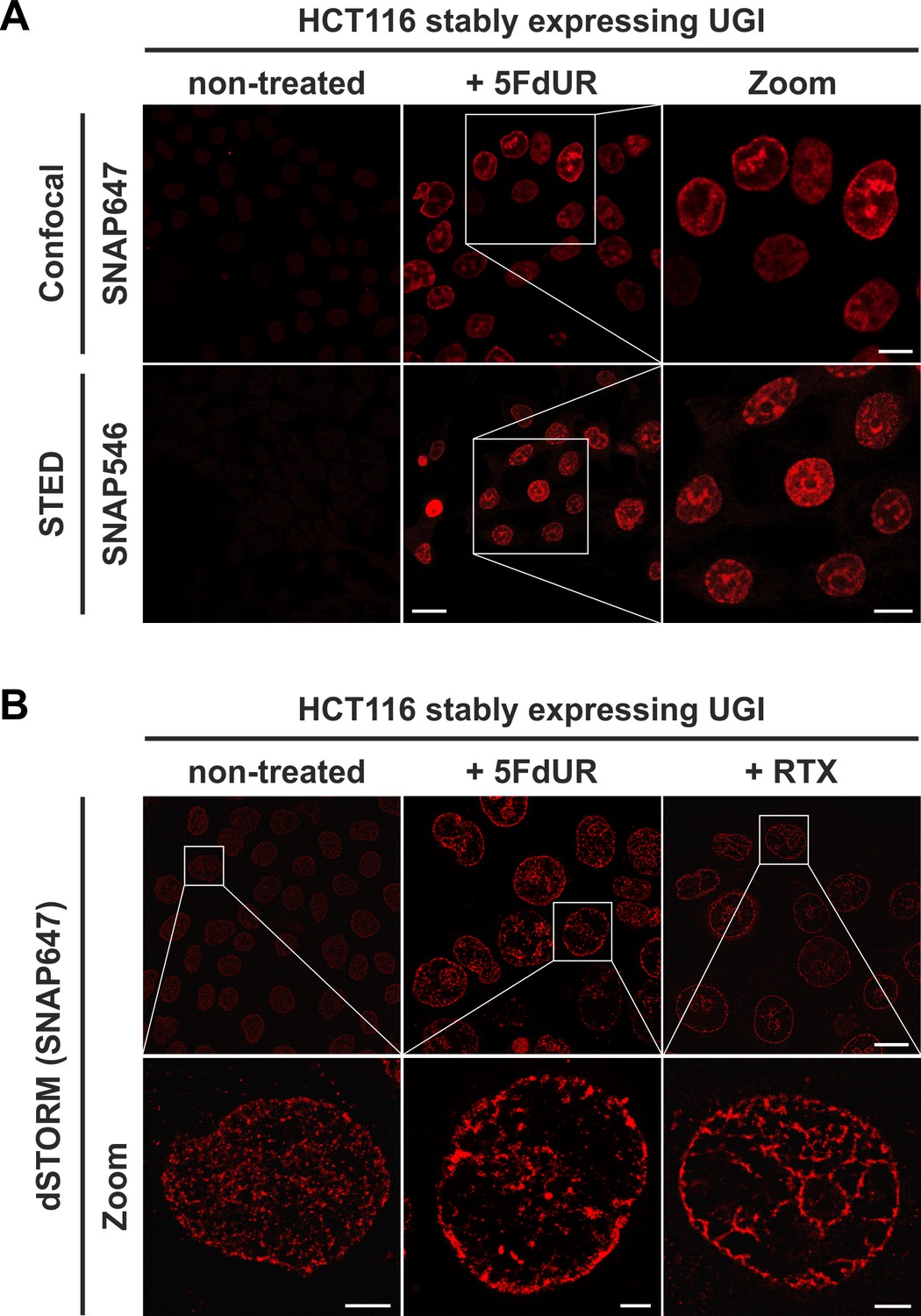
The FLAG-ΔUNG-SNAP sensor enables super-resolution detection of genomic uracil by STED and dSTORM microscopy.
(A) U-DNA staining was performed on non-treated or 5FdUR treated HCT116 cells stably expressing UGI. Different SNAP-tag substrates, SNAP647 for confocal and SNAP546 for super-resolution imaging (STED) were used to label FLAG-ΔUNG-SNAP. Scale bar represents 20 µm for whole images and 10 µm for zoomed sections. (B) dSTORM imaging was performed on non-treated or drug-treated (5FdUR or RTX) HCT116 cells stably expressing UGI to compare the sensitivity of these imaging techniques. U-DNA staining shows a characteristic distribution pattern in cells with elevated uracil levels as compared to non-treated cells. SNAP647 substrate was used to label FLAG-ΔUNG-SNAP. Scale bar represents 10 µm for whole images and 2 µm for zoomed sections.
