ZC3H4 restricts non-coding transcription in human cells
- The Living Systems Institute, University of Exeter, United Kingdom
- The Roslin Institute, Royal (Dick) School of Veterinary Studies, University of Edinburgh, United Kingdom
Figures
Figure 1 with 3 supplements

Proximity labelling of CPSF30-sensitive Pol II interactions by mTurbo.
(a) Schematic of the strategy used to tag CPSF30 with the mini auxin-inducible degron (mAID). Guide RNA-expressing Cas9 plasmid and homology-directed repair (HDR) plasmids are shown and the resulting modification to CPSF30 is represented with each inserted element labelled. (b) Western blot demonstrating CPSF30 depletion. Parental HCT116-TIR1, or CPSF30-mAID cells, were treated ±auxin for 3 hr, then blotted. CPSF30 protein is indicated together with a non-specific product, marked by an asterisk, used as a proxy for protein loading. (c) Metagene analysis of 1795 protein-coding genes demonstrating increased downstream transcription, derived from sequencing nuclear RNA, following auxin treatment (3 hr) of CPSF30-mAID cells. TSS = transcription start site, TES = transcription end site (PAS), read-through signal is normalised against gene body. RPKM is reads per kilobase of transcript, per million mapped reads. Positive and negative signals represent sense and antisense reads, respectively. (d) Schematic of our strategy to identify new factors involved in transcription termination. CPSF30-mAID cells were edited to express Rpb1-mTurbo (blue circle on Pol II). The addition of biotin induces mTurbo-mediated biotinylation (orange haze) of factors proximal to Pol II. CPSF complex is shown as an example of what might be captured by this experiment. (e) Western blot showing streptavidin horseradish peroxidase (HRP) probing of extracts from CPSF30-mAID: RPB1-mTurbo cells. Prior treatment with auxin (3 hr)/biotin (10 min) is indicated. The high molecular weight species in the +biotin samples corresponds in size to Rpb1-mTurbo (*). (f) Heat map detailing proteins with the largest decrease in Pol II interaction. Data underpinning heat map are from mass spectrometry analysis of streptavidin sequestered peptides (±CPSF30) performed in triplicate. Labelling was for 10 min.
Figure 1—figure supplement 1
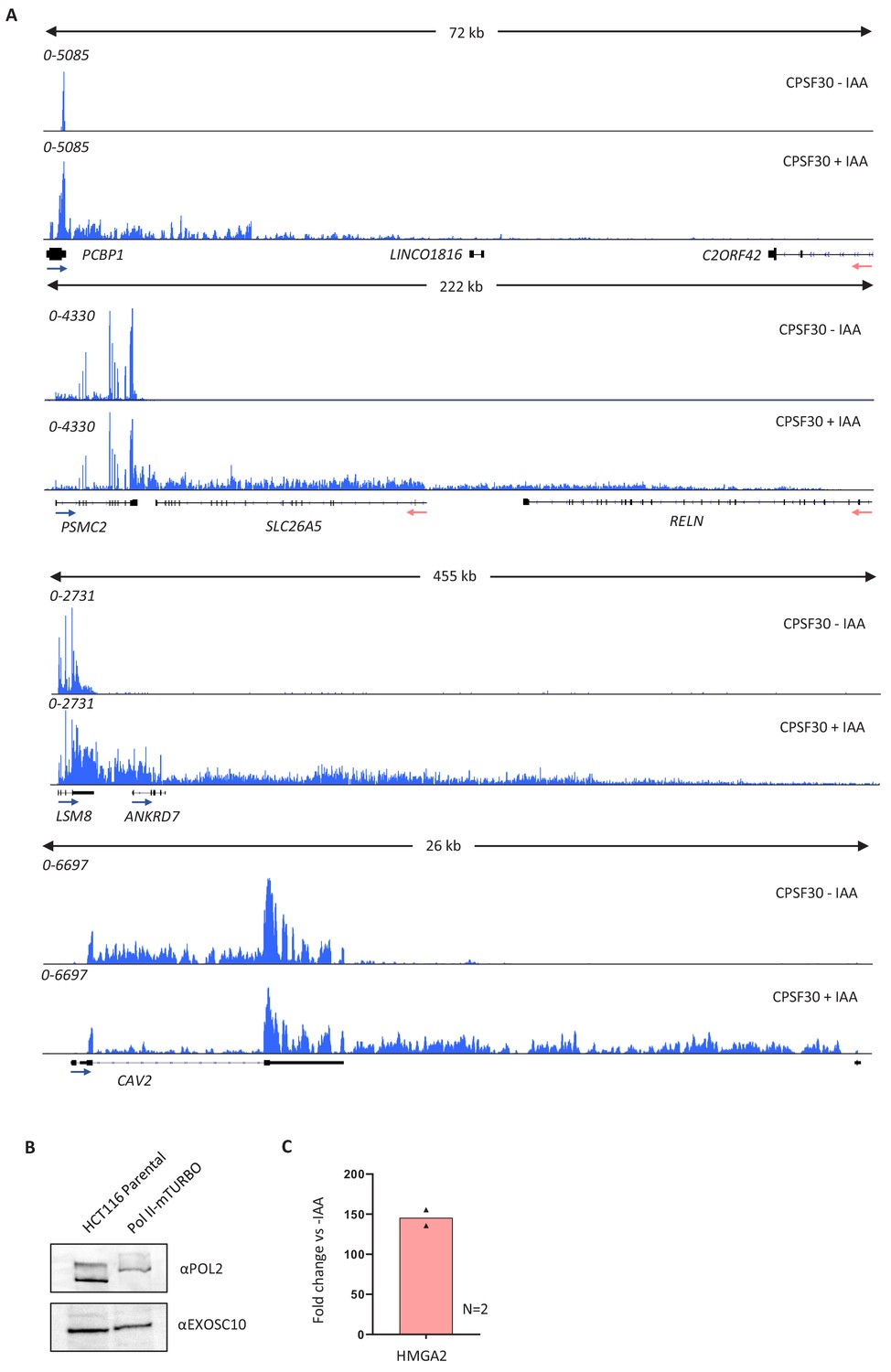
Validation of the CPSF30 transcriptional read-through defect and of tagging RPB1 with mTurbo.
(a) Integrated genomics viewer (IGV) track views of the transcription termination defect at PCBP1, PSMC2, LSM8, and CAV2 genes in the presence (CPSF30-IAA) or absence of (CPSF30+IAA) CPSF30 in CPSF30-mAID cells. Signal is RPKM. (b) Western blot demonstrating bi-allelic modification of RPB1 (Pol II) with mTurbo. The clone employed in Figure 1 is shown against parental HCT116 cells, unmodified at RBP1. The upshift of Pol II signal shows the bi-allelic modification of RPB1. EXOSC10 serves as a loading control. (c) Quantitative reverse transcription and PCR (qRT-PCR) of total RNA isolated from CPSF30-mAID: RPB1- mTurbo cells treated or not with auxin (3 hr). An amplicon located ~10 kb downstream of the HMGA2 PAS was used to assay transcriptional read-through presented as a fold change versus minus auxin after normalising to spliced actin mRNA. n = 2. Individual data points are shown. RPKM = reads per kilobase of transcript, per million mapped reads.
Figure 1—figure supplement 2
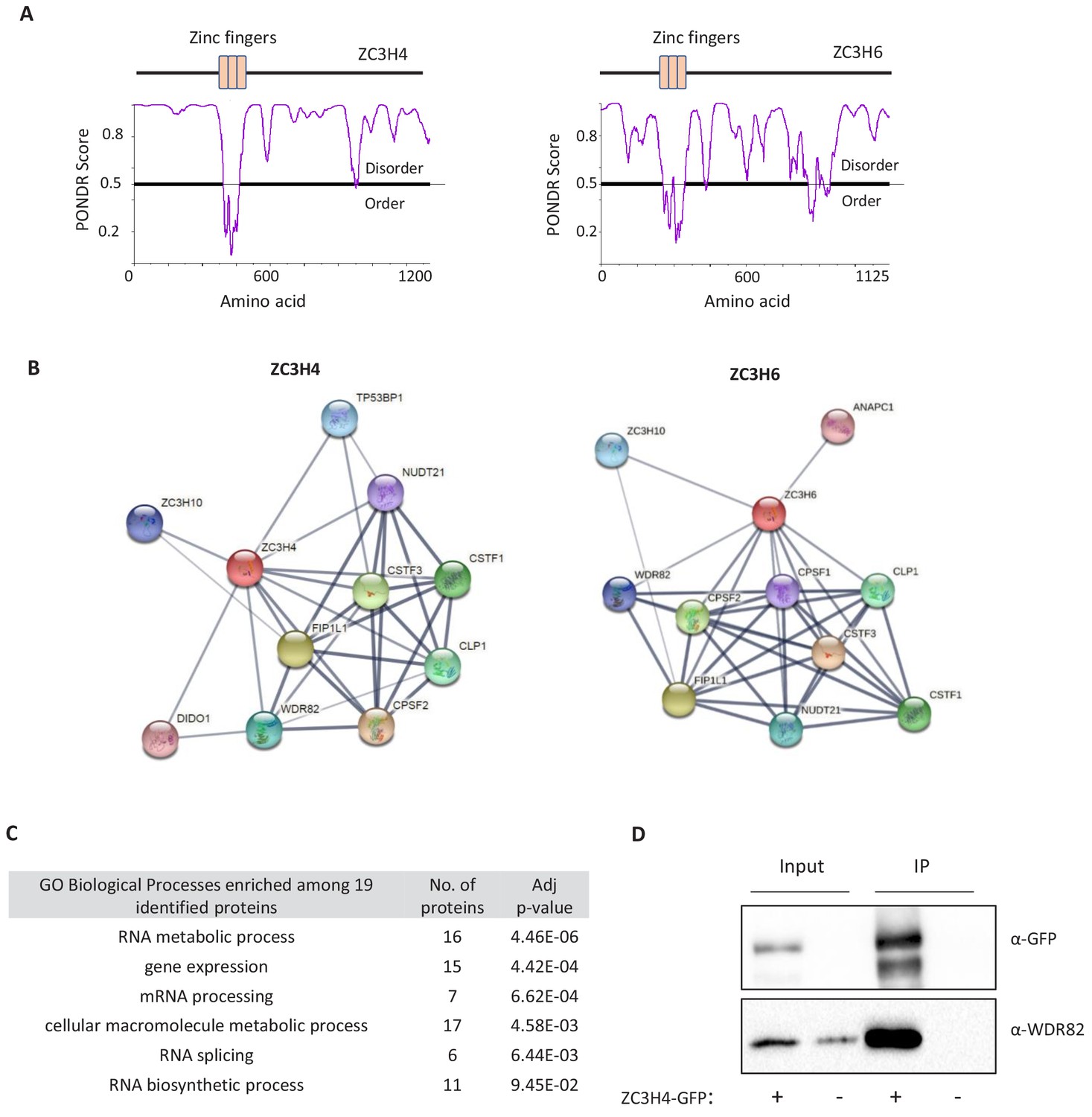
Predicted structures and interactors of ZC3H4 and ZC3H6.
