Targeting a cell surface vitamin D receptor on tumor-associated macrophages in triple-negative breast cancer
- Rutgers Cancer Institute of New Jersey, United States
- Division of Cancer Biology, Department of Radiation Oncology, Rutgers New Jersey Medical School, United States
- Phage Therapy Group, Department of Brain Sciences, Imperial College London, United Kingdom
- The University of Texas M.D. Anderson Cancer Center, United States
- Institute of Metabolism and Cell Death, Helmholtz Zentrum Muenchen, Germany
- Department of Cellular and Molecular Medicine, The University of Arizona Cancer Center, University of Arizona, United States
- Department of Otolaryngology-Head and Neck Surgery, The University of Arizona Cancer Center, University of Arizona, United States
- Division of Cancer Imaging Research, The Russell H Morgan Department of Radiology and Radiological Science, The Johns Hopkins University School of Medicine, United States
- Department of Parasitology, Institute of Biomedical Sciences, University of São Paulo, Brazil
- Division of Hematology/Oncology, Department of Medicine, Rutgers New Jersey Medical School, United States
- Laboratory of Computational Biology, A.C. Camargo Cancer Center, Brazil
- Laboratory of Medical Genomics, A.C. Camargo Cancer Center, Brazil
- Translational Breast Cancer Program, Olivia Newton-John Cancer Research Institute, Australia
- Mathematics in Medicine Program, The Houston Methodist Research Institute, United States
- Department of Oncology and Hematology, University Medical Center Hamburg-Eppendorf, Germany
- Department of Oncology and Hematology, University Medical Center Augsburg, Germany
- Department of Neurology, Harvard Medical School, United States
- Department of Biomedical Engineering, College of Engineering, Wayne State University, United States
- Department of Oncology, School of Medicine, Wayne State University, United States
- Department of Neurosurgery, School of Medicine, Wayne State University, United States
- Robert H Lurie Comprehensive Cancer Center, Feinberg School of Medicine, Northwestern University Chicago, United States
- Department of Breast Medical Oncology, The University of Texas M.D. Anderson Cancer Center, United States
- Research Collaboratory for Structural Bioinformatics Protein Data Bank, San Diego Supercomputer Center, University of California-San Diego, United States
- Research Collaboratory for Structural Bioinformatics Protein Data Bank, Institute for Quantitative Biomedicine, Rutgers, The State University of New Jersey, United States
Figures
Figure 1 with 4 supplements
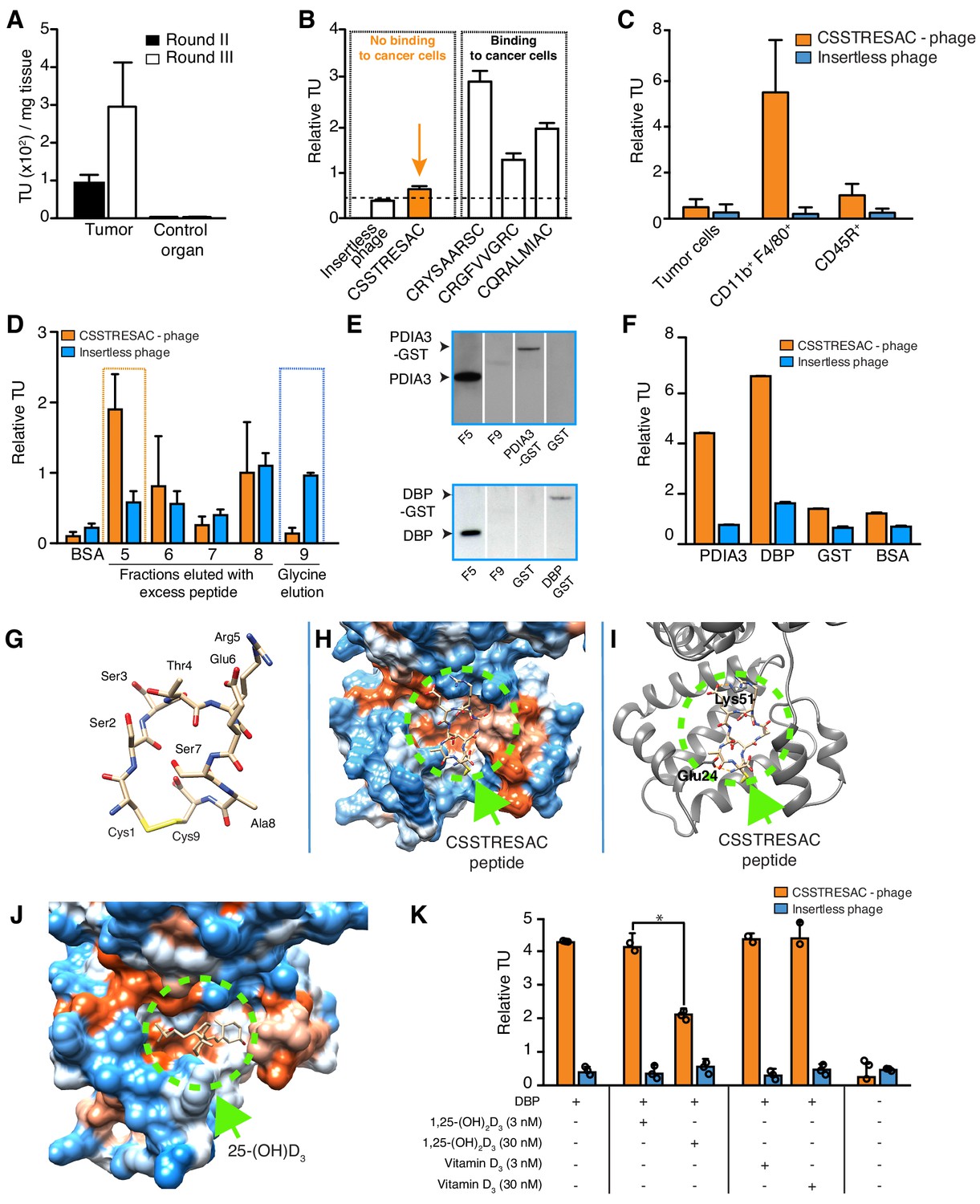
Combinatorial targeting of the tumor cellular microenvironment in a mouse model of TNBC.