Figure 7—figure supplement 1

Representative image for movies in Figure 7—videos 1–4.
Genomic uracil levels of HCT116 cells were elevated with 5FdUR treatment and UGI expression. Videos show the following: confocal Z-stack series of 3 cells (Figure 7—video 1), 3D projection of this confocal series (Figure 7—video 2), Z-stack series of 1 cell acquired by STED microscopy (Figure 7—video 3) and 3D projection of this series acquired by STED microscopy (Figure 7—video 4). The related movies are indicated at the top right corner of the images. Scale bar represents 10 µm.
Figure 7—video 1
Confocal Z-stack series of 3 cells showing uracil distribution.
Figure 7—video 2
3D projection of Figure 7—video 1.
Figure 7—video 3
Z-stack series of 1 cell acquired by STED showing uracil distribution.
Figure 7—video 4
3D projection of Figure 7—video 3.
Figure 8
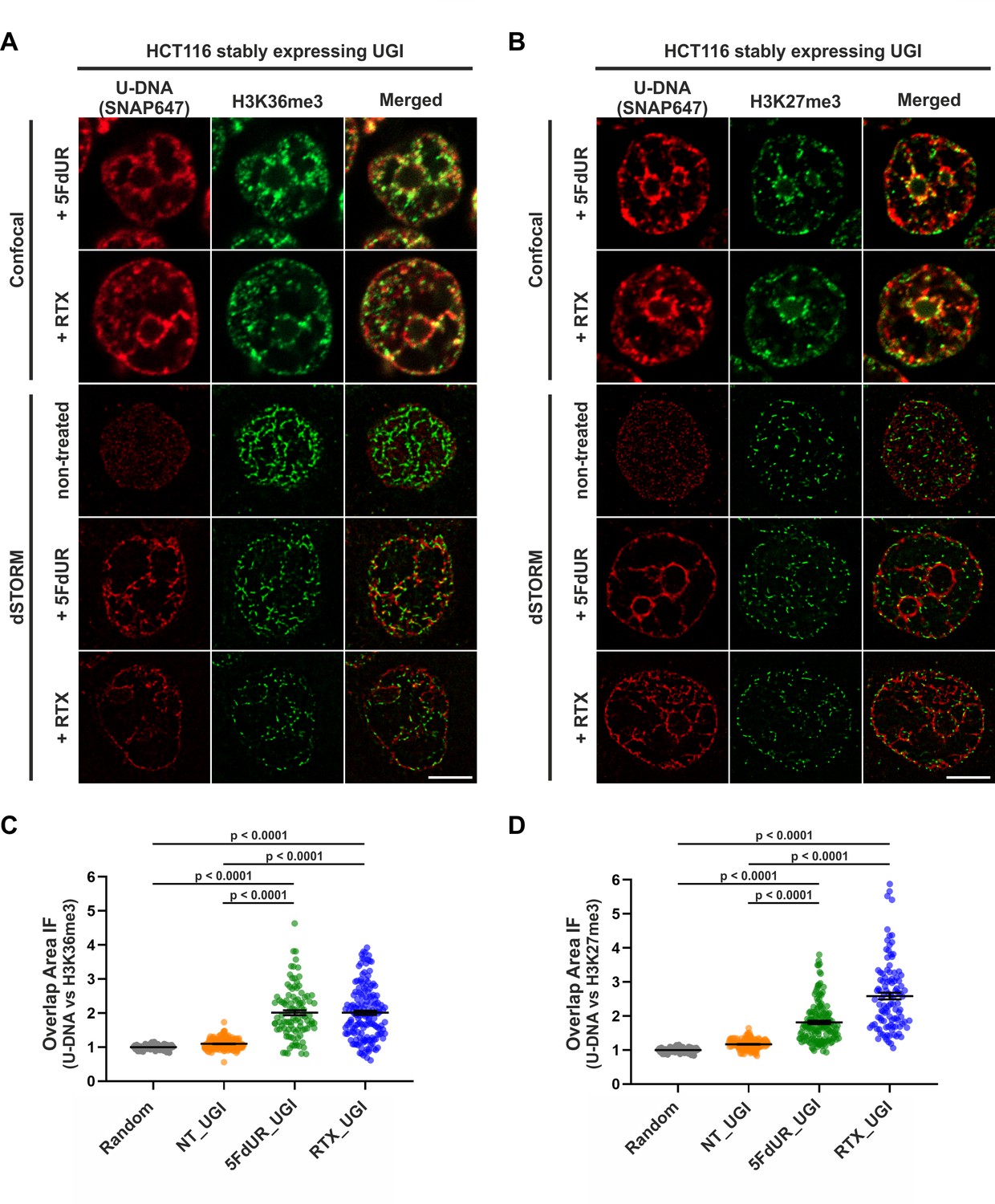
Genomic uracil moieties colocalize with H3K36me3 and H3K27me3 analyzed by super-resolution microscopy.