(a) Schematic of ZC3H4 and ZC3H6 showing the three CCCH zinc finger domains. A predictor of natural disordered region (PONDR) analysis shows that the only ordered region of ZC3H4 coincides with these domains. Graph generated via PONDR.com, set to VSL2. (b) STRING analysis of ZC3H4 and ZC3H6 indicates interactors with 3’ end processing complex members. Image was taken from string.db.org, confidence value was set to medium (0.4). The thickness of lines between nodes is indicative of the confidence in interaction. (c) Proteins that are co-regulated with ZC3H4 according to ProteomeHD (https://www.proteomehd.net/) using a score cut-off set to 0.998. Table shows Gene Ontology (GO) term analysis of the potentially co-regulated factors. (d) Co-immunoprecipitation of WDR82 using ZC3H4-GFP as bait. Blot shows input (5%) and immunoprecpitated material probed with antibodies to WDR82 or GFP. Cells untransfected with ZC3H4-GFP act as a negative control.
Figure 1—figure supplement 3
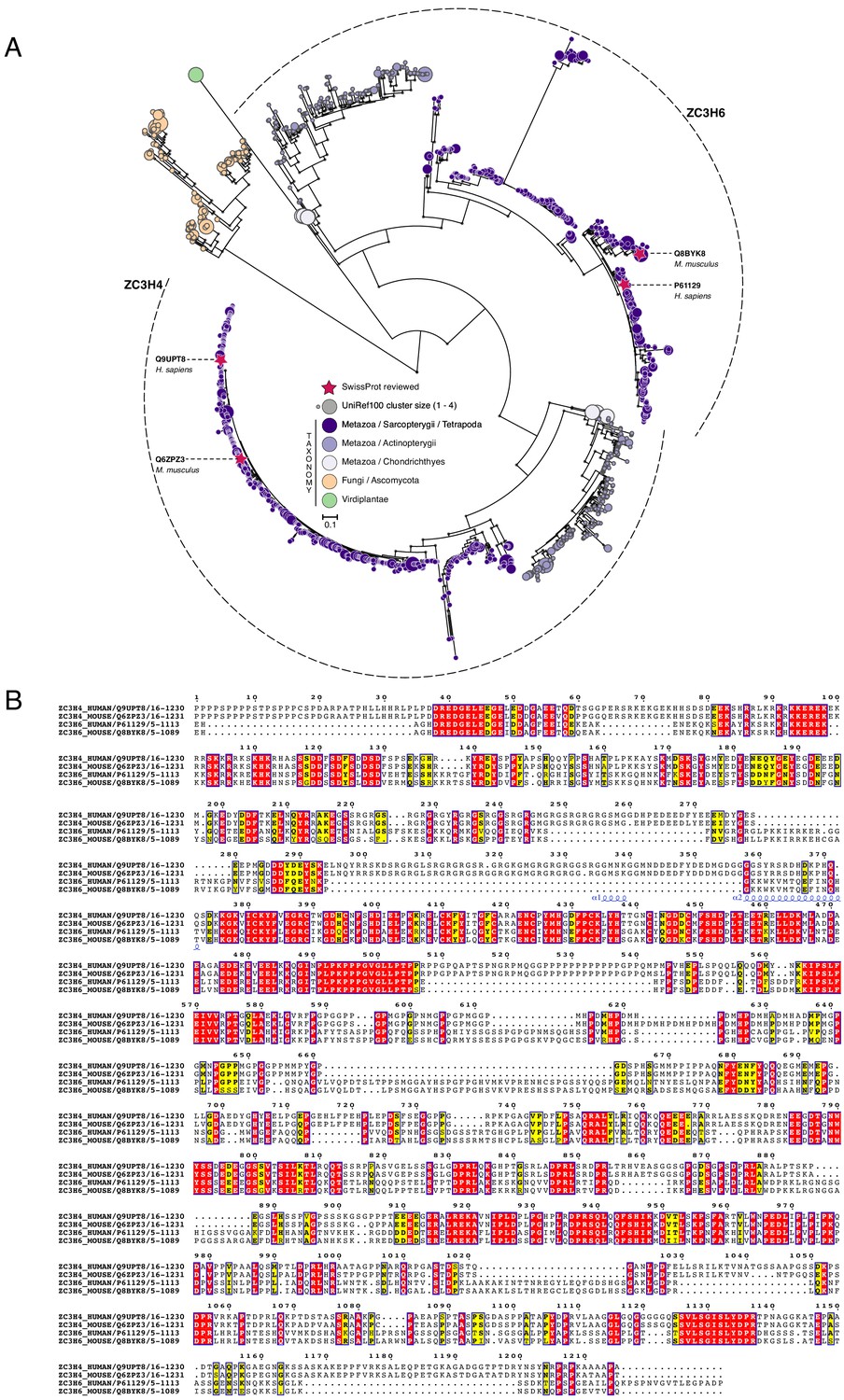
Phylogenetic analysis of ZC3H4 and ZC3H6.
(a) Maximum-likelihood phylogenetic tree of zinc finger CCCH-domains (1513 sequences; 795 parsimony informative sites) inferred under the JTT + R8 model. Clades of ZC3H4-like and ZC3H6-like domains are delimited by dashed lines. CCCH-domains identified using the PANTHER hidden Markov model PTHR13119 against the UniProtKB protein database (non-redundant version: UniRef100; external node size represents protein cluster size). Branch support values ≥90% (based on 1000 ultrafast bootstraps) are indicated by grey circles. Red stars show SwissProt reviewed protein sequences; external nodes are colour-coded according to their taxonomic lineage. Scale bar represents the number of estimated substitutions per site. Virtually all recovered sequences were from metazoan organisms – except for a group of fungal sequences from ascomycetes. The resulting phylogenetic tree shows the dichotomy between the ZC3H4 and ZC3H6 domains, which are found in the same set of organisms. This indicates that they are paralogues and have likely diverged their function following gene duplication. The ancestral gene coding for ZC3H4/6 was likely lost from the non-vertebrates and subsequently underwent a duplication event leading to the ZC3H4- and ZC3H6-like paralogues in vertebrates. Primary data are available in Supplementary file 2 and deposited at Zenodo (https://doi.org/10.5281/zenodo.4637127). (b) Multiple sequence alignment of ZC3H4 and ZC3H6 homologues. PTHR13119 domains from human and mouse SwissProt sequences were aligned using structural information (PDB structure: 2CQE; zinc finger domain; helices are displayed as coils) using TCoffee (Expresso mode). Conserved regions are indicated by blue boxes; identical and similar residues (based on physicochemical properties) are marked in red and yellow, respectively. Sequence identifiers correspond to UniProt/SwissProt accession numbers and to boundaries of identified PTHR13119 domains. Alignment figure was rendered with ESPscript.
Figure 2 with 2 supplements
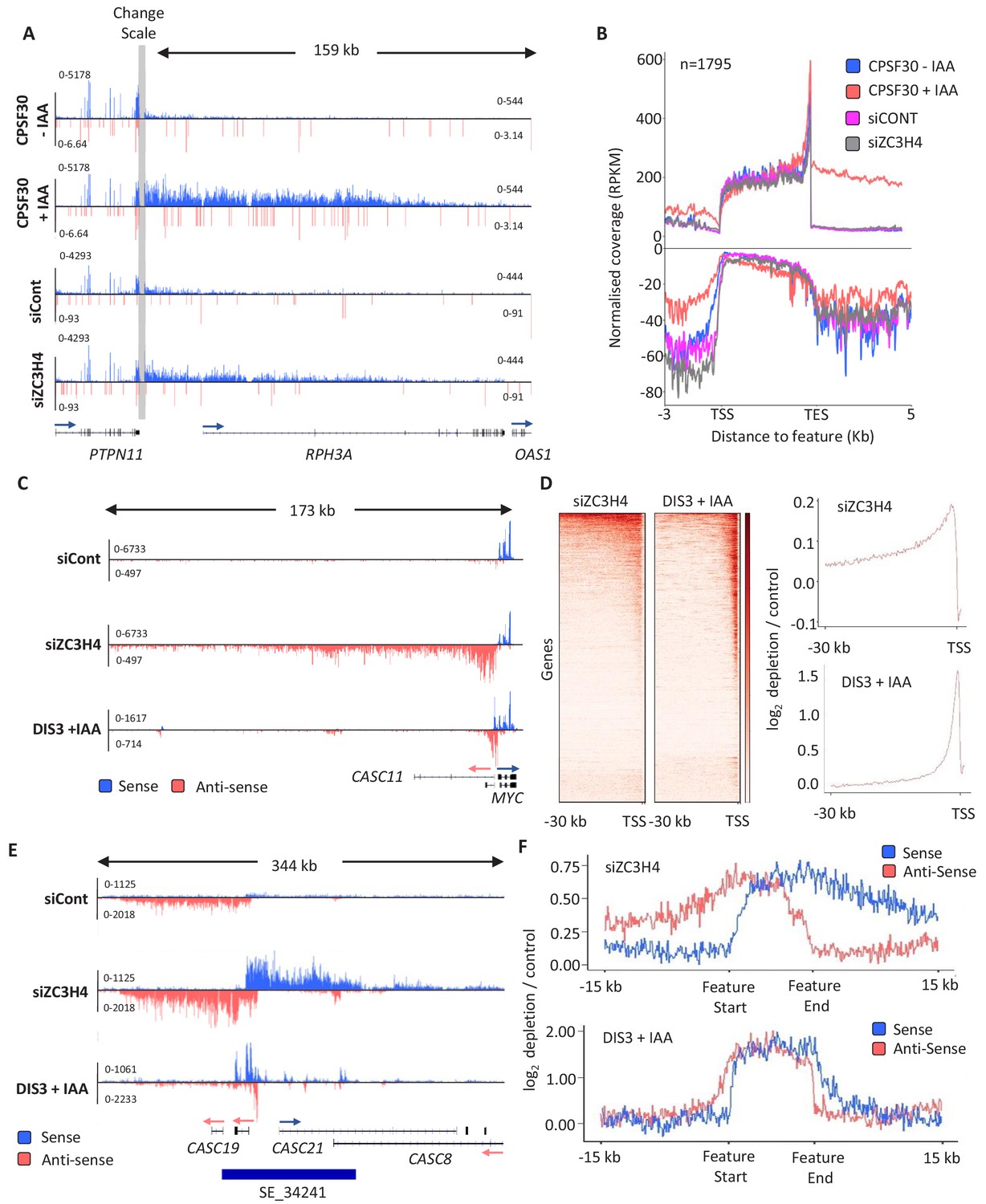
ZC3H4 depletion stabilises unproductive transcripts.
