Simplifying the development of portable, scalable, and reproducible workflows
- Department of Biology, Brigham Young University, United States
- Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, United States
- Department of Medical Oncology and Therapeutics, City of Hope Comprehensive Cancer Institute, United States
Figures
Figure 1
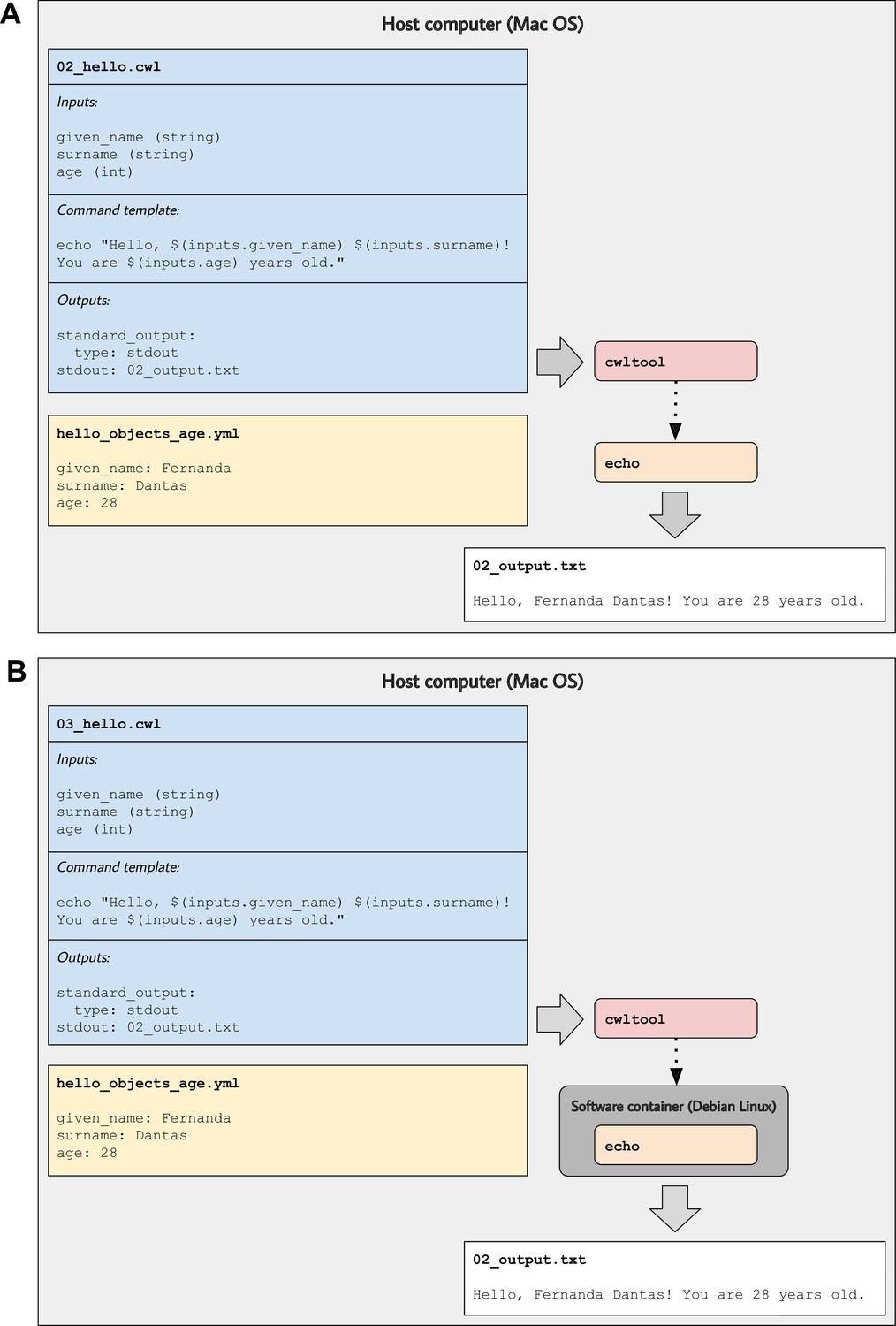
Illustration of tool descriptions for printing simple greetings.