Confocal and dSTORM imaging were performed on non-treated, 5FdUR or RTX treated HCT116 cells stably expressing UGI to compare the localization of genomic uracil residues (red) to histone markers, H3K36me3 (green) (A) or H3K27me3 (green) (B). Scale bar represents 5 μm. The graphs display the cross-pair orrelation analysis between U-DNA and H3K36me3 (C) or H3K27me3 (D) performed on dSTORM images. Overlap is defined as any amount of pixel overlap between segmented objects. Total numbers of analyzed nuclei for H3K36me3 staining (C) were the following: NT_UGI (n = 205), 5FdUR_UGI (n = 101) and RTX_UGI (n = 153) from two independent experiments. Total numbers of analyzed nuclei for H3K27me3 staining (D) were the following: NT_UGI (n = 154), 5FdUR_UGI (n = 151) and RTX_UGI (n = 107) from two independent experiments. Black line denotes the mean of each dataset, and error bars represent standard errors of the mean (SEM). The color code follows the one in Figure 3A. Source data are available in Figure 8—source data 1.
-
Figure 8—source data 1
Interaction factors between U-DNA and selected histone marks, determined in colocalization measurements using dSTORM microscopy.
- https://cdn.elifesciences.org/articles/60498/elife-60498-fig8-data1-v2.xlsx
Appendix 1—figure 1
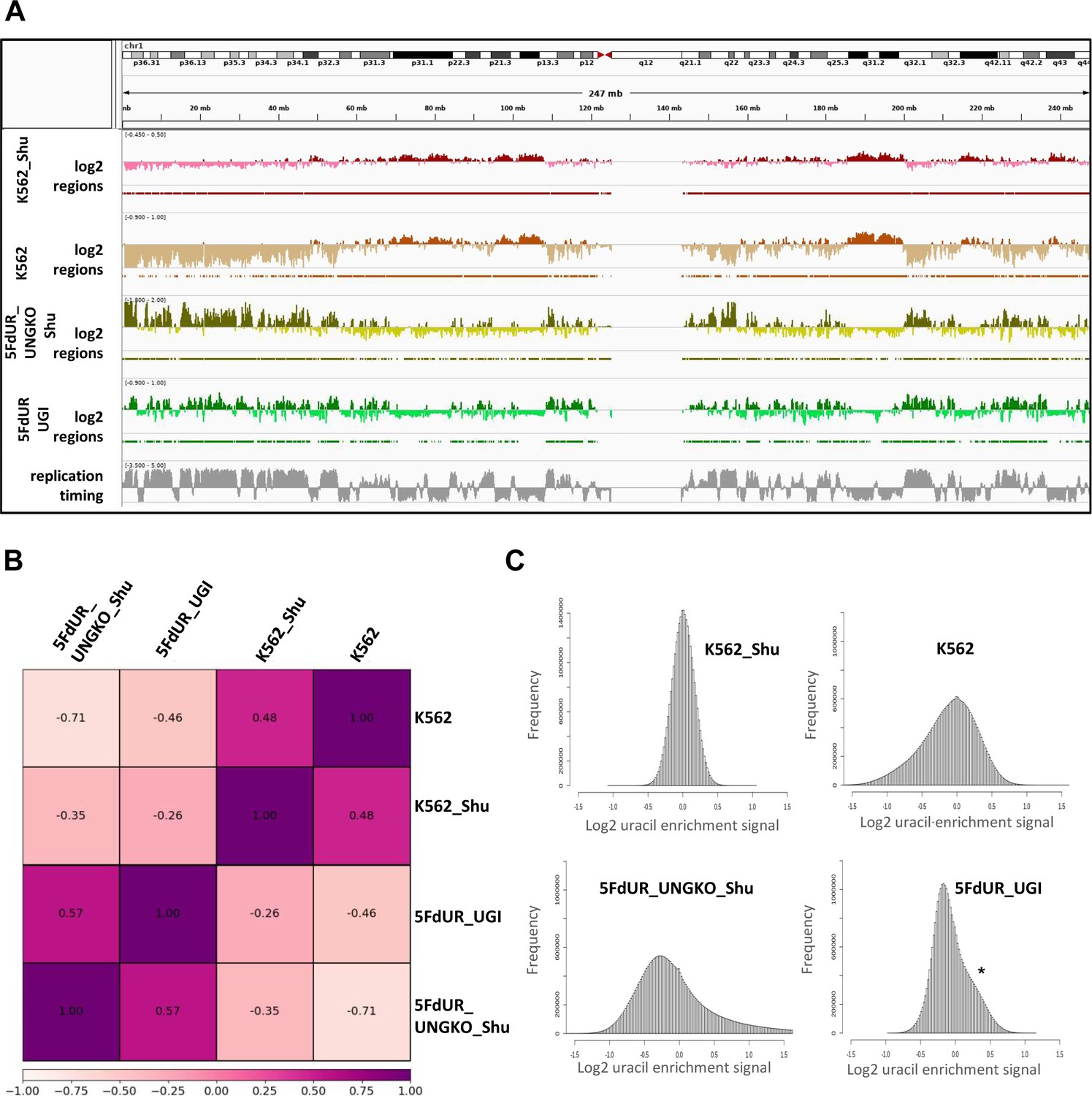
Re-analysis of the published dU-seq data (Shu et al., 2018) reveals that corresponding samples from dU-seq and U-DNA-Seq show similar patterns of uracil distribution.