(a) Integrative Genomics Viewer (IGV) track of the transcription read-through defect at PTPN11 following CPSF30 or ZC3H4 depletion. Blue and red tracks indicate sense/antisense transcripts respectively, grey bar indicates a change in y-axis scale so that comparatively weaker read-through signals can be visualised next to the gene body (left scale for upstream of TES; right for downstream). Y-axis scale is RPKM. (b) Metagene comparison of transcription upstream, across, and downstream of, protein-coding genes in nuclear RNA from CPSF30-mAID cells treated or not with auxin and from HCT116 cells transfected with control or ZC3H4 siRNAs. CPSF30 traces are from the same samples presented in Figure 1C. Positive and negative signals represent sense and antisense reads, respectively. (c) IGV track view of transcription at the MYC PROMPT in RNA-seq samples obtained from control or ZC3H4 siRNA-treated HCT116 cells. We also show a track from HCT116 cells acutely depleted of DIS3-AID (DIS3 + IAA) (Davidson et al., 2019) to highlight the normal extent of this unstable transcript. Y-axis scale is RPKM. (d) Log2 fold change of siZC3H4 vs. siControl or DIS3+ vs. - auxin for RNA upstream of 6057 non-neighbouring, actively transcribed genes, plotted as heat maps. Line graphs are an XY depiction of heat map data. Log2 fold changes are smaller in siZC3H4 samples versus DIS3 depletion because this is an average of all genes in the heat map, a smaller fraction of which are affected by ZC3H4. (e) IGV plot of a known SE upstream of MYC (the location is shown by blue bar under trace). Samples are shown from HCT116 cells treated with control or ZC3H4 siRNAs as well as DIS3-AID cells treated with auxin (the latter from Davidson et al., 2019) to show the normal extent of unstable eRNAs over this region. Y-axis scale is RPKM. (f) Log2 fold change of RNA signal for siZC3H4 vs. siControl or DIS3+ vs. - auxin for 111 SEs. The bed file detailing super-enhancer coordinates in HCT116 cells was taken from dbSUPER.org.
Figure 2—figure supplement 1
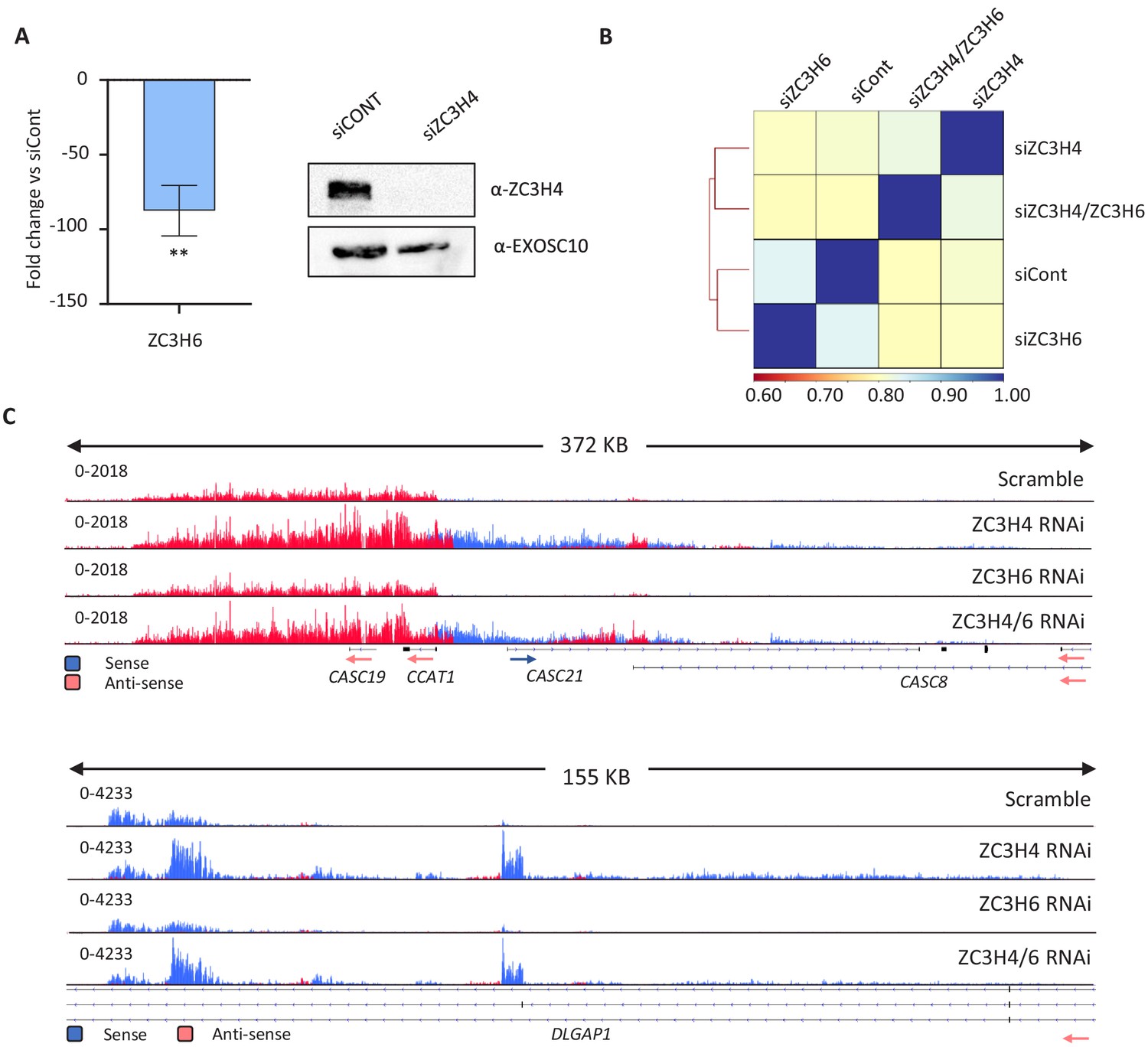
Comparison of ZC3H4 or ZC3H6 depletion, and their co-depletion, via RNA-seq.
(a) qRT-PCR and western blotting evidence of the effectiveness of ZC3H6 and ZC3H4 depletion, respectively. Graph shows fold reduction of ZC3H6 mRNA in cells treated with ZC3H6 siRNAs versus those transfected with control siRNA. N = 3, error bars are SEM. ** is p<0.01. Western blotting of ZC3H4 in HCT116 cells treated with control siRNAs or ZC3H4-targeting siRNAs. The blot was probed with a ZC3H4 antibody revealing strong depletion versus the EXOSC10 loading control. (b) Pearson’s correlation of siControl, siZC3H4, siZC3H6 and their co-depletion (siZC3H4/ZC3H6) from RNAseq BAM files performed by DEEPTOOLS. (c) IGV traces exemplifying two genomic regions with clear RNA accumulation following ZC3H4 depletion. While this is also seen following ZC3H4/ZC3H6 co-depletion, it is not evident following the depletion of ZC3H6 alone. This was generally seen, supporting the correlation analysis in (b). Y-axis scale is RPKM.
Figure 2—figure supplement 2
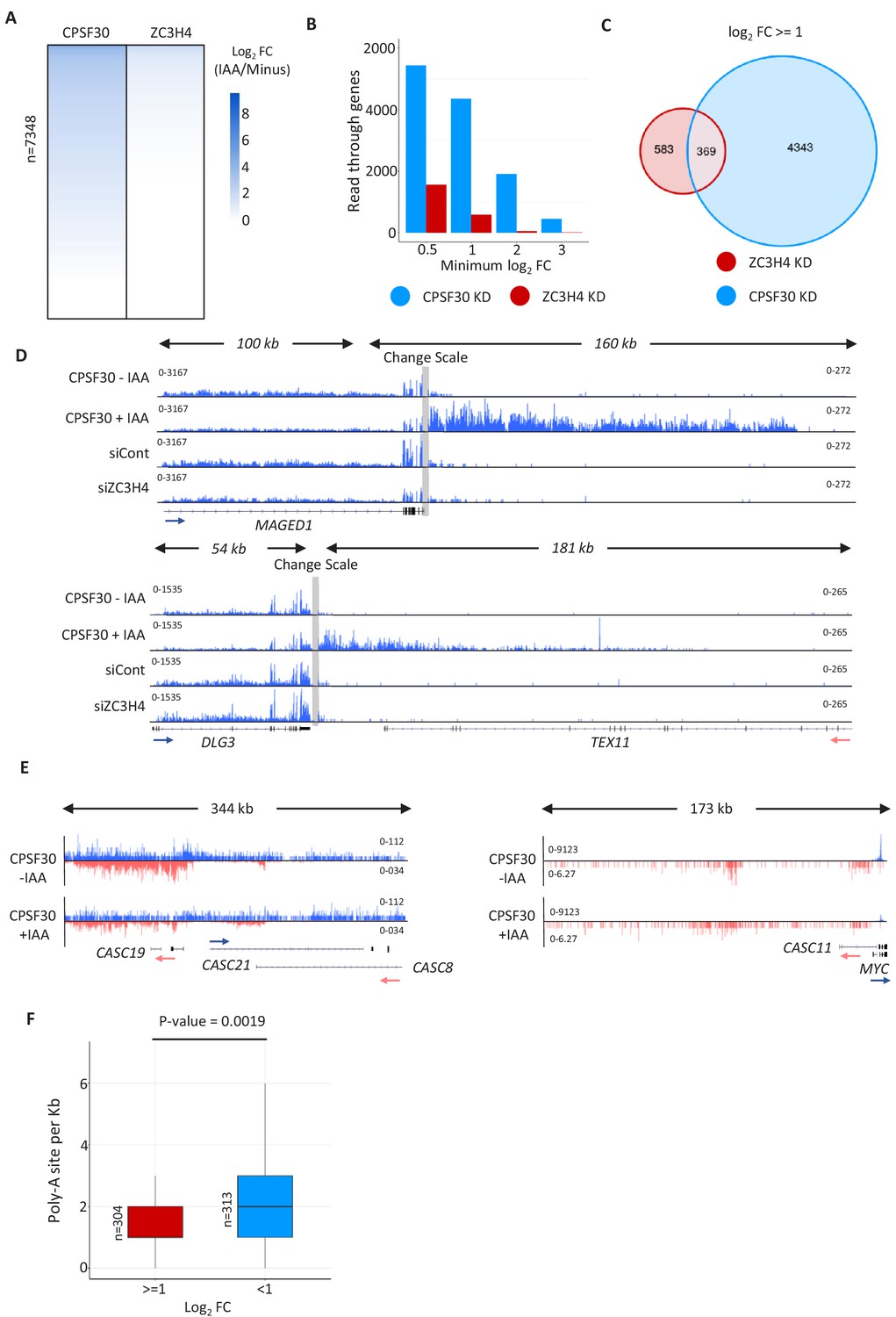
Comparison of CPSF30 and ZC3H4 effects on transcriptional read-through.