(A) A random phage display peptide library displaying CX7C inserts (C, cysteine; X any seven residues) was used in vivo to select peptides homing to the microenvironment of EF43.fgf4-derived mammary tumors. Three sequential rounds of selection resulted in a pool of targeted phage particles with a 300-fold enrichment in the tumor, compared to a control organ (muscle). (B) Binding of individual phage clones to EF43.fgf4 cells was quantified by the counting of transducing units (TU) after host bacterial infection. (C) Binding of CSSTRESAC-phage to EF43.fgf4 tumor cells and non-malignant stromal cell subpopulations isolated from mCherry-expressing EF43.fgf4-derived mammary tumors. (D) Relative binding of the CSSTRESAC-phage or insertless control phage to fractions eluted from a CSSTRESAC-conjugated affinity purification column. BSA was used as negative control protein. (E) Immunoblottings developed with either anti-PDIA3 (top panel) or anti-DBP (lower panel) antibodies show the presence of both affinity-purified proteins in the experimental fraction F#5 but not in the negative control fraction F#9. Human recombinant PDIA3-GST and DBP-GST were used as control for antibody specificity. (F) Phage-binding assay confirms preferential binding of targeted CSSTRESAC-phage to the recombinant human DBP. GST and BSA were used as negative controls. (G) Predicted structure of CSSTRESAC peptide, including a 2.0 Å-disulfide bridge between Cys1 and Cys9, as visualized with UCSF Chimera. (H) Predicted binding conformation and orientation of CSSTRESAC relative to the crystal structure of DBP in a hydrophobicity surface view (PDB ID: 1KW2_A). Orange and blue represent hydrophobic and hydrophilic patches, respectively. (I) Key predicted non-hydrophobic interactions between CSSTRESAC and DBP (PDB ID: 1KW2_A), including a 2.9 Å-salt bridge between Cys1 and Glu24, a 2.9 Å-salt bridge between Glu6 and Lys51, and a 2.9 Å-hydrogen bond between Ala8 and Glu24. CSSTRESAC also blocks access to Tyr48 and Ser92 (Tyr32 and Ser76 in PDB ID: 1J78), which correspond to predicted key residues of DBP interaction with 1,25-(OH)2D3. (J) Crystal structure of 25-(OH)D3 bound to DBP in a hydrophobicity surface view (PDB ID: 1J78). Orange and blue represent hydrophobic and hydrophilic patches, respectively. (K) Binding of CSSTRESAC-phage to DBP is inhibited by the active form of vitamin D [1,25-(OH)2D3], but not by its corresponding vitamin D3 precursor (* represents Student’s t-test, p<0.05).
Figure 1—figure supplement 1
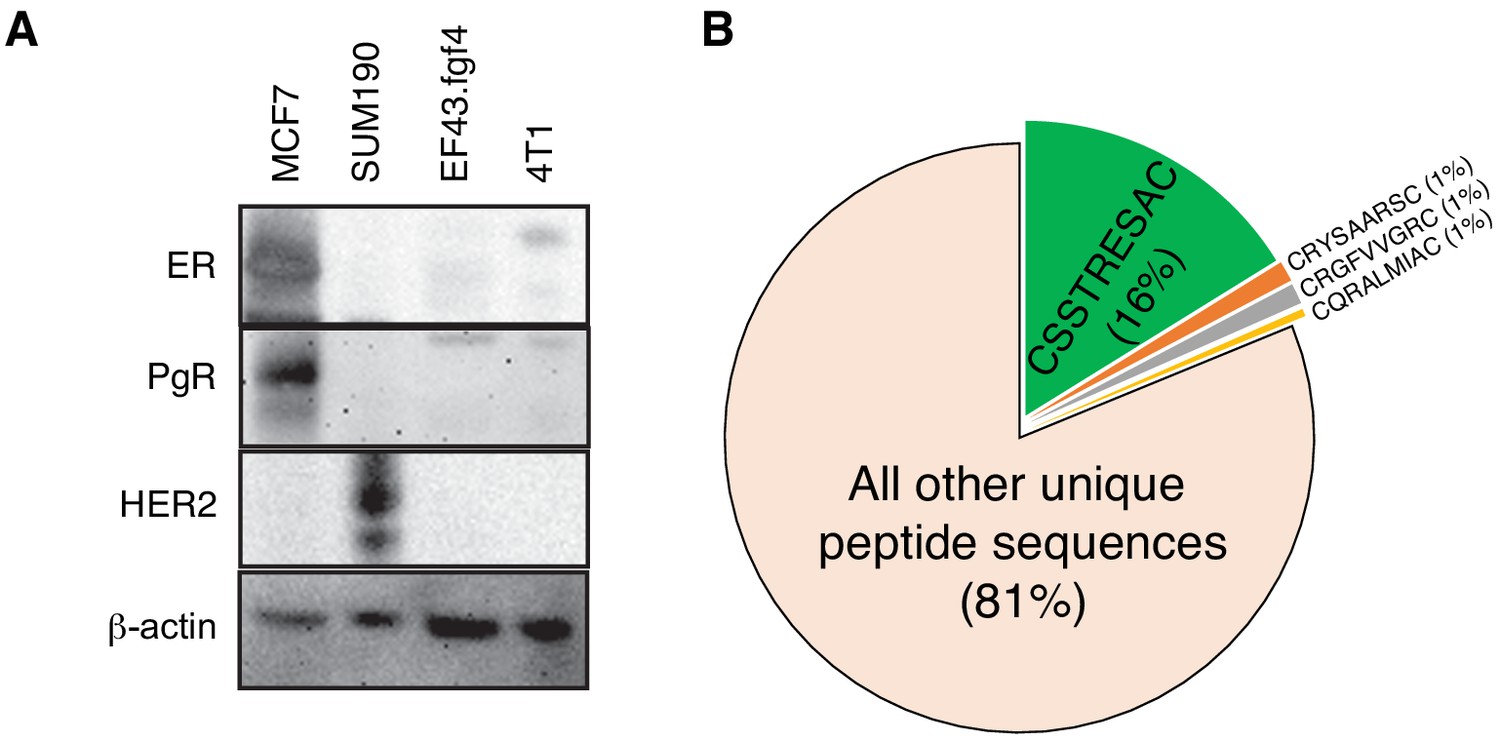
EF43.fgf4-derived tumor is a model of triple negative mammary cancer.
(A) Immunoblotting analysis of estrogen receptor (ER), progesterone receptor (PR) and HER-2/Neu in several breast cancer cell lines confirming that EF43.fgf4 cancer cells are a triple-negative mammary cancer model. (B) Quantitative analysis of peptide sequences obtained from the third round of in vivo phage display library screening in EF43.fgf4-tumor bearing mice.
