Biological controls for standardization and interpretation of adaptive immune receptor repertoire profiling
- University Children’s Hospital and the Children’s Research Center, University of Zurich, Switzerland
- CRTD Center for Regenerative Therapies Dresden, Faculty of Medicine, Technische Universität Dresden, Germany
- Sorbonne Université U959, Immunology-Immunopathology-Immunotherapy (i3), France
- AP-HP Hôpital Pitié-Salpêtrière, Biotherapy (CIC-BTi), France
- Lowance Center for Human Immunology, Emory University School of Medicine, United States
- Perelman School of Medicine, University of Pennsylvania, United States
- University of Genoa, Department of Experimental Medicine, Italy
- The Vanderbilt Vaccine Center, Vanderbilt University Medical Center, United States
- Department of Pediatrics, Vanderbilt University Medical Center, United States
- College of Law, University of Illinois, United States
- Center for Advanced Studies in Biomedical Innovation Law, University of Copenhagen Faculty of Law, Denmark
- Carl R. Woese Institute for Genomic Biology, University of Illinois, United States
- IMGT, The International ImMunoGeneTics Information System (IMGT), Laboratoire d'ImmunoGénétique Moléculaire (LIGM), Institut de Génétique Humaine (IGH), CNRS, University of Montpellier, France
- Laboratoire d'ImmunoGénétique Moléculaire (LIGM) CNRS, University of Montpellier, France