(a) Heat maps showing the effects of CPSF30-mAID or ZC3H4 depletion on read-through beyond protein-coding genes. Coloured scale bar indicates the magnitude of effect (log2 scale). Read-through was scored as a ratio of reads upstream (500 bp) and downstream (1 kb) of the PAS. The lists associated with this heat map are provided in Supplementary file 3. (b) Graph showing the number of protein-coding genes with read-through enhancements of greater than 0.5, 1, 2 or three on a log2 fold scale. This illustrates that the effects of CPSF30-mAID loss are both wider spread and larger than those associated with depletion of ZC3H4. (c) Venn diagram showing the number of genes bioinformatically scored as having increased read-through (log2 fold of 1 or more) following CPSF30-mAID or ZC3H4 loss and those that are common between the two conditions. (d) IGV tracks of individual genes (MAGED1 and DLG3) to exemplify the lack of 3’ termination defect following ZC3H4 depletion. As a control for bone fide read-through, the same tracks are shown in samples obtained from CPSF30-mAID cells treated or not (3 hr) with auxin. Grey bar indicates a change in scale (left scale for upstream of PAS; right for downstream) so that comparatively weaker read-through signals can be visualised next to the gene body. Y-axis scale is RPKM. (e) CPSF30 depletion shows little effect at super-enhancers and PROMPTs. IGV track view of the MYC super-enhancer and PROMPT in RNA-seq data obtained from CPSF30-mAID cells treated or not (3 hr) with auxin. Y-axis scale is RPKM. (f) Plot showing the density of PAS sequences (AWTAAA) in PROMPTs upregulated or unaffected by ZC3H4 loss. Y-axis plots number of PAS sequences/kb.
Figure 3 with 1 supplement
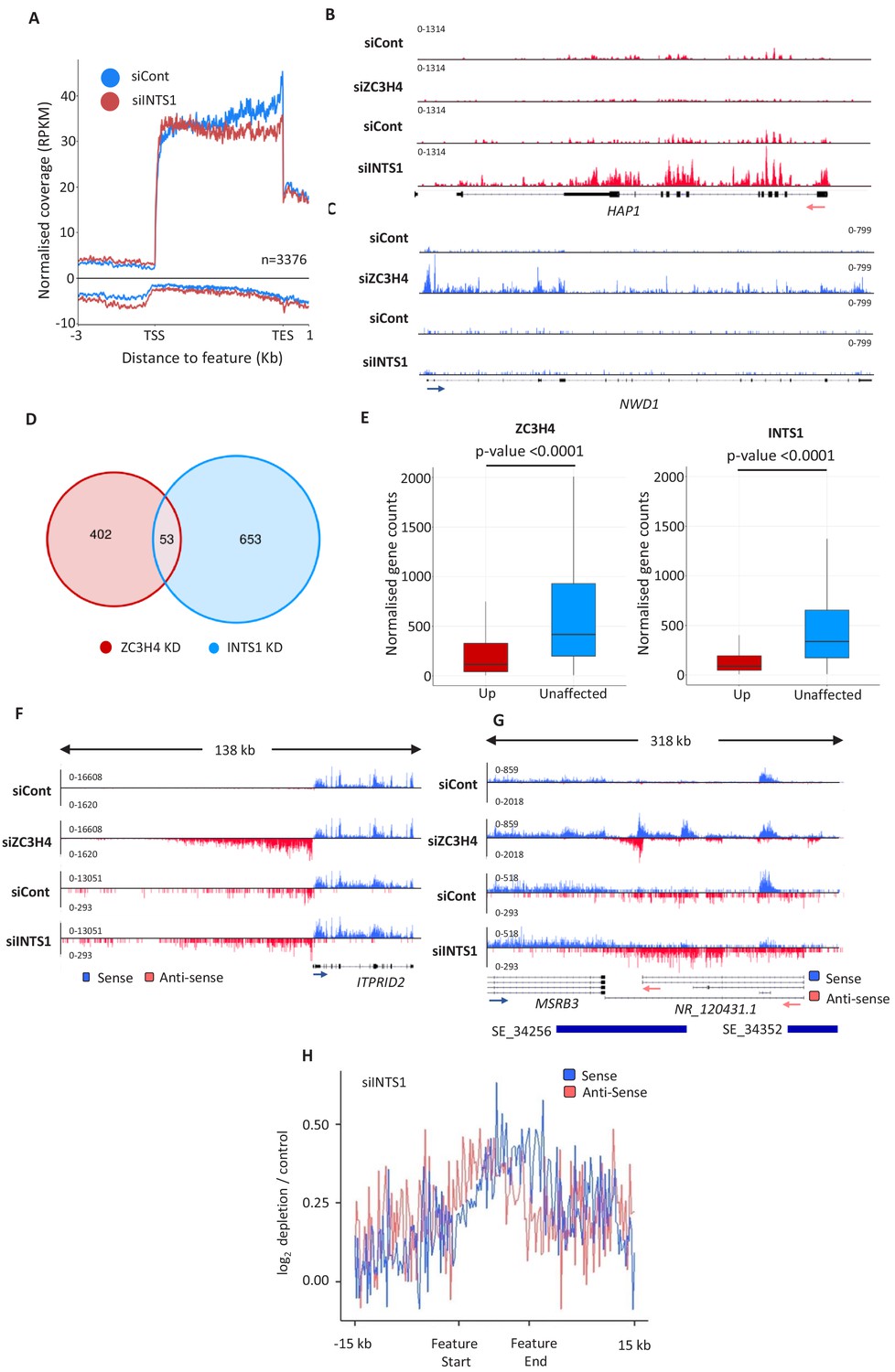
Comparison of ZC3H4 and Integrator effects.
(a) Metagene analysis of chromatin-associated RNA-seq performed on cells treated with control or INTS1-specific siRNA. The plot shows signals upstream, across, and downstream of protein-coding genes. Y-axis scale is RPKM. Positive and negative values represent sense and antisense reads, respectively. (b, c) IGV traces of HAP1 and NWD1 genes derived from chromatin-associated RNA-seq in control and INTS1 siRNA treated samples and nuclear RNA-seq from control or ZC3H4 siRNA treatment. NWD1 transcripts are affected by ZC3H4 but not INTS1, whereas the opposite is true for HAP1 RNAs. Y-axes scales are RPKM. (d) Venn diagram showing the number of mRNAs upregulated ≥2-fold, padj ≤0.05 following ZC3H4 depletion versus INTS1 loss and the overlap between the two sets. Genes that showed increased expression due to transcription read-through from an upstream gene were filtered by assessing coverage over a 1 kb region preceding the TSS, relative to untreated cells. (e) Graphs demonstrating the expression level of mRNA transcripts upregulated (log2FC >1) following ZC3H4 or INTS1 depletion by comparison with transcripts unaffected by loss of either factor. Y-axis shows normalised gene counts (i.e. expression level). (f) Comparison of chromatin-associated RNA-seq in control and INTS1 siRNA treated samples with nuclear RNA-seq derived from control or ZC3H4 siRNA treatment. The ITPRID2 PROMPT is displayed and y-axes are RPKM (note the different scales between ZC3H4 and INTS1 samples). (g) Comparison of chromatin-associated RNA-seq in control and INTS1 siRNA treated samples with nuclear RNA-seq derived from control or ZC3H4 siRNA treatment. The MSRB3 SE is displayed and y-axes are RPKM (note the different scales between INTS1 and ZC3H4 samples). (h) Metaplot of RNA-seq profile over super-enhancers following INTS1 depletion (log2 fold depletion/control over 111 super-enhancer as line graphs). The bed file detailing super-enhancer coordinates in HCT116 cells was taken from dbSUPER.org. RPKM = reads per kilobase of transcript, per million mapped reads, TSS = transcription start site.