In the examples associated with this article, we provide tool descriptions that illustrate how to print custom greetings at the command line. These diagrams illustrate the 02_hello.cwl (A) and 03_hello.cwl (B) examples. In (A), the tool description indicates which inputs that must be specified, along with a template for executing the command; it also indicates that a message will be printed to standard output and that this message should be stored in a file called 02_output.txt. The hello_objects_age.yml input-object file stores values for a particular invocation of the tool. In (A), the cwltool workflow engine uses the host computer’s operating system to execute the tool; thus, the echo command must be supported on that operating system. In (B), the tool description defines a software container environment; thus, cwltool executes the command within a container, which provides the echo command (packaged with the Debian Linux operating system).
Figure 2
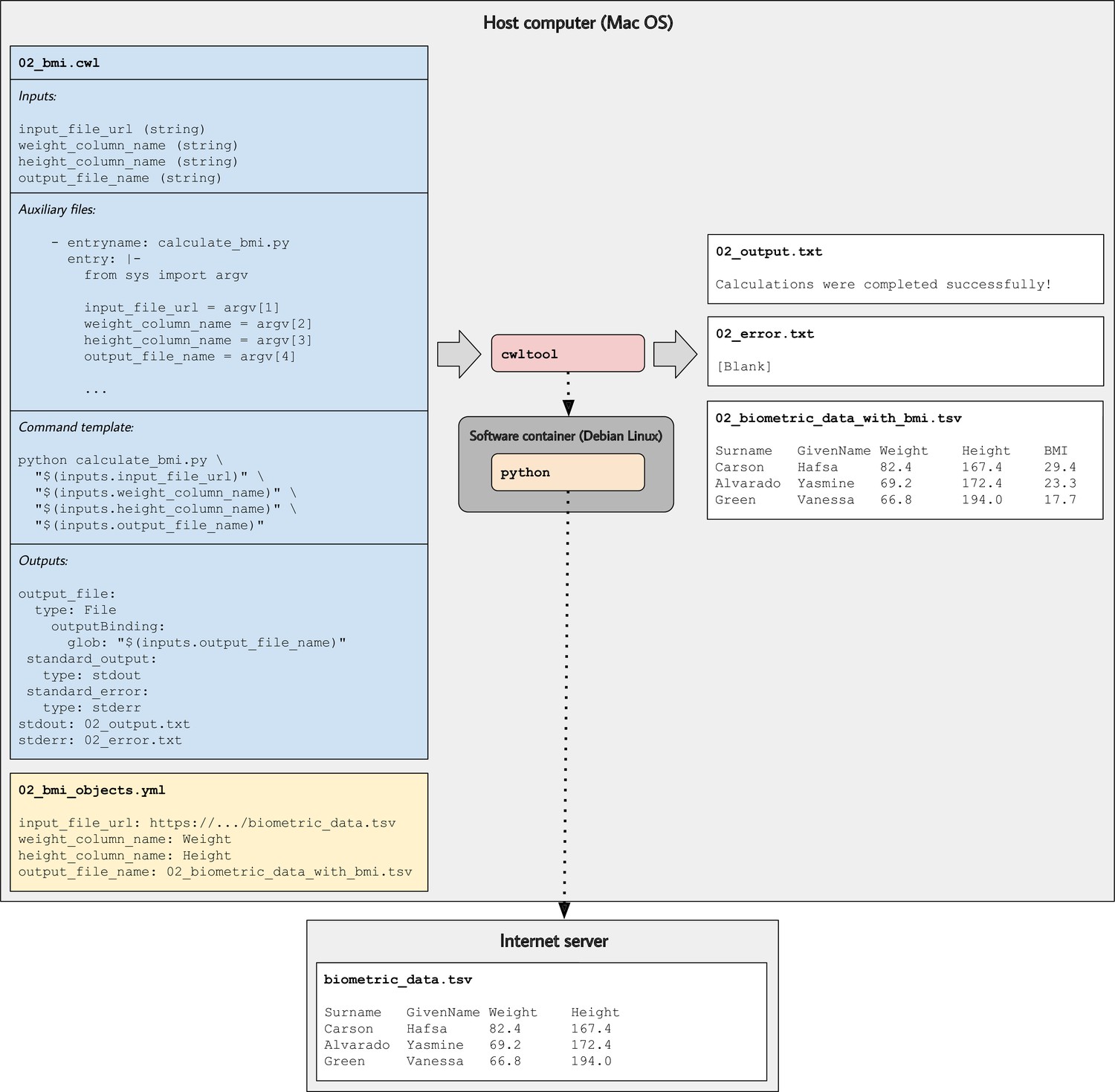
Illustration of tool descriptions for calculating individuals’ body mass index (BMI).