(A) IGV view of dU-seq data (non-treated K562 cells (K562_Shu, wine track), and 5FdUR treated UNG-knock-out HEK293 cells (5FdUR_UNGKO_Shu, olive track)), compared to our own U-DNA-Seq data (non-treated K562 cells (K562, brown track), and 5FdUR treated UNG inhibited HCT116 cells (5FdUR_UGI, green track)) on chromosome 1. Log2 ratio tracks and the derived regions of uracil enrichment are also indicated. The bottom track shows replication timing data (grey) for HCT116 downloaded from Replication Domain database (Weddington et al., 2008). (B) Pearson correlation among dU-seq and U-DNA-Seq log2 ratio tracks calculated from merged replicates. The drug-treated and non-treated samples are well separated again. Pearson correlation between corresponding dU-seq and U-DNA-Seq samples are unexpectedly high, especially considering the cell line difference in case of drug-treated cells, and the overall low signal intensity in case of non-treated K562. (C) Log2 ratio signal distribution of dU-seq and U-DNA-Seq data. The non-treated K562 samples result in a normal like distribution of uracil enrichment signals, while in case of 5FdUR treated cells, these distributions show asymmetry: either a clear shoulder (asterisk), or a more elongated tail towards increased signals in both U-DNA-Seq and dU-seq data, respectively. Source data are available in Appendix 1—figure 1—source data 1.
-
Appendix 1—figure 1—source data 1
Comparison of histograms for the U-DNA signal distributions between dU-seq and U-DNA-Seq data.
- https://cdn.elifesciences.org/articles/60498/elife-60498-app1-fig1-data1-v2.xlsx
Appendix 1—figure 2
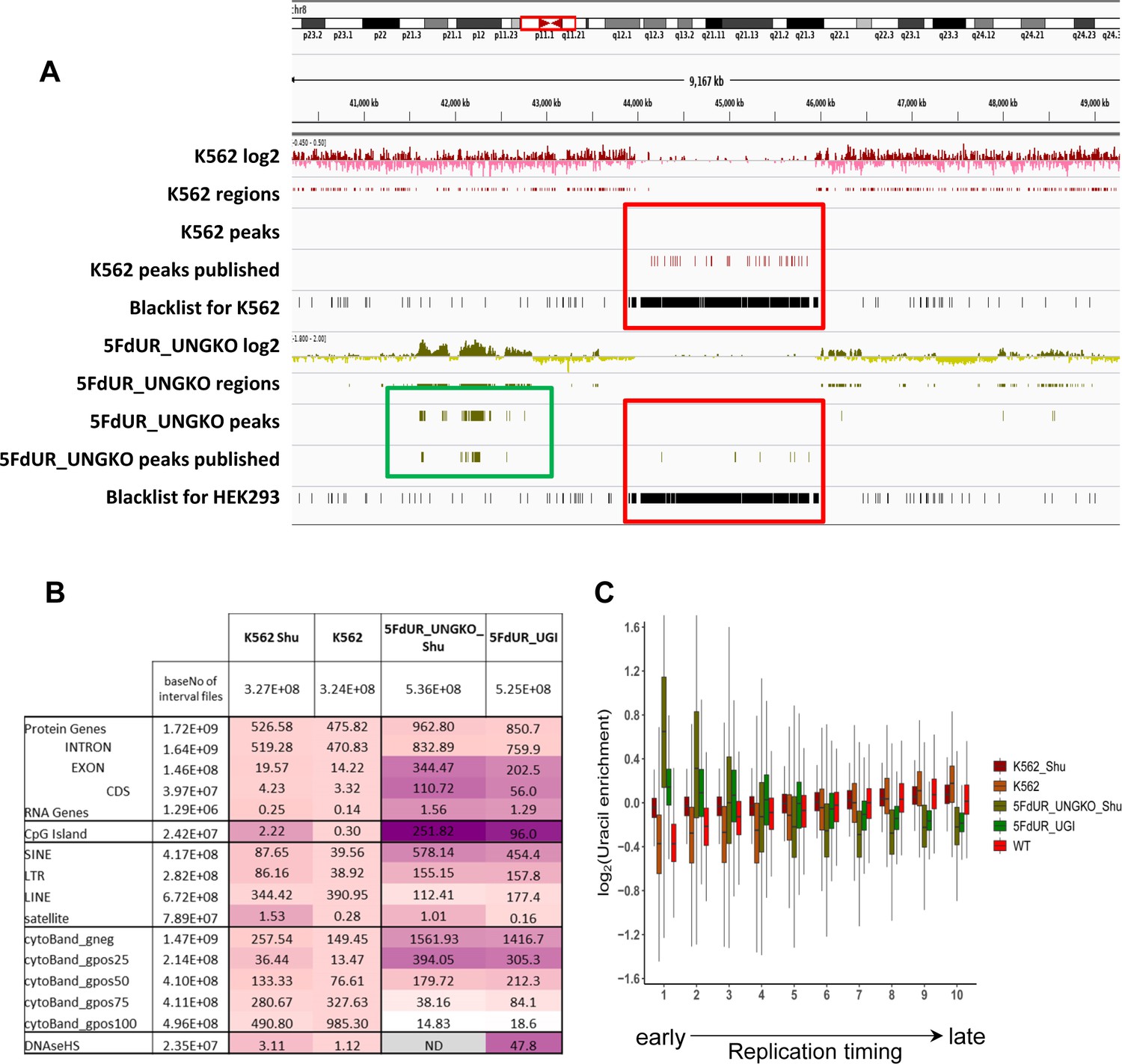
Reinterpretation of dU-seq data.