Figure 1—figure supplement 2
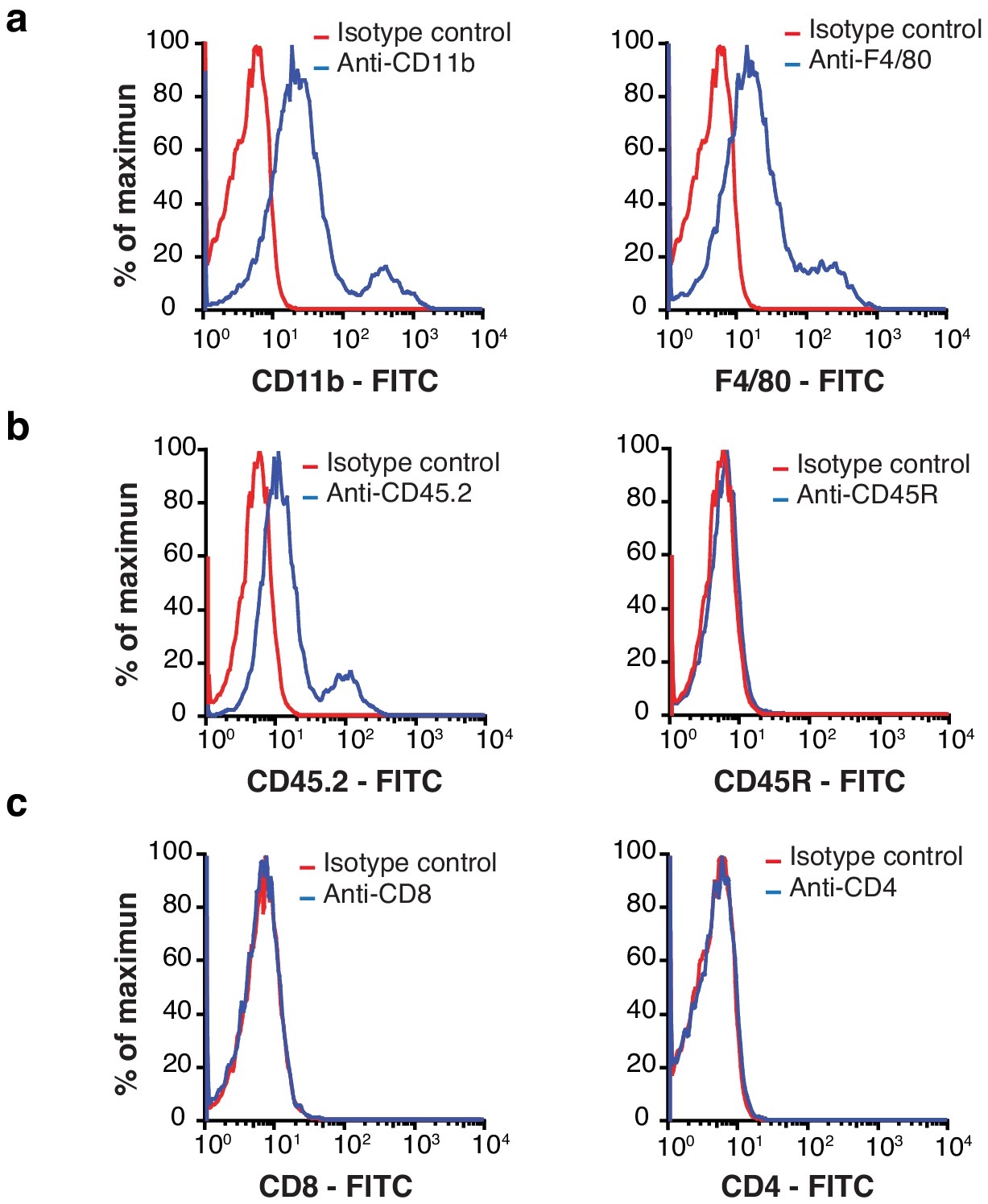
Macrophages are a major component of EF43.fgf4 mammary tumors.
(A – C) Flow cytometry analysis of total cells isolated from mCherry-expressing EF43.fgf4 mammary tumors. A major component of infiltrating non-malignant cells expresses the macrophage markers CD11b and F4/80 (A). (B) B-lymphocytes expressing the common leukocyte antigen CD45.2, and the B-cell lineage marker CD45R were also found, although in small quantities. (C) T-lymphocytes, as identified by the T-cell markers CD8 and CD4 were also tested but not detected.
Figure 1—figure supplement 3
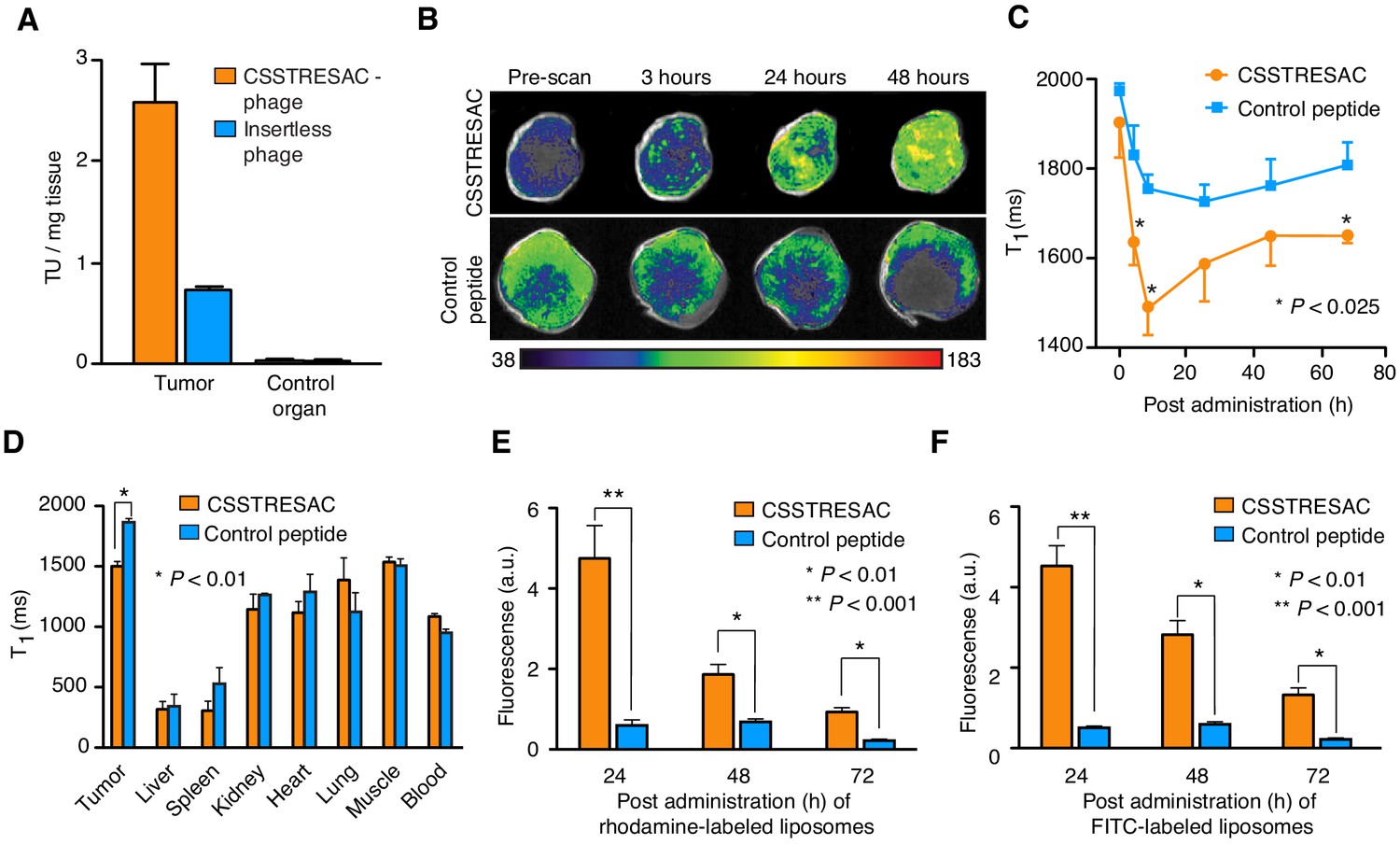
The CSSTRESAC peptide targets breast cancer in various mouse models.