Figure 3—figure supplement 1
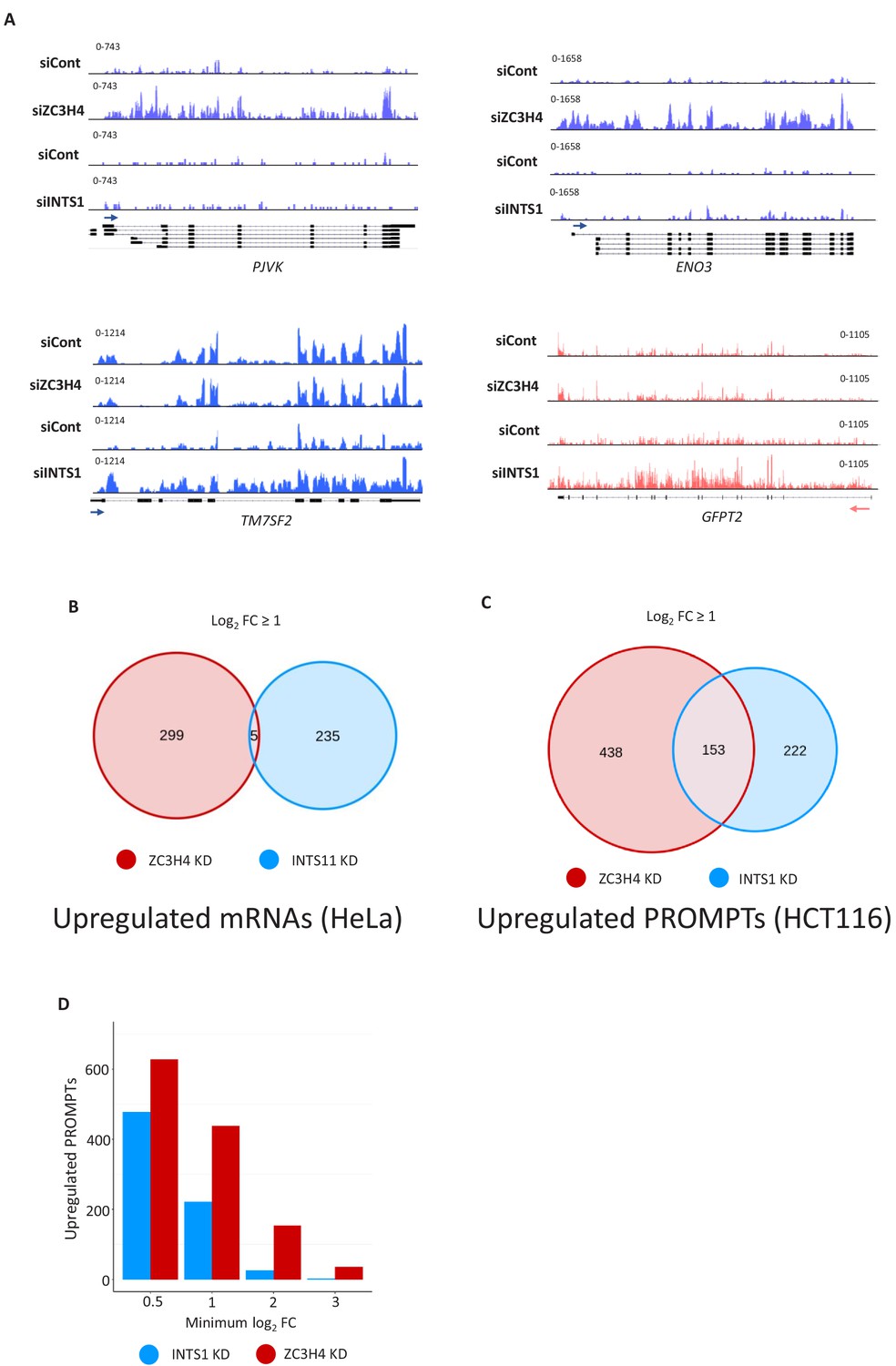
Comparison of ZC3H4 and INTS1 depletion on mRNA and PROMPT transcripts.
(a) IGV snapshots showing examples of protein-coding genes selectively upregulated by ZC3H4 (PJVK and ENO3) or Integrator (TM7SF2 and GFPT2). Y-axis represents RPKM. (b) Venn diagram representing the number of protein-coding transcripts (determined by DESEQ2) that show increased levels in previously published 4sU labelling experiments performed on HeLa cells depleted of INTS11 or ZC3H4 (Austenaa et al., 2021; Lykke-Andersen et al., 2020). Gene lists are provided in Supplementary file 5. Notably, manual curation of this list revealed the presence of false positive hits, especially in the INTS11 data, due to DESEQ2 scoring interference of transcription from neighbouring genes as upregulation. (c) Venn diagram showing the number of PROMPTs showing upregulation (log2 fold of 1 or more) following ZC3H4 or INTS1 loss from HCT116 cells and those that are common between the two conditions. (d) Graph showing the number of PROMPTs enhanced by greater than 0.5, 1, 2, or 3 on a log2 fold scale following the loss of ZC3H4 or INTS1. This illustrates that the effects of ZC3H4 loss are both widely spread and larger than those associated with depletion of INTS1. The list of targets in each case is provided in Supplementary file 6. RPKM = reads per kilobase of transcript, per million mapped reads, PROMPT = promoter upstream transcripts.
Figure 4 with 1 supplement
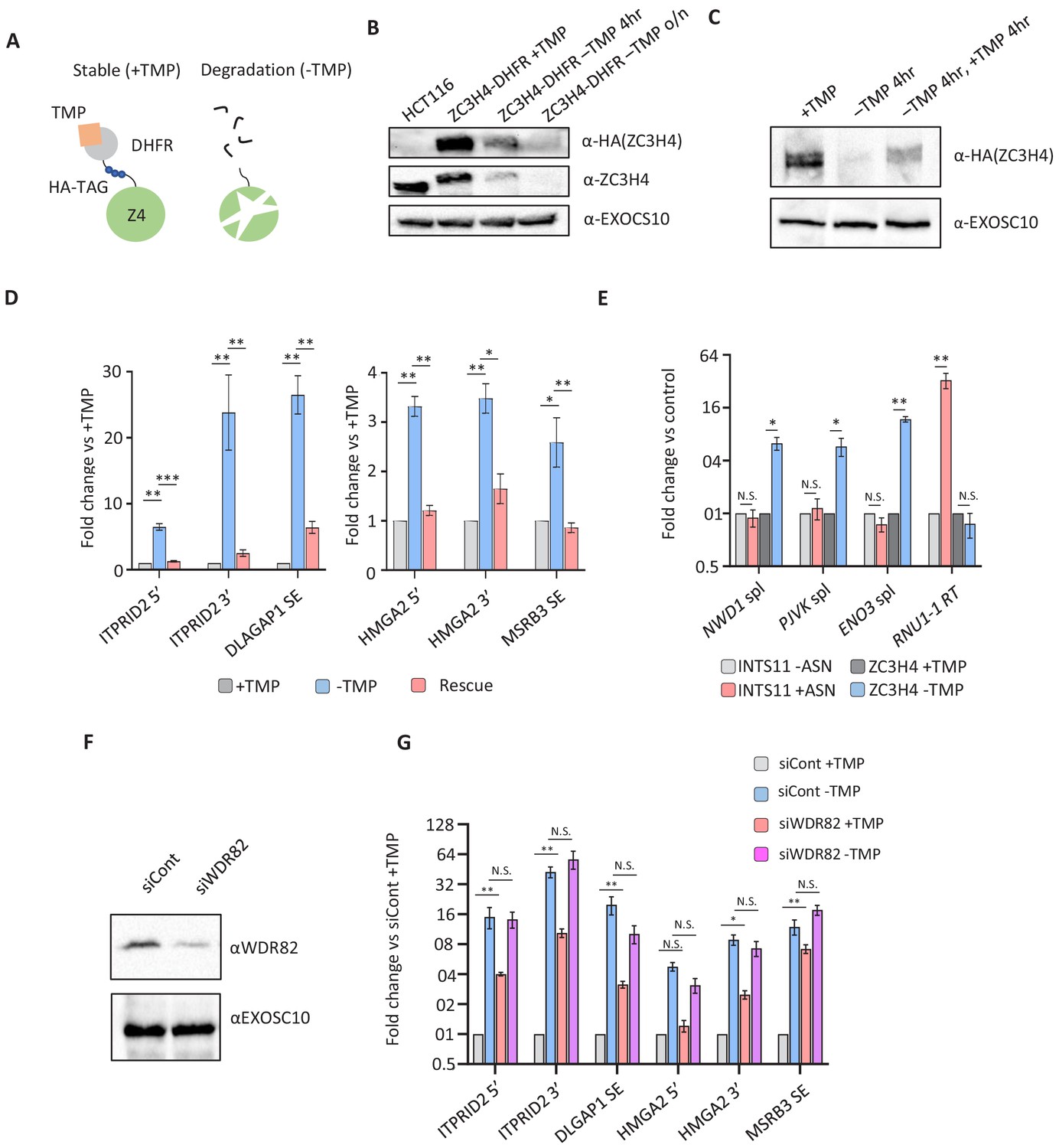
Transcriptional dysregulation following acute ZC3H4 loss.
(a) Schematic detailing how the dihydrofolate reductase (DHFR) degron works. E. coli dihydrofolate reductase (DHFR) is fused to the C-terminus of ZC3H4, which is stabilised by trimethoprim (TMP). When TMP is removed, ZC3H4-DHFR is degraded. (b) Western blot of HCT116 parental and HCT116 ZC3H4-DHFR cells ± TMP. TMP was withdrawn for 4 hr or overnight (o/n), EXOCS10 is used as a loading control, αHA recognises a HA peptide before the DHFR tag, while αZC3H4 recognises native protein. (c) Western blot of ZC3H4-DHFR cells grown under the following conditions: +TMP, –TMP (4 hr), –TMP (4 hr) followed by +TMP (4 hr). ZC3H4-DHFR is detected using αHA, and EXOSC10 is shown as a loading control. (d) qRT-PCR analysis of PROMPT and SE transcripts in ZC3H4-DHFR cells grown under the conditions represented in (c) (rescue refers to –TMP then +TMP for re-establishing ZC3H4). Graph shows fold change versus +TMP following normalisation to spliced actin. N = 3. Error bars are SEM. *, **, and *** denote p<0.05, 0.01, and 0.001, respectively. ITPRID2 5’ and 3’ primers are at approximately −500 bp and −7 kb relative to its TSS. HMGA2 5’ and 3’ primers are at approximately −1.8 kb and −7.1 kb relative to its TSS. (e) qRT-PCR analysis of spliced PJVK, ENO3 and NWD1 mRNAs and RNU1-1 read-through (RT) in ZC3H4-DHFR cells grown with or without (4 hr) TMP and INTS11-SMASh cells grown with or without asunaprevir (ASN; 36 hr to deplete INTS11-SMASh). Graph shows fold change versus control (+TMP for ZC3H4-DHFR samples and –ASN for INTS11-SMASh samples), following normalisation to spliced actin. N = 3. Error bars are SEM. * and ** denote p<0.05 and 0.01, respectively. (f) Western blot of extracts derived from HCT116 cells transfected with control or WDR82-specific siRNAs. The blot shows WDR82 and, as a loading control, EXOSC10. (g) qRT-PCR of PROMPT and SE transcripts in ZC3H4-DHFR cells transfected with control or WDR82 siRNAs before withdrawal, or not, of TMP (14 hr). Graph shows fold change by comparison with control siRNA transfected ZC3H4-DHFR cells maintained in TMP following normalisation to spliced actin transcripts. N = 3. Error bars are SEM. * and ** denote p<0.05 and 0.01, respectively. PROMPT = promoter upstream transcript.