(A) Comparison of the re-analyzed and the published dU-seq data in a representative IGV view of chromosome 8 zoomed to the centromeric region (also Figure 3 in Shu et al., 2018). All centromeric peaks in K562 published for chromosome 8 in Shu et al are found in blacklisted regions (red box). Overall, 75% of their published peaks in K562 are overlapping with blacklisted regions determined by our protocol (Figure 2—figure supplement 2). Accordingly, no peaks were called in the presented region during the re-analysis of the sequencing data (red box). Similarly, in drug-treated samples, published centromeric peaks were not reproducible (red box), while other peaks outside of the centromeres were similar in the published and the re-analyzed data (green box). (B) dU-seq data shows similar correlation to genomic features as compared to the corresponding U-DNA-Seq data. Similarity were measured by bedtools annotate tool and the scores were calculated in the same way as it was in Figure 4C. For each sample, cell type (HCT116 or K562) specific DNase hypersensitive site data were used. For 5FdUR treated HEK293 cells, similarity to DNase hypersensitive site data was not addressed (grey). (C) Correlation between uracil distribution and replication timing were confirmed by dU-seq data as well, although this correlation is weaker than the U-DNA-Seq results (Figure 4D). Replication timing data (bigWig files with 5000 bp binsize) were downloaded from ReplicationDomain database: Int90617792 for HCT116; Int57383924 for HEK293; Int37482971 for K562. Source data are available in Appendix 1—figure 2—source data 1.
-
Appendix 1—figure 2—source data 1
Comparison of dU-seq and U-DNA-Seq data regarding correlation between U-DNA patterns and replication timing.