(A) The MMTV-PyMT mouse model of breast cancer was used to confirm targeting of the CSSTRESAC-phage in vivo. Skeletal muscle was used as a negative control tissue. (B) CSSTRESAC-targeted liposomes loaded with gadolinium (n = 13) or control liposomes (n = 8) were administered iv to MDA-MB-231-bearing mice and MRI measurements (T1-weighted) were acquired at several time points post-administration. An increased contrast enhancement in breast tumor xenografts receiving CSSTRESAC-targeted liposomes was clearly detected after 3 hr of systemic circulation and persisted through 24 and 48 hr (B-C) compared to control liposomes. (C) Quantitative T1 maps showing significantly shorter T1 relaxation times (t-test, p<0.025) for the CSSTRESAC-targeted liposomes at 3, 6, and 72 hr post-administration. (D) Biodistribution of liposomes at 48 hr was examined by measuring the T1 values of excised tumors, control organs, and blood. This ex vivo analysis, along with MRI measurements, confirmed that CSSTRESAC-targeted liposomes preferentially accumulated in MDA-MB-231 breast tumor xenografts, compared to control liposomes. No differences were detected in several control tissues and organs. (E, F) Localization of CSSTRESAC-targeted liposomes in MDA-MB-231 breast tumor xenografts was confirmed by double-fluorescent liposomes in which the ligand CSSTRESAC was labeled with FITC (green), and lipids composing the liposomes were labeled with rhodamine (red). Fluorescence was quantified ex vivo in a Xenogen imaging system with tumor tissue collected at 24, 48, and 72 hr post-iv administration of targeted and control double-fluorescent liposomes. Fluorescence quantification revealed a fivefold higher accumulation of CSSTRESAC-targeted liposomes in tumors as fast as 24 hr post-administration relative to control liposomes. The differential accumulation of CSSTRESAC-targeted liposomes in MDA-MB-231 breast tumor xenografts became less pronounced, yet still statistically significant (t-test, p<0.01), at 48 and 72 hr.
Figure 1—figure supplement 4
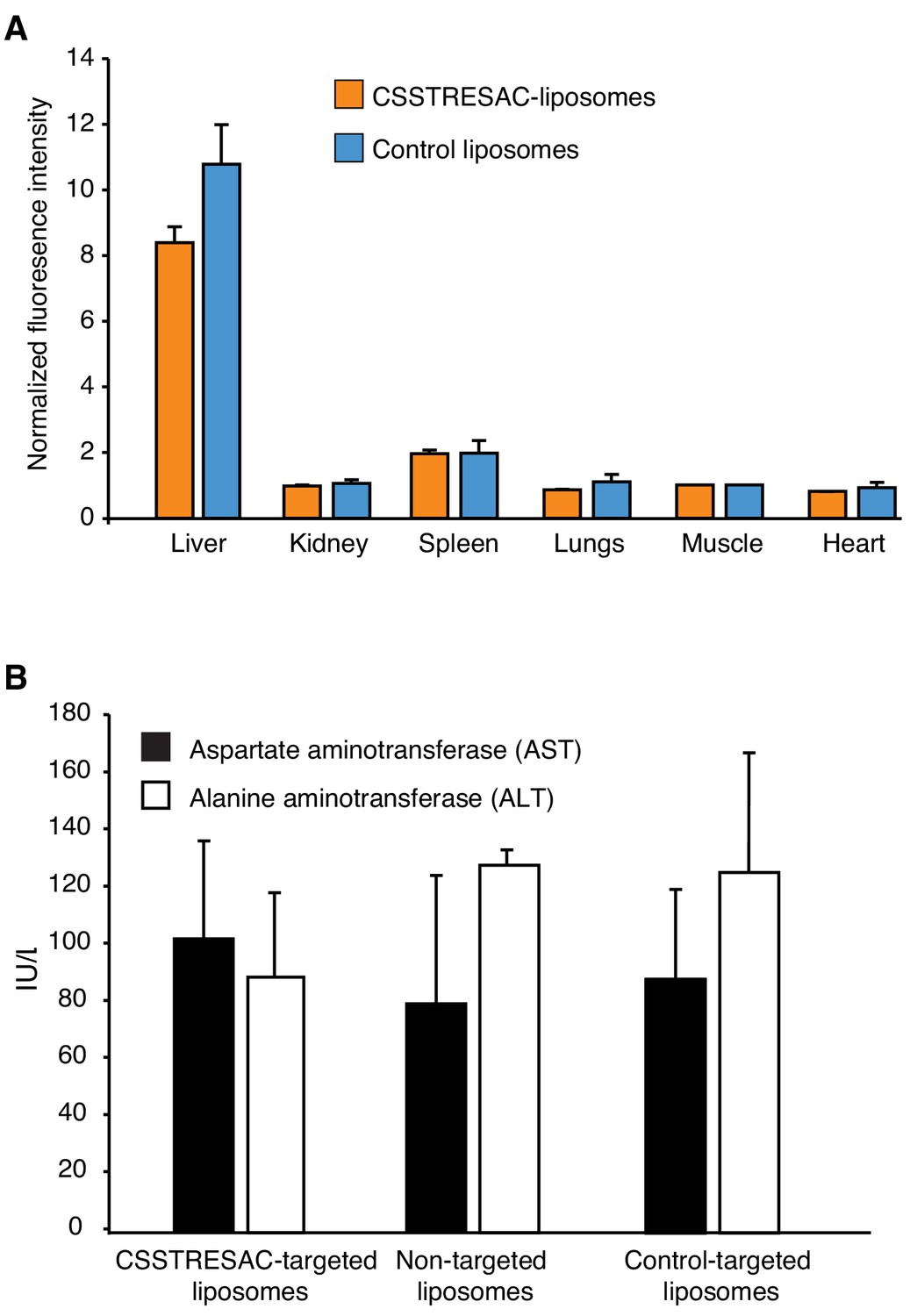
CSSTRESAC-targeted liposomes do not cause toxicity in mice.