In the examples associated with this article, we provide tool descriptions that illustrate how to calculate BMI values based on individuals’ weights and heights stored in a tab-separated value file. This diagram illustrates the 02_bmi.cwl example. The tool description indicates the expected inputs. In this case, the URL of a data file must be provided. That file must contain a column that stores weights (in kilograms) and a column that stores heights (in centimeters). In the input-objects file (02_bmi_objects.yml), the user specifies the names of these columns. The final input is the name of an output file that will be generated. This file will store the original data and a new column with the calculated BMI value for each individual. As the tool executes, Python (within a software container) downloads the input file, performs the calculations, generates the output file, and stores the standard output and standard error in text files.
Figure 3
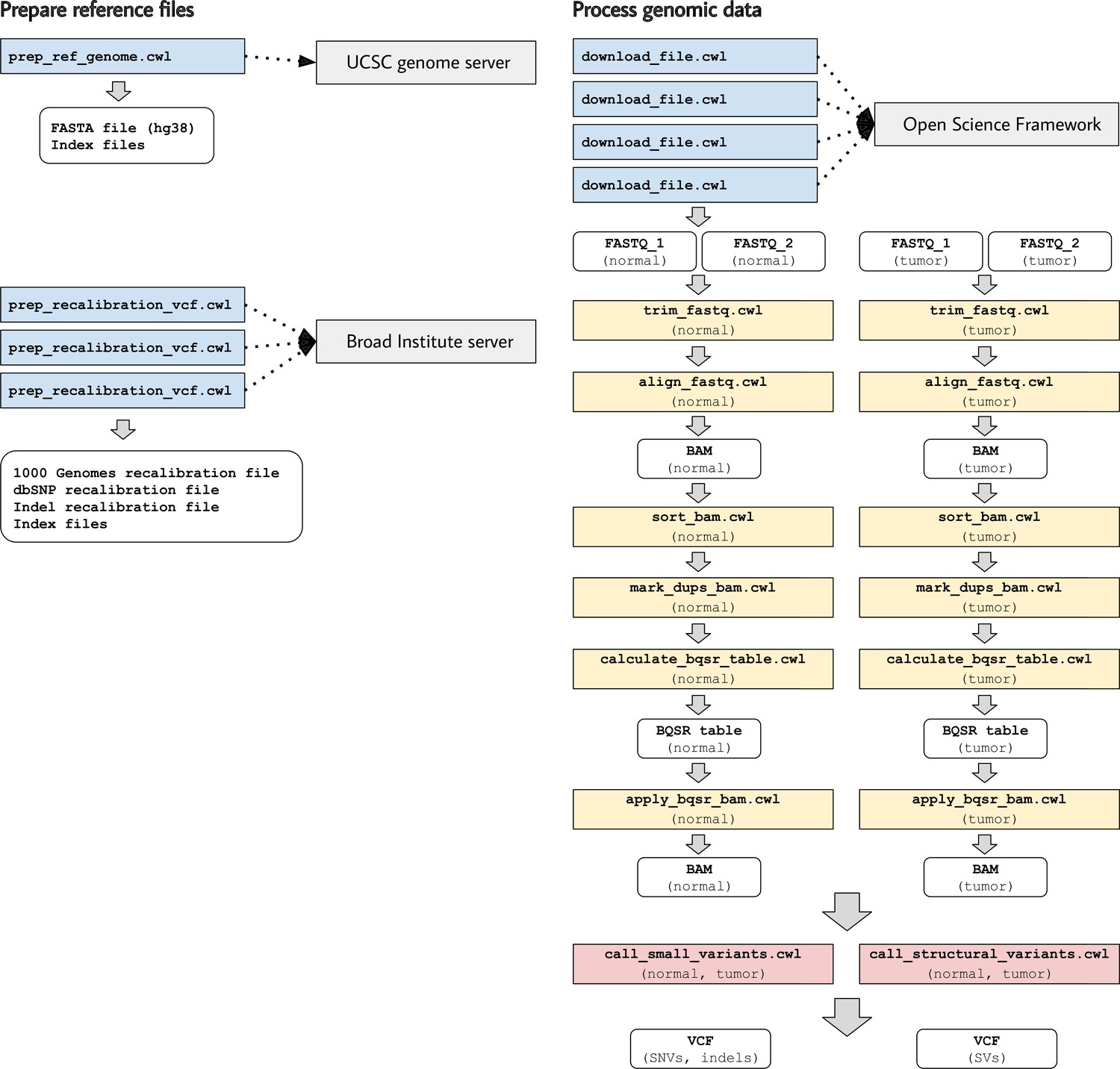
Illustration of tool descriptions for calling somatic variants from a cancer genome.
In the examples associated with this article, we provide tool descriptions that illustrate how to call somatic variants from second-generation sequencing data for a cancer genome (compared against a normal genome from the same patient). This process requires execution of 11 distinct tools in a defined succession of steps (a workflow). Two tools (prep_ref_genome.cwl and prep_recalibration_vcf.cwl) prepare reference files associated with a given human reference genome. These tools download data files from public Internet servers and then create index files and standardize contig identifiers. The third