- Institut de Génétique Humaine (IGH), CNRS, University of Montpellier, France
- Takara Bio USA, Inc., United States
Figures
Figure 1 with 1 supplement
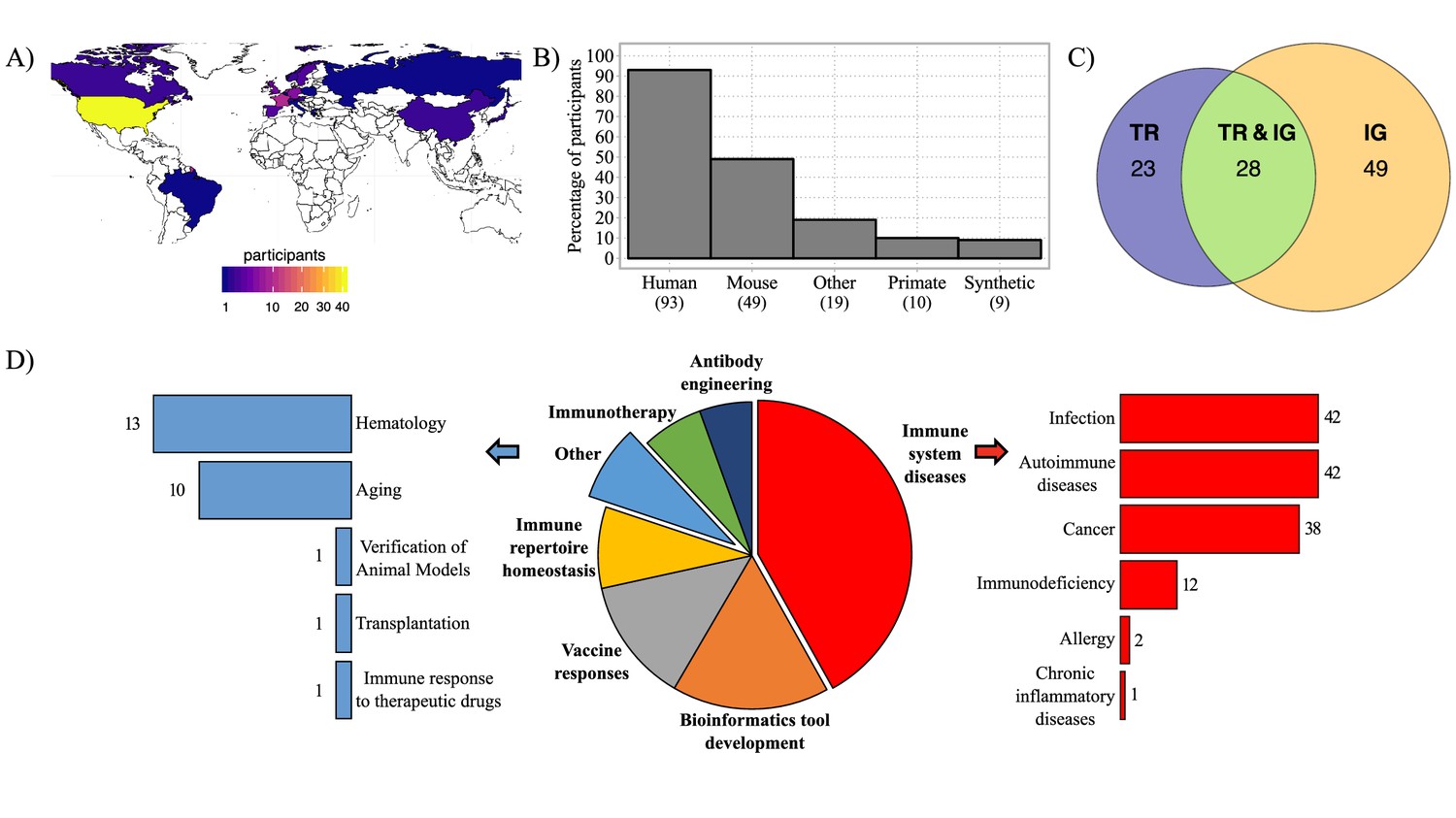
Geographic distribution of survey participants and their AIRR-seq research interests.
(A) Map with geographic distribution of survey participants. (B) Histogram showing the principal studied organisms among the participants. The ‘Other’ category includes rat, ferret, rabbit, goat, pig, canine, bovis, cattle, chicken, fish, teleost, salmon, zebrafish, other fish species, transgenic animals. (C) Venn diagram representing the percentage of participants according to their interest in AIRR template type. (D) Pie-chart representing the distribution of survey participants according to their research interest(s). Immune system diseases and other categories are described in more detail in the bar plots (right and left). Numbers of respondents for each category are shown next to the bars.
Figure 1—figure supplement 1
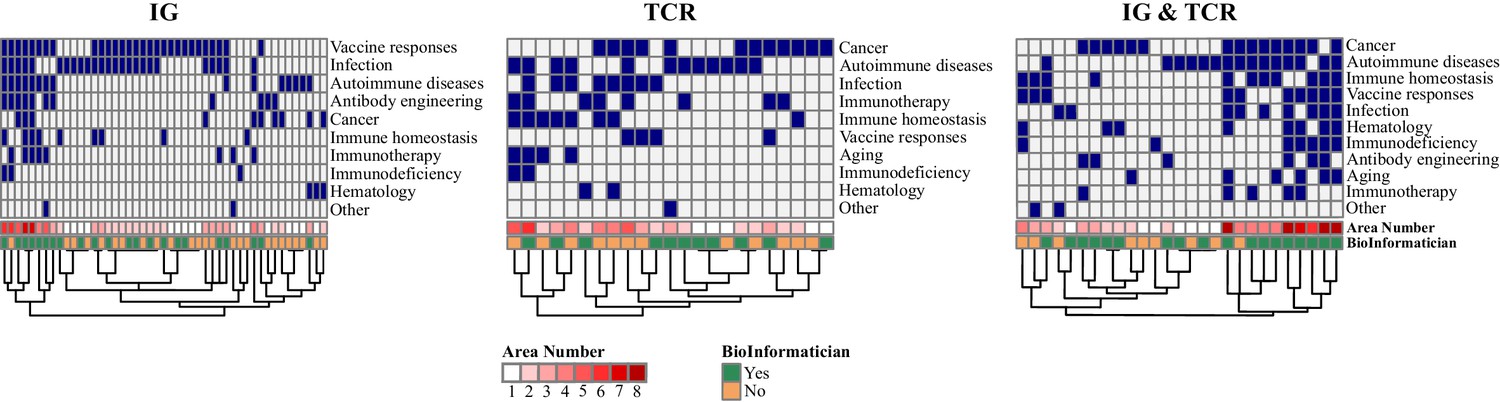
Heatmaps of the areas of study depending on the interest.
Each column is a participant group and the colors represent the area studied (blue: area studied, white: area not studied). For each heatmap, areas are ordered by decreasing frequency depending on the number of participants in each group. Information about the number of areas per participant and bioinformatic skills are added below (Chi2: p=0.0064).