Figure 4—figure supplement 1
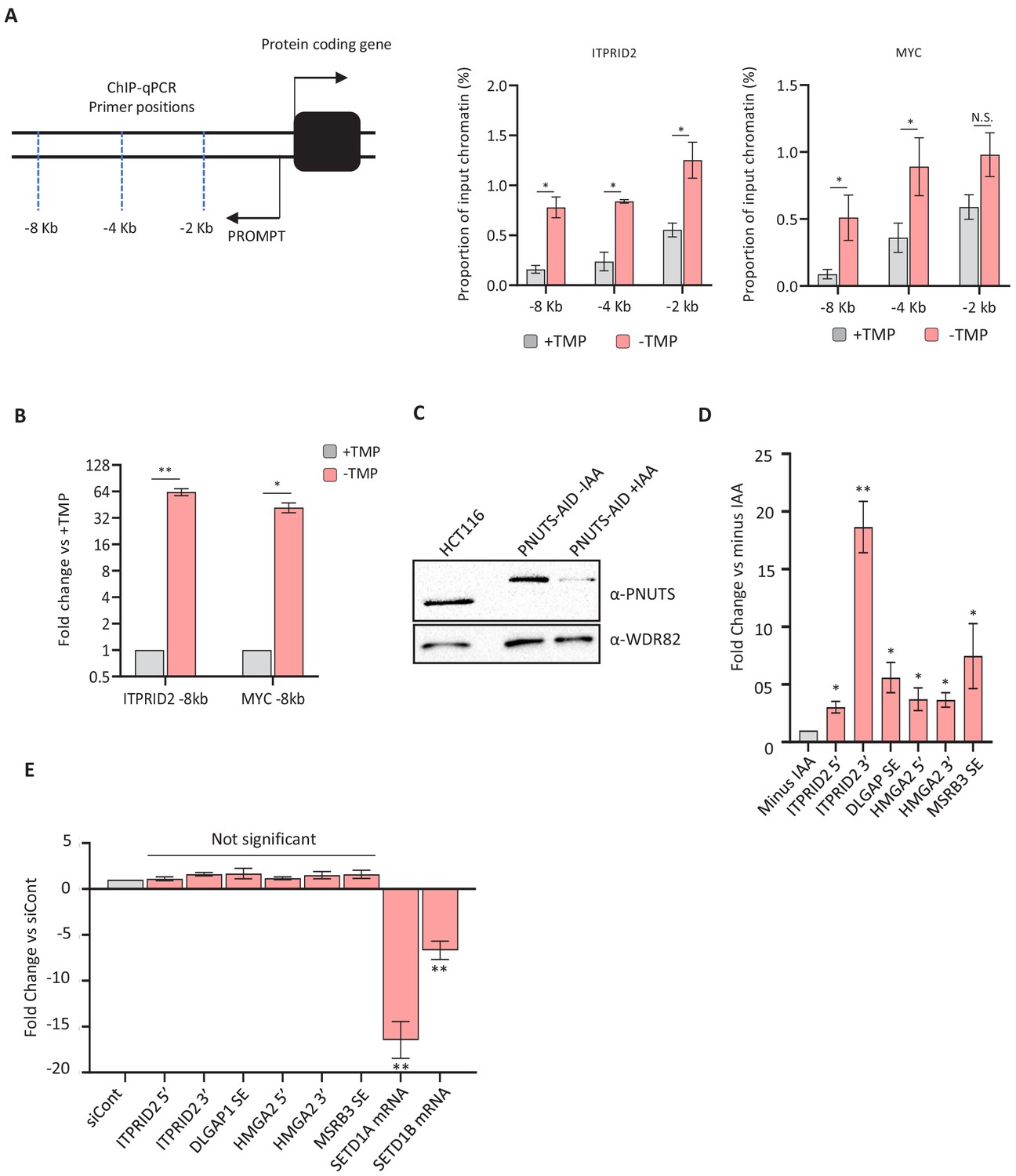
ZC3H4, PNUTS and SETD1A/B effects on PROMPT and SE transcripts/transcription.
(a) Pol II ChIP over ITPRID2 and MYC PROMPT regions. Graphs plot Pol II occupancy as a percentage of input at amplicons ~2, 4, and 8 kb upstream of each gene. N = 4. Error bars are SEM. * denotes p<0.05. The schematic illustrates the approximate location of each primer pair. (b) qRT-PCR analysis of extended ITPRID2 and MYC PROMPTs (−8 kb amplicons) in chromatin-associated RNA isolated from ZC3H4-DHFR cells grown with or without (4 hr) TMP. Y-axis displays fold change versus +TMP following normalisation to spliced actin levels. n = 3. Error bars are SEM. * and ** denote p<0.05 and 0.01, respectively. (c) Western blot showing acute depletion of PNUTS (the nuclear targeting subunit of PP1 phosphatase), tagged with an auxin-inducible degron (PNUTS-AID). Blot shows extracts from unmodified HCT116 cells and PNUTS-AID cells that were treated or not with auxin (3 hr). WDR82 is used as a loading control. (d) qRT-PCR of PROMPT and SE transcripts in PNUTS-AID cells treated or not with auxin (3 hr). Graph shows fold change by comparison with non-auxin treated cells following normalisation to spliced actin transcripts. N = 3. Error bars are SEM. * and ** denote p<0.05 and 0.01, respectively. (e) qRT-PCR analysis of PROMPT and SE transcripts in HCT116 cells treated with control siRNAs or siRNAs against both SETD1A and B. Quantitation shows fold change versus cells transfected with control siRNAs following normalisation to spliced actin levels. Note that the PROMPT and SE targets that are increased following ZC3H4 loss show relatively little change following depletion of SETD1A and B. The success of the RNAi is indicated by the strong reduction of SETD1A and B mRNAs. N = 3. Error bars show SEM. ** denotes p<0.01. PROMPT = promoter upstream transcript TMP = trimethoprim.
Figure 5 with 1 supplement
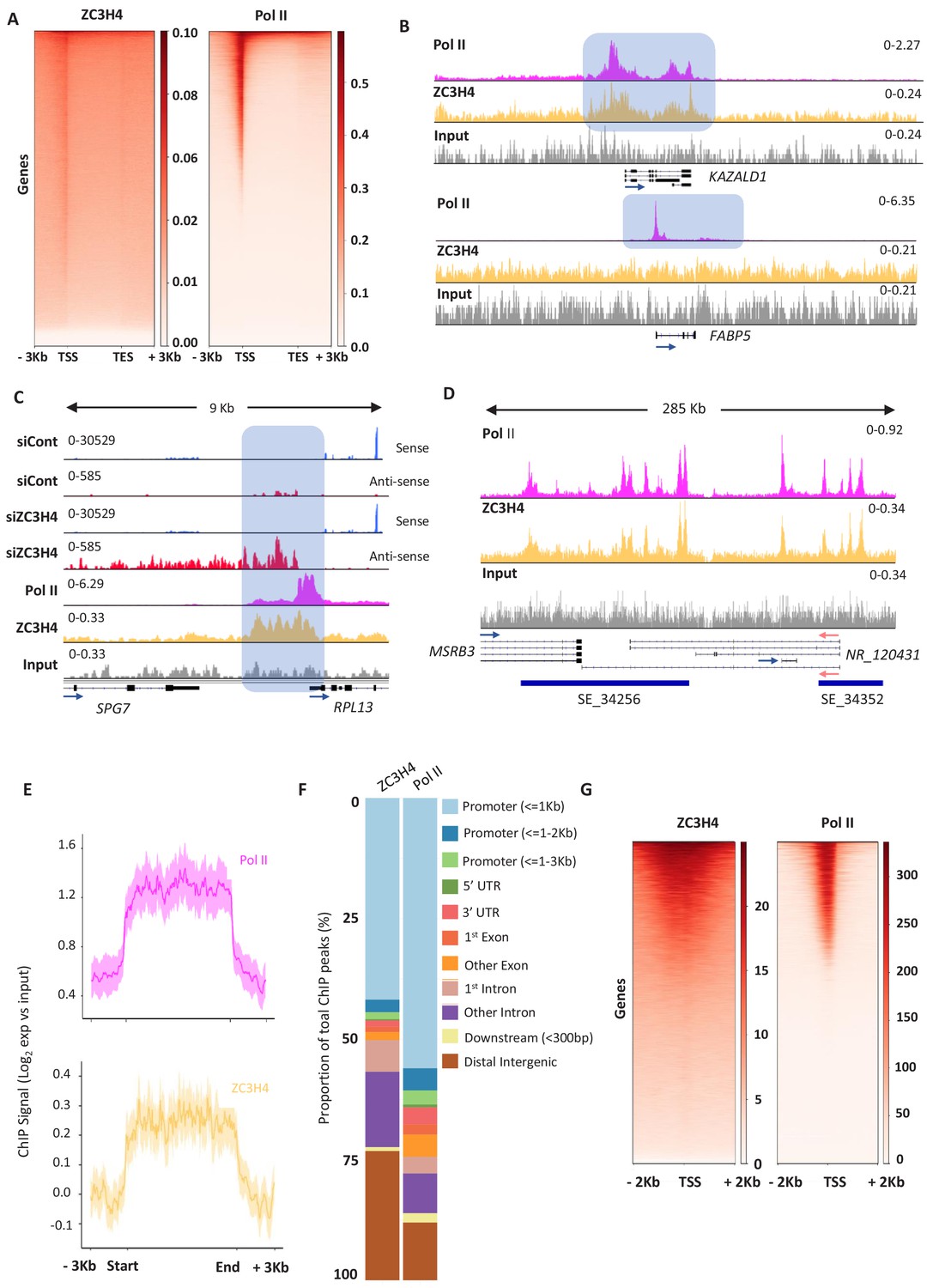
ZC3H4 occupies regions where transcription is affected by its absence.