- https://cdn.elifesciences.org/articles/60498/elife-60498-app1-fig2-data1-v2.xlsx
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (bacteriophage PBS2) | UGI | Mol et al., 1995 | UniProtKB - P14739 | UNG inhibitor protein encoded in Bacillus subtilis bacteriophage PBS2 |
| Antibody | Anti-FLAG M2, mouse monoclonal | Sigma | F3165 | (1:10000) |
| Antibody | anti-H3K36me3, rabbit monoclonal | Cell Signaling Technologies | 4909T | (1:8000) for ICC |
| Antibody | anti-H3K27me3, rabbit monoclonal | Cell Signaling Technologies | 9733T | (1:6000) for ICC |
| Antibody | anti-γH2AX, rabbit monoclonal | Sigma | 05–636 | (1:500) for measuring DSBs in flow cytometry |
| Cell line (Homo sapiens) | 293T | Yvonne Jones (Cancer Research UK, Oxford, UK) | maintained in Dulbecco’s modified Eagle’s medium completed with PenStrep and FBS | |
| Cell line (Homo sapiens) | K562 | European Collection of Cell Cultures | maintained in RPMI 1640 (GlutaMAX Supplement, HEPES) Medium (Gibco), completed with PenStrep and FBS | |
| Cell line (Homo sapiens) | HCT116 | European Collection of Cell Cultures | maintained in McCoy’s 5A medium, completed with PenStrep and FBS | |
| Cell line (Homo sapiens) | HCT116+ch3 sub-line | C. Richard Boland (Baylor University, Dallas, Texas, US) | sub-line of HCT116: MLH1 restored, MMR is functional | |
| Cell line (Homo sapiens) | HCT116+ hUGI/EGFP | This paper | sub-line of HCT116: stable expressing UGI (Materials and methods: Generation of UGI-expressing stable cell lines) | |
| Cell line (Homo sapiens) | HCT116+ch3+ hUGI/EGFP | This paper | sub-line of HCT116+ch3: stable expressing UGI (Materials and methods: Generation of UGI-expressing stable cell lines) | |
| Strain, strain background (Escherichia coli) | XL1-Blue | Stratagene | ||
| Strain, strain background (Escherichia coli) | CJ236 [dut-, ung-] | NEB | E. coli strain for preparation of the uracil-containing DNA | |
| Strain, strain background (Escherichia coli) | BL21(DE3) ung-151 | Samuel E Bennett (Oregon State University, Corvallis, US) | E. coli strain for expression of ΔUNG sensor constructs | |
| Sequence-based reagent | actin-for | Sigma-Aldrich, Ho et al., 2016 | 5’-CCTCATGGCCTTGTCACAC-3’ | |
| Sequence-based reagent | actin-rev | Sigma-Aldrich, Ho et al., 2016 | 5’-GCCCTTTCTCACTGGTTCTCT-3’ | |
| Sequence-based reagent | pET15b-For | Sigma-Aldrich | 5’-CATATGCTCGAGGATCCGGC-3’ | |
| Sequence-based reagent | pET15b-Rev | Sigma-Aldrich | 5’-TCATCGATAAGCTTTAATGCGGT-3’ | |
| Sequence-based reagent | Spin-Fw | Sigma-Aldrich | 5’- ACCGGCATAACCAAGCCTAT-3’ | |
| Sequence-based reagent | Spin-Rev | Sigma-Aldrich | 5’- ACAATGCGCTCATCGTCATC-3’ | |
| Recombinant DNA reagent | pLGC-hUGI/EGFP | Michael D Wyatt (South Carolina College of Pharmacy, University of South Carolina, US) | for producing sub-lines stably expressing UGI | |
| Recombinant DNA reagent | pSNAPf | NEB | N9183S | to clone the FLAG-ΔUNG-SNAP construct |
| Peptide, recombinant protein | FLAG-ΔUNG-SNAP | This paper | produced in E. coli BL21(DE3) ung-151 (Materials and methods: Plasmid constructs and cloning of the FLAG-ΔUNG-SNAP construct) | |
| Peptide, recombinant protein | 1xFLAG-ΔUNG | Róna et al., 2016 | produced in E. coli BL21(DE3) ung-151 | |
| Peptide, recombinant protein | 3xFLAG-ΔUNG | Róna et al., 2016 | produced in E. coli BL21(DE3) ung-151 | |
| Peptide, recombinant protein | FLAG-ΔUNG-DsRed | Róna et al., 2016 | produced in E. coli BL21(DE3) ung-151 | |
| Commercial assay, kit | Quick-DNA Miniprep Plus Kit | Zymo Research | D4069 | for genomic DNA preparation |
| Commercial assay, kit | NucleoSpin Gel and PCR Clean-up Kit | MACHEREY-NAGEL GmbH and Co. KG | 740609.25 | for IP DNA purification |
| Commercial assay, kit | NGS including library preparation | Novogene | Novaseq 6000, 20 GB, 150 PE | as a service |