Figure 2 with 1 supplement
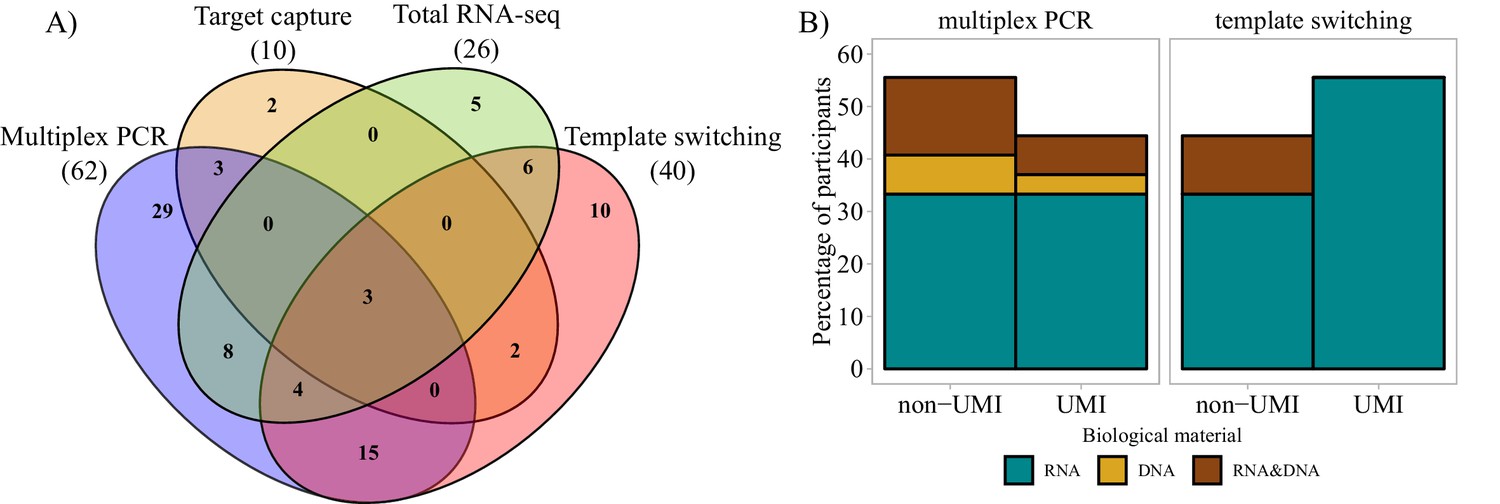
Molecular approaches used in bulk sequencing.
(A) Venn diagram representing the most important molecular approaches used and their usage and sharing among participants. Numbers of respondents in each of the four main categories are shown in parentheses. (B) Bar plots representing biological material used and molecular barcoding proportion for the two major molecular biology approaches (multiplex PCR and template switching). Only the answers of respondents who used one technology exclusively are shown (Multiplex PCR: n = 29; Template switching: n = 10). UMI = unique molecular identifier.