(a) ZC3H4 ChIP profile over protein-coding genes is similar to Pol II. Heat map representation of ZC3H4 and Pol II ChIP-seq occupancy over the gene body ±3 kb. (b) ZC3H4 occupies fewer promoters than Pol II. IGV track view of ZC3H4 and Pol II occupancy over KAZALD1 and FABP5 genes, Pol II is present at both genes, while ZC3H4 is only present at KAZALD1. Scale is counts per million (CPM). Shaded blue box shows peak of Pol II and ZC3H4 at KAZALD1 and of Pol II over FABP5. (c) RNA-seq (HCT116 cells treated with control or ZC3H4 siRNA) and ChIP-seq (Pol II, ZC3H4 and input) profiles at RPL13. ZC3H4 occupancy is focused more on the PROMPT transcript region (blue box) than the TSS where, in contrast, the Pol II signal is maximal. RNA-seq scale is RPKM and ChIP-seq is CPM. (d) ZC3H4 ChIP occupancy mirrors Pol II at super-enhancers. IGV track view of ZC3H4 and Pol II occupancy over the SE at the MSRB3 locus. HCT116 super-enhancer gene track is from dbSUPER and depicted as blue bars. (e) Log2 fold change of ZC3H4 and Pol II vs. input at SEs shown as a line graph. Halo denotes 95% confidence level. (f) ChIPseeker analysis of peak distribution of ZC3H4 and Pol II. Occupancy regions are colour-coded and the number of ChIP peaks expressed as a proportion of 100%. (g) Heat map showing Pol II and ZC3H4 ChIP occupancy in HEPG2 cells obtained via the ENCODE consortium. Occupancy ±2 kb of the TSS is shown. RPKM = reads per kilobase of transcript, per million mapped reads, ChIP-seq = chromatin immunoprecipitation and sequencing, CPM = counts per million, TSS = transcription start site.
Figure 5—figure supplement 1
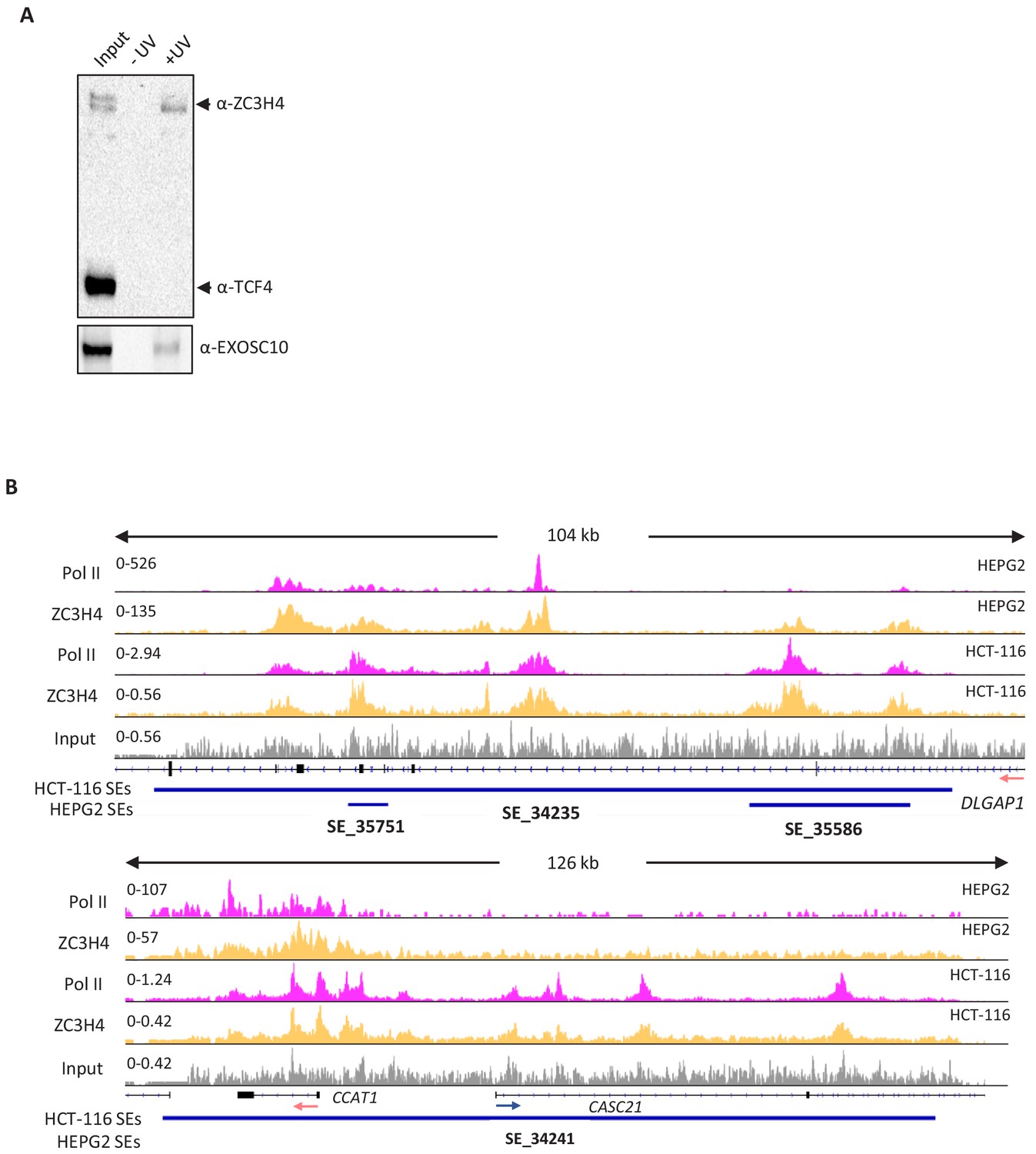
ZC3H4 RNA binding in HCT116 cells and ChIP-seq comparison of its occupancy of super-enhancers in HCT116 and HEPG2 cells.
(a) XRNAX analysis of ZC3H4 RNA binding in HCT116 cells. Samples show input and those isolated following UV treatment or not. Bands representing each protein are labelled accordingly. ZC3H4 is recovered in a UV-dependent manner indicating that it is directly bound to RNA in cells. The same is true of EXOSC10 that, as an exoribonuclease, acts as a positive control. TCF4 is a DNA binding transcription factor and acts as a negative control. (b) ZC3H4 only marks transcribed super-enhancers. IGV track view of ZC3H4 and Pol II occupancy at two different super-enhancers, DLAGAP1 present in both HEPG2 and HCT116 (top tracks) and the MYC super-enhancer (bottom tracks) that is only present in HCT116 cells. HEPG2 and HCT116 super-enhancer annotation is under each track as blue bars and was obtained from dbSUPER. Y-axis shows CPM. CPM = counts per million.
Figure 6 with 1 supplement

Directed recruitment of ZC3H4 recapitulates its effects on endogenous targets.
(a) Schematic of the MS2 system. A reporter plasmid (MS2hp-IRES-GFP) expressing a GFP transcript with 6 x MS2 hairpins upstream of an IRES and GFP gene. ZC3H4-MS2 or MS2-GFP can be specifically tethered to the MS2 hairpins to assess consequent effects on transcription/RNA output. Positions of primer pairs used in qRT-PCR experiments elsewhere in the figure are indicated by labelled horizontal lines under reporter. POI is protein of interest. (b) qRT-PCR analysis of total RNA isolated from MS2hp-IRES-GFP transfected cells co-transfected with MS2-GFP, ZC3H4-GFP, or ZC3H4-MS2. The level of reporter RNA is plotted (‘UP’ amplicon) as a percentage of that obtained in the MS2-GFP sample following normalisation to spliced actin. N = 3. Error bars are SEM. * denotes p<0.05. (c) qRT-PCR analysis of chromatin-associated RNA isolated from MS2hp-IRES-GFP transfected cells co-transfected with either MS2-GFP or ZC3H4-MS2. The level of reporter RNA upstream of the MS2 hairpins (UP) and transcripts yet to be cleaved at the BGH poly(A) site (BGH UC) are plotted as a percentage of that obtained in the MS2-GFP sample following normalisation to spliced actin. N = 3. Error bars are SEM. * denotes p<0.05. (d) qRT-PCR analysis of total RNA isolated from MS2hp-IRES-GFP transfected DIS3-AID cells co-transfected with either MS2-GFP or ZC3H4-MS2 – simultaneously treated or not with auxin to deplete DIS3 (14 hr in total). The graph shows the ratio of RNA species recovered upstream (UP) versus downstream (DOWN) of the MS2 hairpins. N = 4. Error bars are SEM. * denotes p<0.05. (e) Schematic detailing an interplay between ZC3H4 and DIS3 that sees transcription stop and nascent RNA degraded (f) Colony formation assay of ZC3H4-DHFR cells grown in the presence or absence of TMP. Cells were grown for 10 days before crystal violet staining.
Figure 6—figure supplement 1
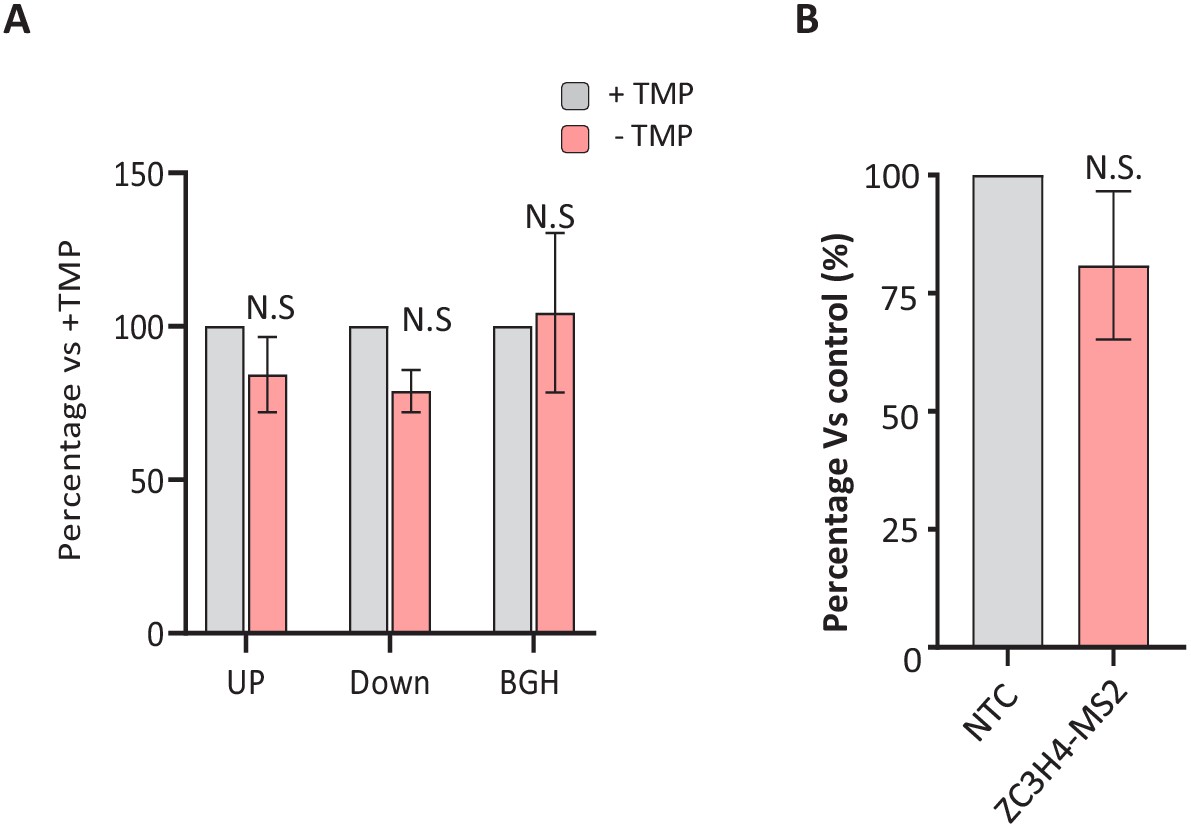
Control experiments for the specificity of ZC3H4 tethering effects.