tool (download_file.cwl) downloads FASTQ files from an Internet server. The remaining tools process the normal and tumor sequences separately before comparing the tumor genome against the normal genome to identify single-nucleotide variants, indels, and structural variants.
Figure 4

Examples of command templates used in Common Workflow Language (CWL) tool descriptions.
These examples illustrate diverse types of command templates for configuring execution of CWL tools. In each example, placeholders are used for inputs. When the tools are executed, the placeholders are replaced with input-object values. (A) A simple greeting is printed to standard output. (B) An R script (stored as an auxiliary file within the tool description) is executed; this script performs a differential-expression analysis using the DESeq2 package. (C) The bwa software aligns FASTQ files to a reference genome and pipes the output to samtools; the output is then converted to BAM format. This example illustrates a scenario in which two complementary software packages are used to perform a data-analysis task. Although these packages could be incorporated into distinct CWL tools, we combine them because read alignment and BAM conversion are typically performed jointly. (D) The sambamba software sorts and then indexes a BAM file. (E) The Delly software identifies structural variants in a cancer genome. Delly can be configured to exclude telomere and centromere regions as well as unplaced contigs. This example downloads an exclusion file, invokes Delly, and converts the output to VCF format. Examples (D) and (E) illustrate additional scenarios in which related tasks are executed as practical units.