| Commercial assay, kit | 5-Bromo-2′-deoxy-uridine (BrdU) Labeling and Detection Kit I | Roche, Sigma | 11296736001 | |
| Chemical compound, drug | Anti-FLAG M2 agarose beads | Sigma | A2220 | for U-DNA-IP |
| Chemical compound, drug | Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | 88802 | for ChIP |
| Chemical compound, drug | 5-fluoro-2′-deoxyuridine (5FdUR) | Sigma | F0503 | Thymidylate synthase inhibitor, treatment: 20 µM, 48 hr |
| Chemical compound, drug | raltitrexed (RTX) | Sigma | R9156 | Thymidylate synthase inhibitor, treatment: 100 nM, 48 hr |
| Chemical compound, drug | SNAP-Surface Alexa Fluor 546 | NEB | S9132S | SNAP substrate for superresolution imaging |
| Chemical compound, drug | SNAP-Surface Alexa Fluor 647 | NEB | S9136S | SNAP substrate for superresolution imaging |
| Software, algorithm | ImageJ (Fiji) | National Institutes of Health | for densitometry, and image processing | |
| Software, algorithm | Huygens STED Deconvolution Wizard | Huygens Software | superresolution image analyzing software package | |
| Software, algorithm | BWA | Li and Durbin, 2010 | short sequencing read aligner software | |
| Softwares, algorithm | deepTools package | Ramírez et al., 2016 | NGS data processing tools | |
| Software, algorithm | bedtools package | Quinlan and Hall, 2010 | tools for analyzing interval files | |
| Software, algorithm | GIGGLE search | Layer et al., 2018. | search tool for similarity screening in large set of interval files | |
| Software, algorithm | Segway software package | Chan et al., 2018; Hoffman et al., 2012 | machine learning software for genome segmentation | |
| Software, algorithm | Integrated Genome Viewer (IGV) | Thorvaldsdóttir et al., 2013. | tool for visualisation of many types of processed NGS data |
Appendix 1—table 1
Statistics on pre-processing of dU-seq data.
Samples from the study of Shu et al., 2018 (non-treated K562 (K562), and 5FdUR treated UNG-knock-out HEK293 cells (5FdUR_UNGKO HEK293)) are compared to our samples (5FdUR treated UGI-expressing HCT116 cells (5FdUR_UGI), and non-treated wild type K562 cells (K562), the same data as in Supplementary file 1-table 3). In case of dU-seq samples, inputs are genomic DNA fragmented and treated according to the dU-seq protocol, also containing additional spike-in sequences; controls are pulled-down in a mock experiment excluding UNG treatment; while PD means the pull-down samples according to the dU-seq protocol. Number of raw reads means read number before starting alignment (the sum of the mapped and unmapped reads). Uniquely mapped read means that MAPQ is not zero. The dU-seq and the U-DNA-Seq samples markedly differ in the ratio of mapped (*) and unmapped (*) reads due to the spike-in DNA applied in dU-seq only.
| Sample | Replicates | Number of raw reads | Number of mapped* reads | Unmapped* reads | Uniquely mapped reads | Uniquely mapped reads after blacklisting | |||
|---|---|---|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | ||||
| K562 input | SRR5572773 | 9,59,22,009 | 9,06,63,272 | 52,58,737 | 5.48 | 8,40,45,278 | 87.62 | 8,21,92,353 | 85.69 |
| SRR5572774 | 9,50,30,662 | 8,98,16,930 | 52,13,732 | 5.49 | 8,33,06,810 | 87.66 | 8,14,57,577 | 85.72 | |
| K562 Control | SRR5572775 | 7,63,94,870 | 4,61,89,867 | 3,02,05,003 | 39.54 | 4,13,85,042 | 54.17 | 4,04,05,241 | 52.89 |
| SRR5572776 | 7,89,62,287 | 4,10,53,994 | 3,79,08,293 | 48.01 | 3,62,60,497 | 45.92 | 3,53,93,512 | 44.82 | |
| K562 PD | SRR5572777 | 8,74,66,276 | 5,41,13,837 | 3,33,52,439 | 38.13 | 4,84,46,026 | 55.39 | 4,73,24,075 | 54.11 |
| SRR5572778 | 8,24,99,155 | 5,29,29,849 | 2,95,69,306 | 35.84 | 4,75,55,693 | 57.64 | 4,64,66,915 | 56.32 | |
| 5FdUR_UNGKO | SRR5998406 | 12,56,31,380 | 10,83,20,783 | 1,73,10,597 | 13.78 | 10,10,27,428 | 80.42 | 9,90,12,730 | 78.81 |