Figure 2—figure supplement 1
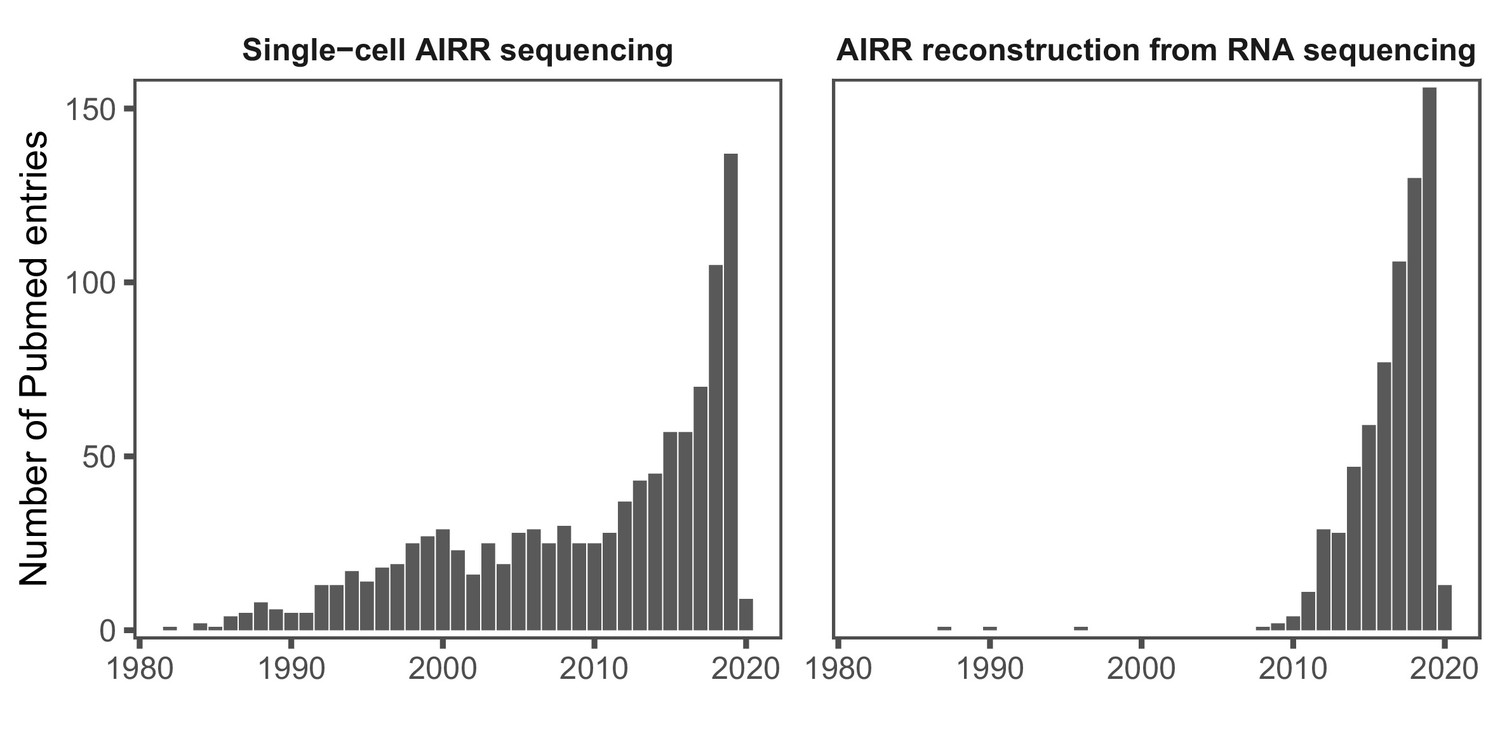
Yearly number of PubMed entries referring to single-cell AIRR sequencing (left panel) and bioinformatic AIRR reconstruction from RNA-seq studies (right panel) 1980–2020 (via https://www.ncbi.nlm.nih.gov/pubmed/, accessed on 16 January 2020).
Search query (left panel): (‘single cell’[Title/Abstract] OR ‘10X’[Title/Abstract]) AND (‘VDJ’[Title/Abstract] OR ‘TCR’[Title/Abstract] OR ‘BCR’[Title/Abstract] OR ‘b cell receptor’[Title/Abstract] OR ‘t cell receptor’[Title/Abstract] OR ‘repertoire’[Title/Abstract]) Search query (right panel): (‘rna-seq’[Title/Abstract] OR ‘rna sequencing’[Title/Abstract]) AND (‘VDJ’[Title/Abstract] OR ‘TCR’[Title/Abstract] OR ‘BCR’[Title/Abstract] OR ‘b cell receptor’[Title/Abstract] OR ‘t cell receptor’[Title/Abstract] OR ‘repertoire’[Title/Abstract]).