(A) Fluorescence imaging experiments were performed on several control organs and normalized to a defined control tissue (skeletal muscle). Liposome uptake was low in all organs except the liver, a well-known biological phenomenon due to the relatively large size and cationic charge of liposomes (targeted or control), and their non-specific uptake by the reticuloendothelial system. (B) None of the liposome preparations caused liver toxicity as confirmed by levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) measured in serum of treated mice.
Figure 2 with 1 supplement
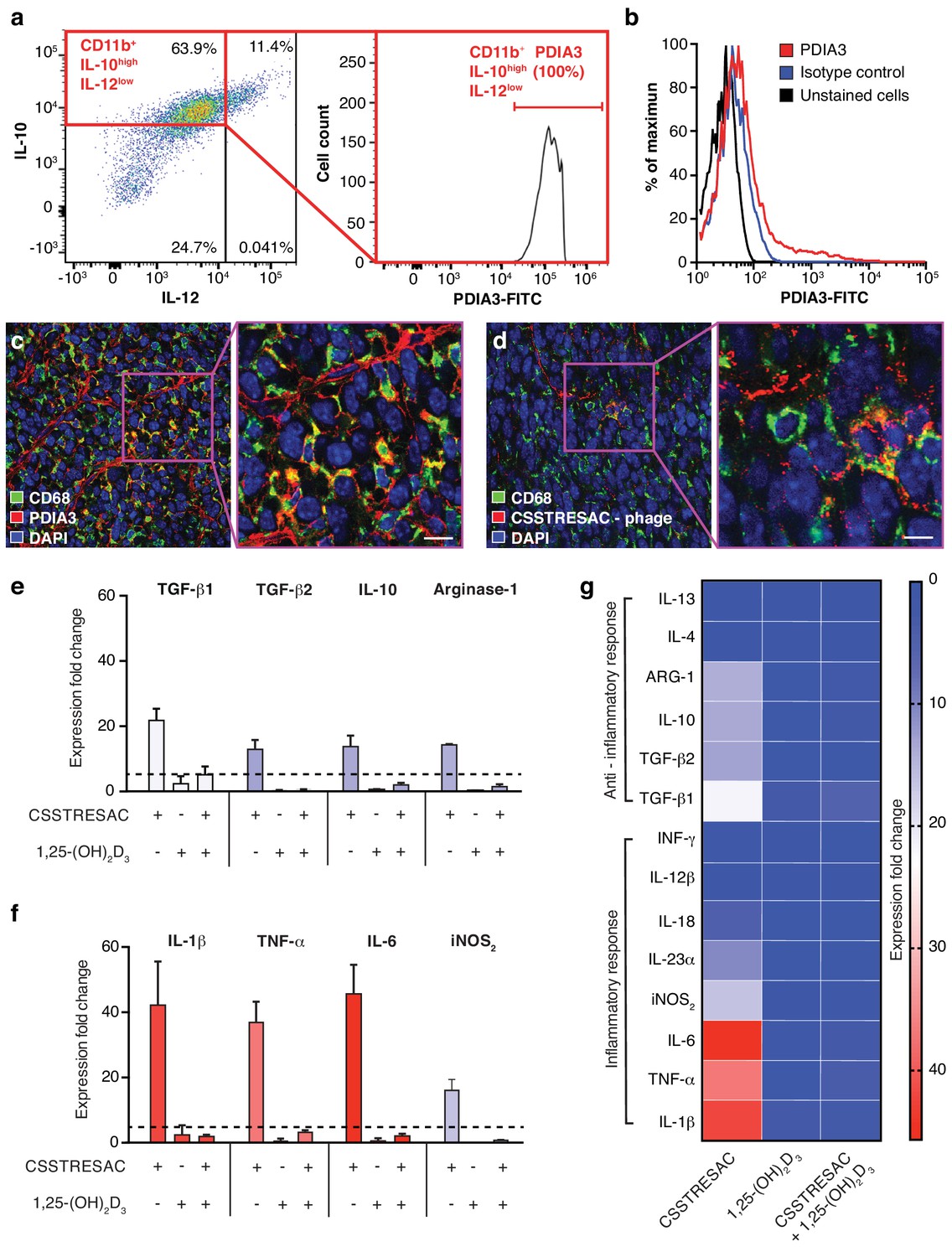
PDIA3 is present on the surface of TAM.
(A) FACS analysis of total TAM isolated from EF43.fgf4-derived mammary tumors shows high levels of PDIA3 expression in a subpopulation of F4/80+CD11b+IL10highIL12low TAM. (B) EF43.fgf4 cells do not express detectable levels of PDIA3 on their surface. (C-D) PDIA3 expression in TAM and co-localization with the pan-macrophage marker CD68 as detected by immunofluorescence of tumor tissue sections from tumor-bearing mice administered iv with anti-PDIA3 antibody (C) or CSSTRESAC-phage (D). (E-G) Purified TAM from EF43.fgf4 mammary tumors were established in culture and treated with either the soluble CSSTRESAC peptide, 1,25-(OH)2D3, or both. Controls included untreated cells, and cells treated with vehicle. Expression of anti-inflammatory (E and G) or pro-inflammatory (F and G) cytokines in CD11b+F4/80+ TAM was assessed by quantitative real-time PCR. Graphics represent expression fold-change relative to control cells.
Figure 2—figure supplement 1

PDIA3 is accessible through the systemic circulation.