Figure 5
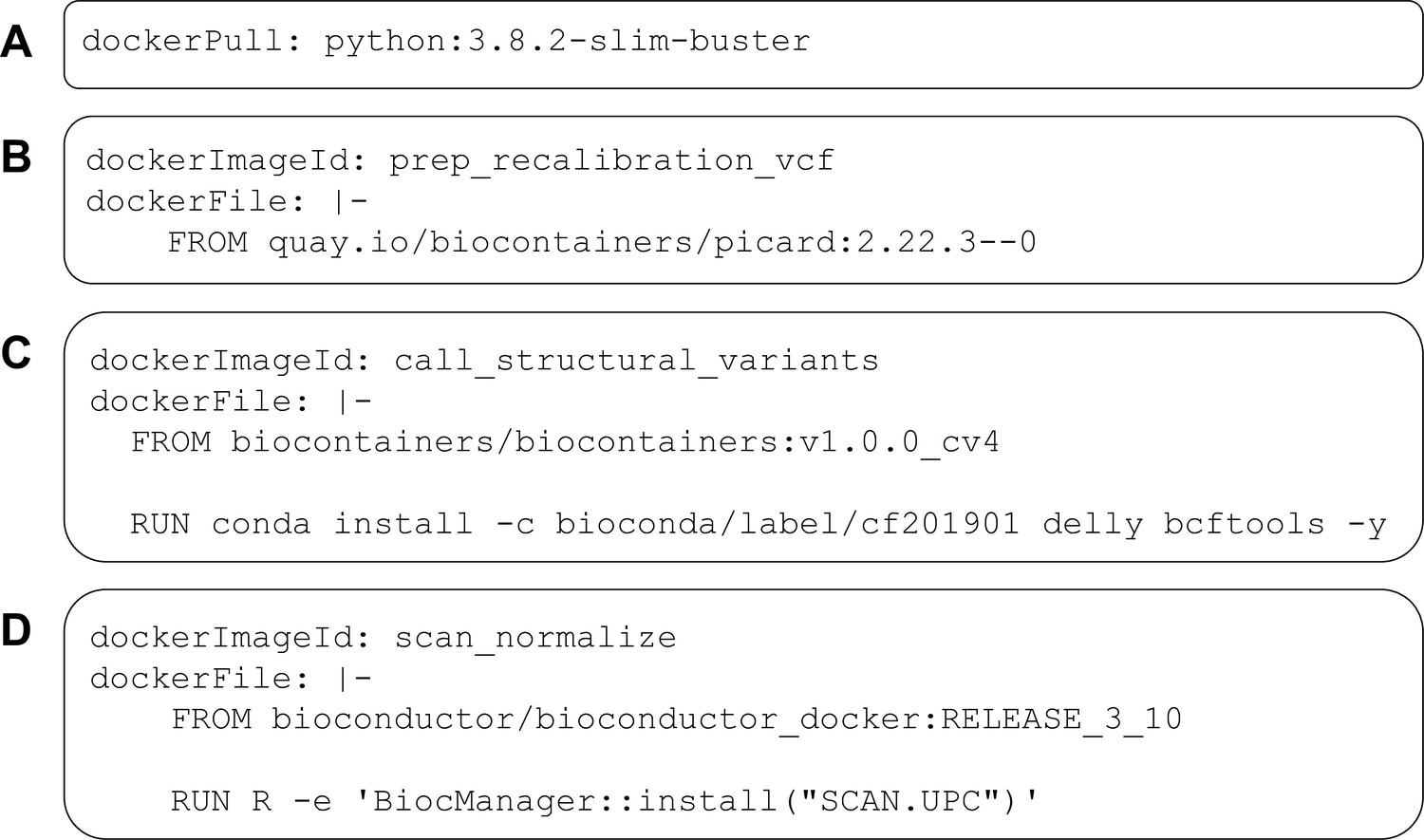
Examples of DockerRequirement specifications used in Common Workflow Language (CWL) tool descriptions.
These examples illustrate diverse ways to configure CWL tools to be executed in software containers. In (A), a container image is pulled from Docker Hub; this image encapsulates a minimal (‘slim’) version of Debian Linux 10.3 (‘buster’) and includes the Python 3.9 interpreter. In (B), the contents of a Dockerfile are included within the CWL description. In this case, the Dockerfile is simple—it pulls an existing image from https://quay.io. This image is provided as part of the BioContainers project and includes the Picard Tools software. (C) uses a base image from BioContainers and the Bioconda package manager to install the Delly and bcftools software within the image. (D) uses a base image from Bioconductor and executes R code to install the SCAN.UPC package within the image.
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Simplifying the development of portable, scalable, and reproducible workflows
eLife 10:e71069.
https://doi.org/10.7554/eLife.71069




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}