(a) qRT-PCR of ZC3H4-DHFR cells transfected with MS2-IRES-GFP before growth in the presence or, to deplete ZC3H4, absence of TMP (4 hr). Graph shows percentage of each amplicon following TMP removal relative to that found in the presence of TMP following normalisation to spliced actin transcripts. N = 3. Error bars show SEM. * denotes p<0.05. (b) qRT-PCR of HCT116 cells transfected with IRES-GFP and either a control beta-globin plasmid (NTC) or ZC3H4-MS2. The graph shows the percentage of GFP RNA versus control following normalisation to spliced actin transcripts. N = 3. Error bars show SEM. TMP = trimethoprim, UP = upstream, BGH = bovine growth hormone.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (human) | HCT116- CPSF30-mAID | In-house | This paper | |
| Cell line (human) | HCT116- CPSF30-mAID:RPB1-mTurbo | In-house | This paper | |
| Cell line (human) | HCT116- ZC3H4-HA-DHFR | In-house | This paper | |
| Cell line (human) | HCT116- DIS3-AID | In-house | PMID:30840897 | |
| Cell line (human) | HCT116-PNUTS-AID | In-house | This paper | |
| Cell line (human) | HCT116-INTS11-SMASh | In-house | PMID:33113359 | |
| Recombinant DNA reagent | 3xHA-mTurbo-NLS_pCDNA3 | Addgene | RRID #:Addgene_107172 | |
| Recombinant DNA reagent | px300 | Addgene | RRID #:Addgene_42230 | |
| Recombinant DNA reagent | ZC3H4- pcDNA3.1(+)-C-eGFP | Genscript | Custom synthesis | ENTS00000253048 |
| Recombinant DNA reagent | pSL-MS2-6x | Addgene | RRID #:Addgene_27118 | |
| Recombinant DNA reagent | pcDNA3.1(+)IRES GFP | Addgene | RRID #:Addgene_51406 | |
| Antibody | CPSF30 | Bethyl | RRID #:AB_2780000 Cat #: A301-585A-T | (1:1000) |
| Antibody | RNA Pol II | Abcam | RRID #:AB_306327 Cat #: ab817 | Now discontinued at abcam (1:1000 for western blot. 4–5 ug used for ChIP qPCR and –seq, respectively) |
| Antibody | PNUTS | Bethyl | RRID #:AB_2779219 Cat #: A300-439A-T | (1:1000) |
| Antibody | WDR82 | Cell Signalling | RRID #:AB_2800319 Clone: D2I3B Cat #: 99715 | (1:1000) |
| Antibody | EXOSC10 | Santa Cruz | RRID #:AB_10990273 Cat #: sc-374595 | (1:2000) |
| Antibody | ZC3H4 | Atlas Antibodies | RRID #:AB_10795495 Cat #: HPA040934 | (1:1000) |
| Antibody | HA tag | Roche | RRID #:AB_390918 Clone: 3f10 Cat #: 11867423001 | (1:2000) |
| Antibody | GFP | Chromotek | Clone: PABG1 Cat #: PABG1-100 RRID #:AB_2749857 | (1:2000) |
| Antibody | TCF4/TCF7L2 | Cell Signalling | RRID #:AB_2199816 Clone: C48H11 Cat #: 2569 | (1:1000) |
| Chemical compound drug | TMP | Sigma | Cat #: T7883 | |
| Chemical compound drug | IAA | Sigma | Cat #: 12886 | |
| Commercial assay, kit | Lipofectamine RNAiMax | Life Technologies | Cat #: 13778075 | |
| Commercial assay, kit | JetPRIME | PolyPlus | Cat #: 114–01 | |
| Commercial assay, kit | Streptavidin Sepharose High Performance slurry | GE Healthcare | Cat #: GE28-9857-38 | |
| Commercial assay, kit | GFP TRAP magnetic agarose | Chromotek | RRID #:AB_2827592 Cat #: gtd-100 | |
| Commercial assay, kit | Dynabeads α-mouse | Life Technologies | RRID #:AB_2783640 Cat #: 11201D | |
| Commercial assay, kit | Dynabeads α-rabbit | Life Technologies | RRID #:AB_2783009 Cat #: 11203D | |
| Commercial assay, kit | SimpleChIP Plus Enzymatic Chromatin kit | Cell Signalling | Cat #: 9005 | |
| Commercial assay, kit | TruSeq Stranded Total RNA Library Prep Kit | Illumina | Cat #: 20020596 | |
| Commercial assay, kit | NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | Cat #: E7645S | |
| Commercial assay, kit | Ribo-Zero Gold rRNA removal kit | Illumina | Cat #: 20040526 | |
| Commercial assay, kit | Ampure XP beads | Beckman Coulter | Cat #: A63880 | |
| Commercial assay, kit | RNAClean XP Beads | Beckman Coulter | Cat #: A63987 | |
| Software, algorithm | BamTools | Barnett et al., 2011 | RRID #:SCR_015987 | v2.4.0 |
| Software, algorithm | BEDtools | Quinlan and Hall, 2010 | RRID #:SCR_006646 | v2.26.1 |
| Software, algorithm | Bioconductor | https://bioconductor.org/ | RRID #:SCR_006442 | v3.11 |
| Software, algorithm | DeepTools | Ramírez et al., 2014 | RRID #:SCR_016366 | v3.3.0 |
| Software, algorithm | DESeq2 | Love et al., 2014 | RRID #:SCR_015687 | v1.28.1 |
| Software, algorithm | featureCounts | Liao et al., 2013; Liao et al., 2014 | RRID #:SCR_012919 | v2.0.0 |
| Software, algorithm | FIMO | Grant et al., 2011 | RRID #:SCR_001783 | v5.3.3 |
| Software, algorithm | genomicRanges | http://bioconductor.org/packages/release/bioc/html/GenomicRanges | RRID #:SCR_000025 | v1.40.0 |
| Software, algorithm | ggplot2 | https://cran.r-project.org/web/packages/ggplot2 | RRID #:SCR_014601 | v3.3.3 |
| Software, algorithm | Hisat2 | Kim et al., 2015 | RRID #:SCR_015530 | v2.1.0 |
| Software, algorithm | IGV | Robinson et al., 2011 | RRID #:SCR_011793 | v2.8.2 |
| Software, algorithm | MACS2 | Zhang et al., 2008 | RRID #:SCR_013291 | v2.2.6 |
| Software, algorithm | pheatmap | https://cran.r-project.org/web/packages/pheatmap | RRID #:SCR_016418 | v1.0.12 |
| Software, algorithm | R | https://cran.r-project.org/ | NA | v4.0.4 |
| Software, algorithm | Rstudio | https://rstudio.com/ | RRID #:SCR_000432 | v1.3.1093 |
| Software, algorithm | rtracklayer | https://bioconductor.org/packages/release/bioc/html/rtracklayer | NA | v1.48.0 |
| Software, algorithm | SAMTools | Li et al., 2009 | RRID #:SCR_002105 | v.1.11 |
| Software, algorithm | Trim_galore! | https://github.com/FelixKrueger/TrimGalore/ | RRID #:SCR_011847 | v.0.6.5dev |
Additional files
-
Source data 1
Values (average, SEM, p-value) of data underpinning graphs within the paper.
- https://cdn.elifesciences.org/articles/67305/elife-67305-data1-v2.xlsx
-
Source data 2
Uncropped western blot images.
- https://cdn.elifesciences.org/articles/67305/elife-67305-data2-v2.pdf
-
Supplementary file 1
Mass spectrometry data associated with the Pol II-miniTurbo experiment.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp1-v2.xlsx
-
Supplementary file 2
Underpinning data for phylogenetic analyses.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp2-v2.csv
-
Supplementary file 3
Log2 fold changes in read-through following CPSF30 or ZC3H4 depletion from HCT116 cells.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp3-v2.xlsx
-
Supplementary file 4
List of mRNAs that are upregulated following ZC3H4 depletion (nuclear RNA-seq) or INTS1 depletion (chromatin-associated RNA-seq) in HCT116 cells.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp4-v2.xlsx
-
Supplementary file 5
List of mRNAs that are upregulated following ZC3H4 depletion (4sU RNA-seq; Austenaa et al., 2021) or INTS11 depletion (4sU TT-seq; Lykke-Andersen et al., 2020) in HeLa cells.
Genes in each set were manually checked for upregulation (TRUE) or possible artefacts – primarily transcription from surrounding region into a gene that is consequently scored as upregulated (FALSE).
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp5-v2.xlsx
-
Supplementary file 6
List of PROMPTs that are upregulated following ZC3H4 depletion (nuclear RNA-seq) or INTS1 depletion (chromatin-associated RNA-seq) in HCT116 cells.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp6-v2.xlsx
-
Supplementary file 7
Table of oligonucleotide and DNA sequences used in this study.
- https://cdn.elifesciences.org/articles/67305/elife-67305-supp7-v2.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/67305/elife-67305-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
ZC3H4 restricts non-coding transcription in human cells
eLife 10:e67305.
https://doi.org/10.7554/eLife.67305




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}