(A) An anti-PDIA3 antibody was administered iv into EF43.fgf4 tumor-bearing mice and was allowed to circulate for 5 min in deeply anesthetized mice before whole body perfusion through the heart. The anti-PDIA3 antibody preferentially targeted the tumor, indicating that PDIA3 is systemically accessible. (B) The F4/80 pan-macrophage marker was used as a positive control for TAM identification. (C) TAM isolated from EF43.fgf4 mammary tumors were established in culture and treated with the soluble CSSTRESAC peptide, 1,25-(OH)2D3, or both. Expression of cytokine genes was evaluated by quantitative real-time PCR. Graphics represent expression fold change relative to untreated cells.
Figure 3 with 2 supplements
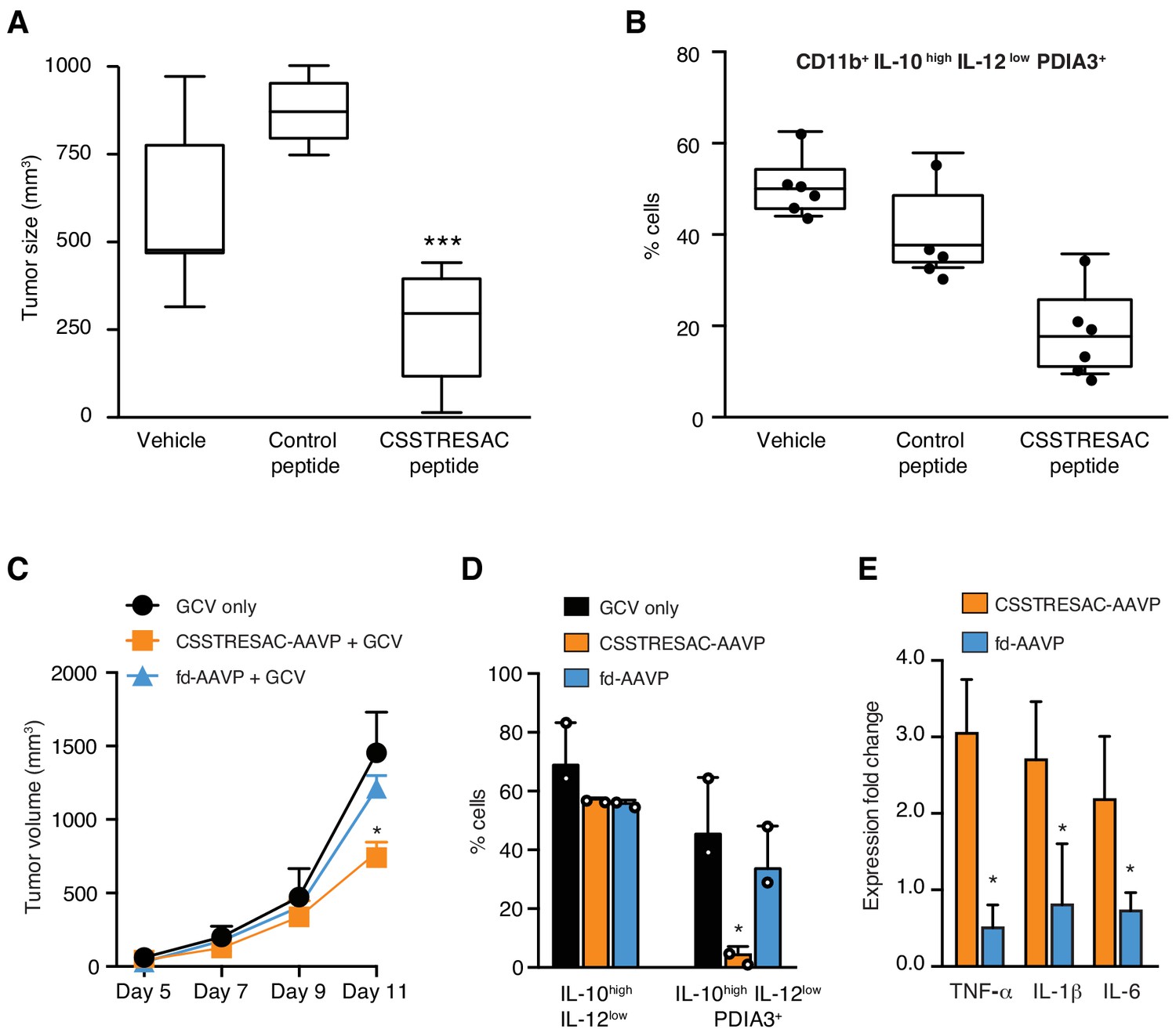
Targeted therapy delays growth of EF43.fgf4-derived mammary tumors.
(A) Therapeutic effect of systemic treatment of EF43.fgf4 tumor-bearing mice with soluble CSSTRESAC peptide (n = 10 each experimental cohort, details in Materials and methods). An unrelated control peptide and vehicle served as negative controls. Tumor sizes were measured by digital caliper 1 week after treatment initiation, and every other day afterwards. *** represents p<0.001. (B) Treatment of tumor-bearing mice with CSSTRESAC reduces the number of PDIA3-expressing TAM (F4/80+CD11b+IL-10highIL-12lowPDIA3+). The TAM population is represented as percentage of total non-malignant cells, as determined by flow cytometry. (C) Gene therapy with CSSTRESAC-AAVP-HSVtk plus GCV delays tumor growth. Mice cohorts with size-matched EF43.fgf4 mammary tumors received a single systemic iv administration of targeted CSSTRESAC-AAVP-HSVtk (5 × 1010 TU) or control fd-AAVP-HSVtk. Mice received daily doses of GCV (80 mg/kg/day) starting at day 7 post AAVP-HSVtk administration until the end of the experiment. * represents p<0.05. (D) Flow cytometry confirms reduction of F4/80+CD11b+IL-10highIL-12lowPDIA3+ TAM in tumors from CSSTRESAC-AAVP-HSVtk-treated mice. (E) Cytokine production by macrophages from tumors of mice treated with CSSTRESAC-AAVP-HSVtk or control groups. * represents p<0.05. Results are reported as expression fold-change relative to control group (set to 1).
Figure 3—figure supplement 1

CSSTRESAC peptide targets macrophage in vivo.
(A) FACS analysis confirming reduction of F4/80+CD11b+IL-10highIL-12lowPDIA3+ TAM in the CSSTRESAC-treated cohort, compared to a negative control cohort (n = 8 tumor-bearing mice per cohort). (B) CD163 expression in tumor tissue sections showing reduced number of monocytes and macrophages in mice treated with soluble CSSTRESAC compared to a control peptide.