Figure 3
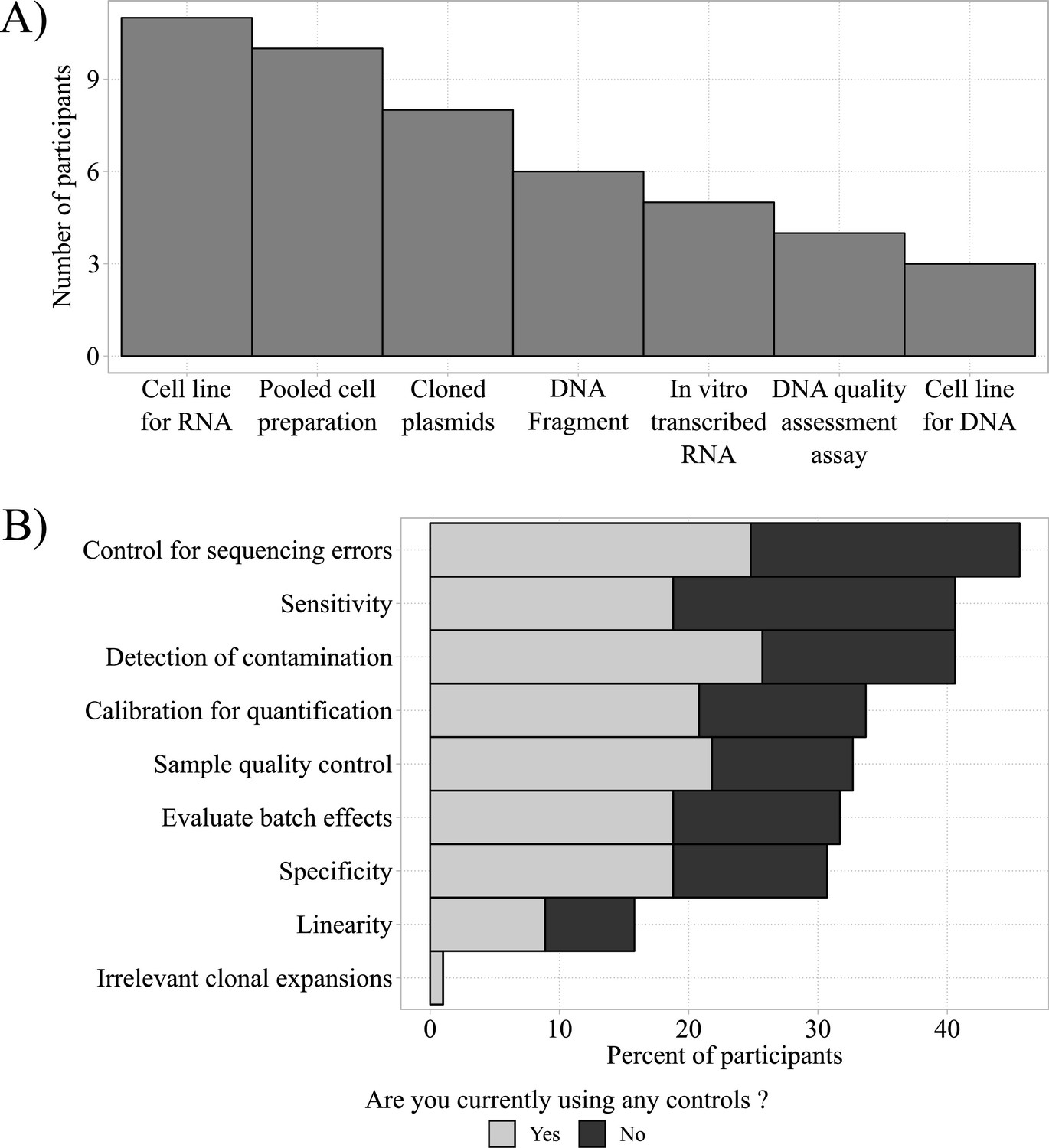
Homebrew controls and their desired applications.
(A) Most frequently used homebrew controls (total n = 47). (B) Total frequencies of desired applications of homebrew controls for respondents currently using (gray bars; total n = 47) and currently not using controls (black bars; total n = 42).
Tables
Table 1
Current AIRR-seq methods and their typical use(s).
Bulk gDNA, bulk cDNA, and single-cell cDNA-based sequencing methods are compared with respect to their general features, uses, methods, and potential issues. Each is ranked using a semi-quantitative scale (from ‘+++” for best to ‘-” for worst or non-existent).