| HEK293 input | SRR5998407 | 7,03,49,101 | 6,10,39,638 | 93,09,463 | 13.23 | 5,69,70,384 | 80.98 | 5,58,07,073 | 79.33 |
| 5FdUR_UNGKO | SRR5998408 | 11,36,54,134 | 6,26,79,292 | 5,09,74,842 | 44.85 | 5,53,33,969 | 48.69 | 5,41,29,569 | 47.63 |
| HEK293 Control | SRR5998409 | 12,91,96,940 | 5,80,03,222 | 7,11,93,718 | 55.10 | 4,97,06,846 | 38.47 | 4,86,00,418 | 37.62 |
| 5FdUR_UNGKO | SRR5998410 | 8,00,35,762 | 6,79,39,558 | 1,20,96,204 | 15.11 | 6,34,53,866 | 79.28 | 6,21,84,497 | 77.70 |
| HEK293 PD | SRR6026694 | 6,62,42,483 | 5,63,03,837 | 99,38,646 | 15.00 | 5,26,53,804 | 79.49 | 5,15,98,007 | 77.89 |
| 5FdUR_UGI input | 5FdUR1_son | 12,87,06,895 | 12,86,69,770 | 37,125 | 0.03 | 12,24,76,766 | 95.16 | 11,85,58,597 | 92.12 |
| 5FdUR1_son | 20,19,26,203 | 20,15,60,665 | 3,65,538 | 0.18 | 19,30,86,643 | 95.62 | 18,47,56,297 | 91.50 | |
| 5FdUR_UGI enriched | 5FdUR1_IP | 15,05,96,242 | 15,05,22,522 | 73,720 | 0.05 | 14,45,54,269 | 95.99 | 14,15,82,874 | 94.01 |
| 5FdUR2_IP | 13,86,51,760 | 13,84,10,833 | 2,40,927 | 0.17 | 13,32,00,761 | 96.07 | 12,85,84,894 | 92.74 | |
| K562 input | K562_son | 10,61,37,622 | 10,58,75,437 | 2,62,185 | 0.25 | 10,03,26,105 | 94.52 | 9,75,04,876 | 91.87 |
| K562 enriched | K562_IP | 10,94,90,393 | 10,93,06,854 | 1,83,539 | 0.17 | 10,53,10,296 | 96.18 | 10,21,17,055 | 93.27 |
Additional files
-
Supplementary file 1
Detailed analysis pipeline – methods of U-DNA-Seq data analysis.
List of the investigated samples (table 1); list of applied tools (table 2); pre-processing including blacklisting and additional statistics (table 3); and methods to determine uracil enrichment pattern. All applied processing steps are given in generalized command lines.
- https://cdn.elifesciences.org/articles/60498/elife-60498-supp1-v2.pdf
-
Supplementary file 2
IGV views of log2 ratio and regions of uracil enrichment on all the chromosomes.
- https://cdn.elifesciences.org/articles/60498/elife-60498-supp2-v2.pdf
-
Supplementary file 3
Genome-wide analysis of uracil-DNA pattern comparing to ChIP-seq data and DNA accessibility data using either GIGGLE search or the Segway genome segmentation tool.
Database information, applied command lines, full results of GIGGLE search (table 1), details of our own ChIP-seq (table 2), and list of files for Segway analysis (table 3) are provided.
- https://cdn.elifesciences.org/articles/60498/elife-60498-supp3-v2.pdf
-
Supplementary file 4
Genome-wide analysis of uracil-DNA pattern comparing to other genomic features using bedtools annotate.
Database information, applied command lines, full results (table 1), and calculation of replication timing scores and AT content on genomic segments (from the Segway analysis) are provided.
- https://cdn.elifesciences.org/articles/60498/elife-60498-supp4-v2.pdf
-
Supplementary file 5
Detailed comparison of U-DNA pattern to replication timing data (R script).
- https://cdn.elifesciences.org/articles/60498/elife-60498-supp5-v2.pdf
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/60498/elife-60498-transrepform-v2.pdf
-
Appendix 1—figure 1—source data 1
Comparison of histograms for the U-DNA signal distributions between dU-seq and U-DNA-Seq data.
- https://cdn.elifesciences.org/articles/60498/elife-60498-app1-fig1-data1-v2.xlsx
-
Appendix 1—figure 2—source data 1
Comparison of dU-seq and U-DNA-Seq data regarding correlation between U-DNA patterns and replication timing.
- https://cdn.elifesciences.org/articles/60498/elife-60498-app1-fig2-data1-v2.xlsx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genome-wide alterations of uracil distribution patterns in human DNA upon chemotherapeutic treatments
eLife 9:e60498.
https://doi.org/10.7554/eLife.60498




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}