Figure 3—figure supplement 2
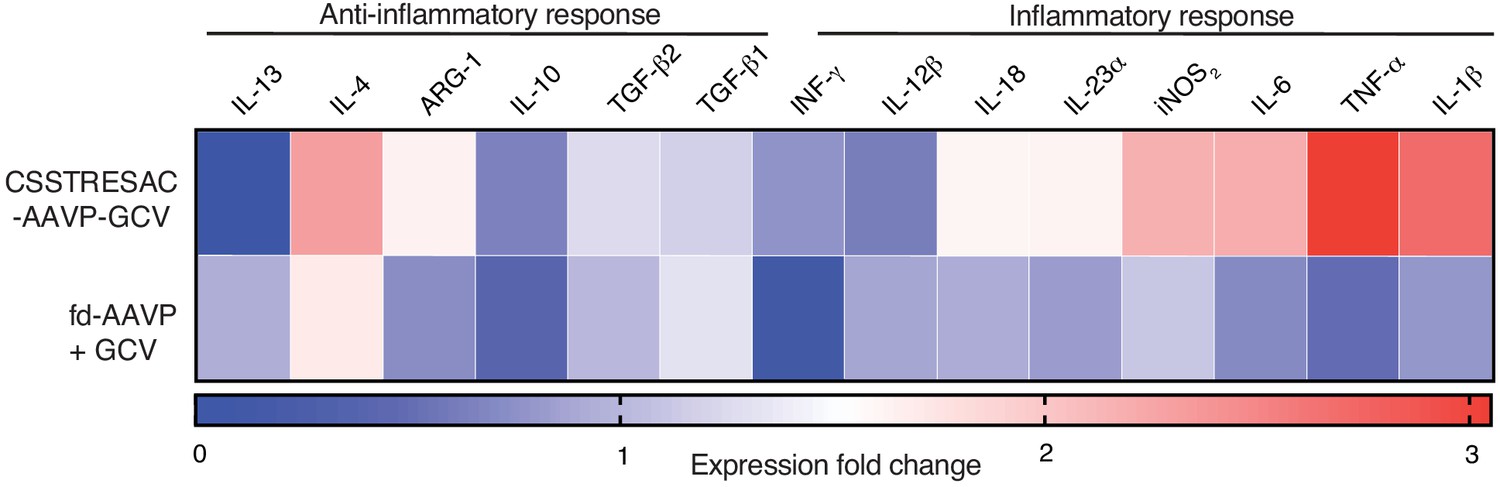
Heat-map representing a more extensive cytokine profile of F4/80+CD11b+IL-10highIL-12lowPDIA3+ TAM isolated from tumors of treated and control groups.
Figure 4 with 1 supplement

Mechanistic mathematical model of tumor growth inhibition upon treatment with soluble CSSTRESAC and competitive antagonism by 1,25-(OH)2D3.
(A) System interactions captured by a mechanistic mathematical model. Upper panel shows the non-linear regression of the tumor growth model upon treatment of tumor-bearing mice with soluble CSSTRESAC. Error bar represents mean ± standard deviation (S.D.) of the data shown in Figure 3A. Lower panel shows the projected temporal evolution of the tumor volume without infusion (μ = 0) and with infusion (μ = 0.75) in a simulated human clinical trial. (B) A schematic representation of the working hypothesis. The complex CSSTRESAC-DBP binds PDIA3 and eliminates PDIA3-expressing TAM from the tumor microenvironment (through an unknown mechanism), resulting in a pro-inflammatory local response and inhibition of tumor growth. Because 1,25-(OH)2D3 may compete out the effects of CSSTRESAC, binding to PDIA3-expressing TAM in the presence of 1,25-(OH)2D3 may be abrogated, and tumor cells can continue to grow.
Figure 4—figure supplement 1
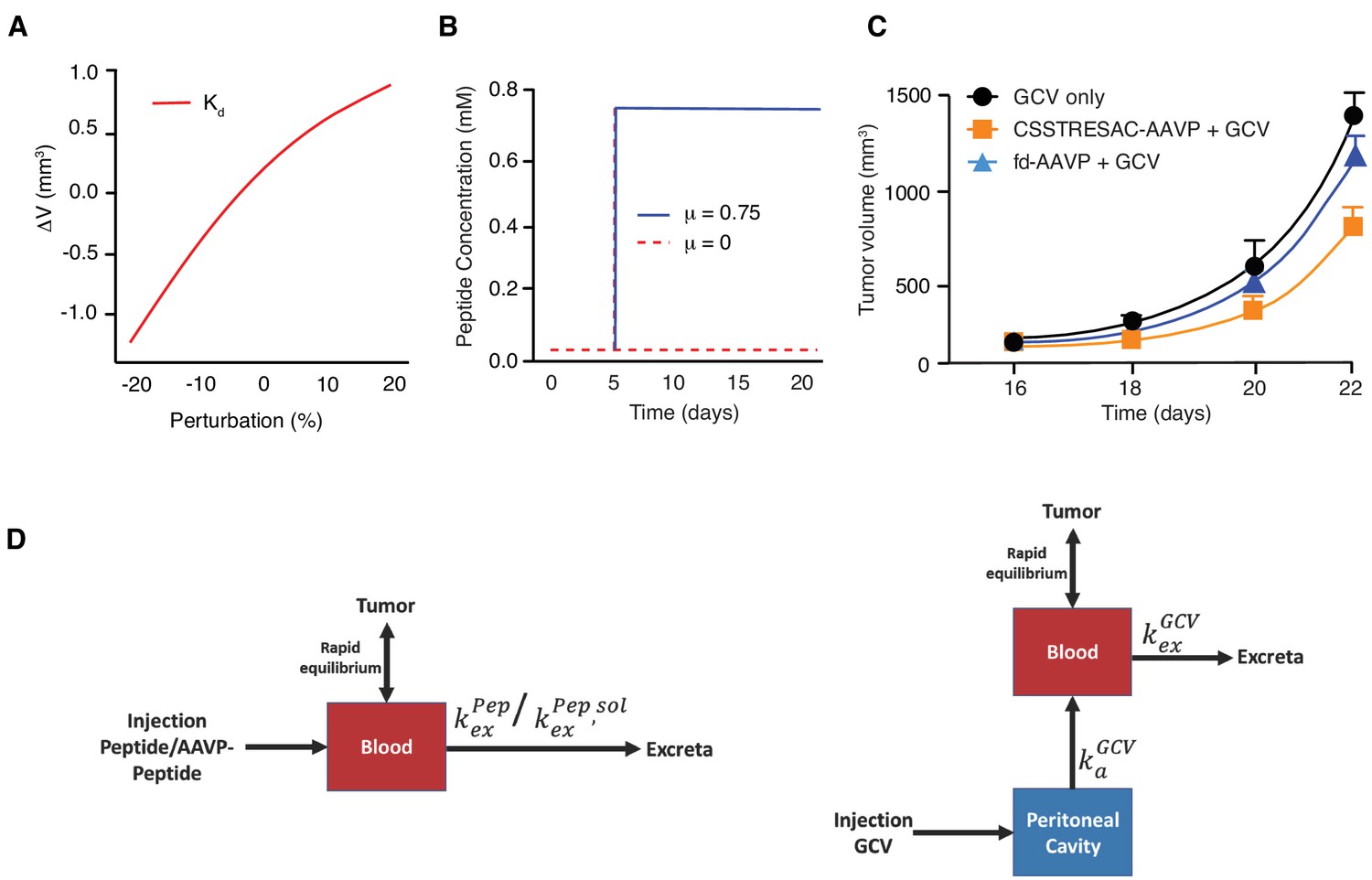
Mathematical modeling of CSSTRESAC peptide distribution in tumor-bearing mice.