| Bulk gDNA sequencing | Bulk cDNA sequencing | Single-cell cDNA sequencing | ||
|---|---|---|---|---|
| General Features | PCR method | Multiplex | Multiplex and 5' RACE | Multiplex and 5' RACE |
| Cell number | 102–106 | 102–106 | 102–103 | |
| Sample throughput | Low-high | Low-moderate | Low | |
| Length of receptor sequences | 100–600 bp | 150–600 bp | 700–800 bp | |
| Availability of commercial kits and service providers | ++ | +++ | + | |
| Uses | Gene usage | ++ | ++ | + |
| CDR3 length and properties | ++ | ++ | + | |
| Somatic hypermutation (for IG) | ++ | ++ | + | |
| Repertoire diversity | ++ | ++ | +/- | |
| Clonal expansion | +++ | ++ | + | |
| Clonal evolution | ++ | +++ | ++ | |
| Tracking of clonotypes | +++ | ++ | + | |
| Clinical use (e.g., MRD detection) | ++ | +/- | - | |
| Unbiased detection of unproductive rearrangements | ++ | - | - | |
| Inference of germline | ++ | + | +/- | |
| Determination of constant gene | - | ++ | + | |
| Structural annotation | +/- | ++ | + | |
| Linkage of both antigen receptor chains | +/- | +/- | ++ | |
| Direct combination of AIRR-seq with single-cell immunophenotype (e.g., transcriptome or cell surface protein expression) | - | - | ++ | |
| Characterization of clonotype full antigen receptor/Functional testing | - | +/- | ++ | |
| Rare clonotype detection | ++ | ++ | +/- | |
| Methods | Simplicity of workflow (library preparation) | +++ | ++ | + |
| Cost for library preparation commercial kits (per sample) | Low | Moderate | High | |
| Fidelity in sequences | Moderate | High | High | |
| Molecular barcoding (correcting PCR/sequencing error) | +/- | ++ | ++ | |
| Potential Issues | V-gene amplification bias | ++ | + | +/- |
| V-gene annotation issues | ++ | + | + | |
| PCR and sequencing error | ++ | + | +/- | |
| Difficulty with translation of copy number to cells | +/- | ++ | +/- | |
| Degradation of template | + | ++ | ++ |
-
bp = base pairs; CDR3 = complementarity determining region 3; MRD = minimal residual disease; RACE = rapid amplification of cDNA ends; V = variable.
Table 2
Concerns and expected errors introduced during AIRR-seq workflows and possible controls to detect them.
A typical workflow consists of 5 steps: Sample collection > Extraction > Amplification > Sequencing > Analysis.
| Concern | Mechanism(s) | Example of potential controls |
|---|---|---|
| Sequence errors | Enzyme errors (RT, DNA polymerase); Sequencing errors | UMIs for bioinformatic error correction; Spike-in controls with defined sequences to evaluate error rates |
| Sensitivity | Enzymatic inefficiencies (RT or PCR conditions/polymerase); Sample collection size (e.g., cell input number, purity); Sequencing depth | Spike-in controls (synthetic or cellular) at known concentrations |
| Specificity | Enzyme bias (RT, DNA polymerase); Analysis pipelines (annotation, error correction) | Spike-in controls with defined sequences to identify overall V/D/J gene amplification bias |
| Detection of contamination | Bench-level cross contamination (sample mixing or PCR contamination) or barcode jumping during sequencing | Unique spike-in (synthetic) for each sample; UDIs for sequencing barcode crosstalk |
| Sample quality control | Sample collection or nucleic acid purification | Identified by spectroscopy or agarose electrophoresis |
| Evaluate batch effects | Subtle differences introduced at all stages of the workflow | Spike-in controls (synthetic or cellular); Parallel biological (clonal or complex) sample |
| Linearity/accuracy of clonotype quantification | Enzymatic inefficiencies (RT or PCR conditions); Analytical error correction | Spike-in controls (synthetic or cellular) at known concentrations |
| Reproducibility/Batch effects | All stages | Spike-in controls (synthetic or cellular); Parallel biological (clonal or complex) sample; Comparison of replicate amplifications of the same sample; Comparison of sequences generated on the same sample in different sequencing runs |
| Data processing | Database/annotation limitations; filtering; error correction; collapsing/consensus algorithms | Spike-in controls (synthetic or cellular); Parallel biological (clonal or complex) sample |
-
RT, reverse transcriptase; UMIs, unique molecular identifiers; UDIs, unique dual indices.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Biological controls for standardization and interpretation of adaptive immune receptor repertoire profiling
eLife 10:e66274.
https://doi.org/10.7554/eLife.66274




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}