(A) Change in tumor volume with respect to the reference volume versus the dissociation constant Kd. (B) Peptide concentration kinetics without infusion (μ = 0) and with infusion (μ = 0.75). (C) Numerical results of tumor growth model corresponding to Figure 3c. Error bars represent mean ±S.D. of the data shown in Figure 3c. (D) Schematic of one-compartment pharmacokinetic model following (left panel) iv administration of peptide or AAVP-peptide, and (right panel) ip administration of GCV.
Figure 5
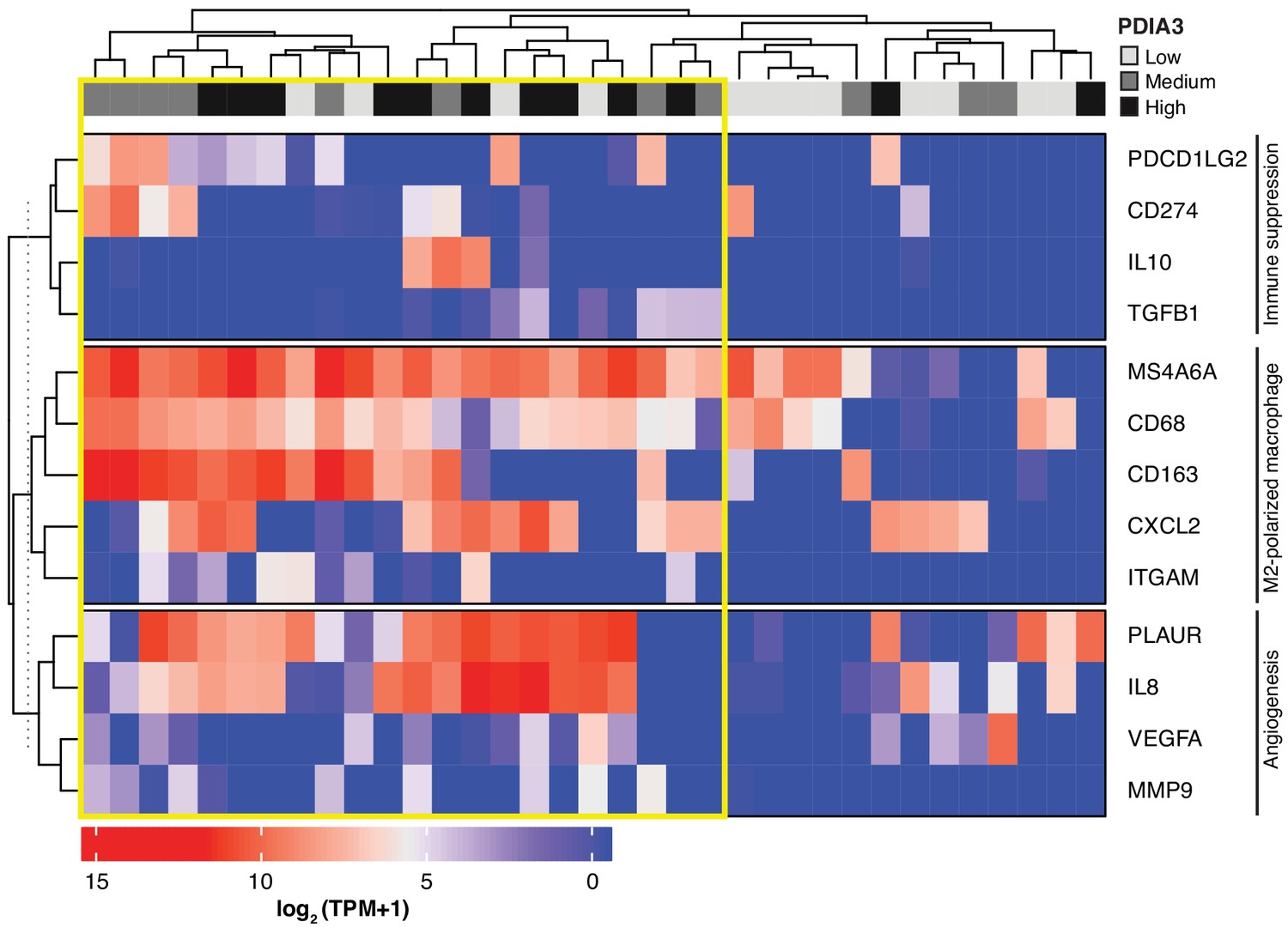
Heat-map of PDIA3 gene expression in pre-defined myeloid cells from human TNBC.
The heat map shows a strong association with the expression of genes characteristic of M2-polarized macrophage, markers of immunosuppression and angiogenesis (i.e. poor prognosis). The yellow box highlights cells with the highest expression of PDIA3.
Additional files
-
Supplementary file 1
MS/MS analysis of CSSTRESAC-binding proteins.
- https://cdn.elifesciences.org/articles/65145/elife-65145-supp1-v1.xlsx
-
Supplementary file 2
Computer parameters of the mathematical model.
- https://cdn.elifesciences.org/articles/65145/elife-65145-supp2-v1.docx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/65145/elife-65145-transrepform-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Targeting a cell surface vitamin D receptor on tumor-associated macrophages in triple-negative breast cancer
eLife 10:e65145.
https://doi.org/10.7554/eLife.65145




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}