The right time for senescence
- Instituto de Medicina Molecular – João Lobo Antunes, Faculdade de Medicina da Universidade de Lisboa, Portugal
- CEDOC, NOVA Medical School, Faculdade de Ciências Médicas da Universidade Nova de Lisboa, Portugal
- Instituto de Medicina Molecular – João Lobo Antunes e Instituto de Histologia e Biologia do Desenvolvimento, Faculdade de Medicina da Universidade de Lisboa, Portugal
Figures
Figure 1
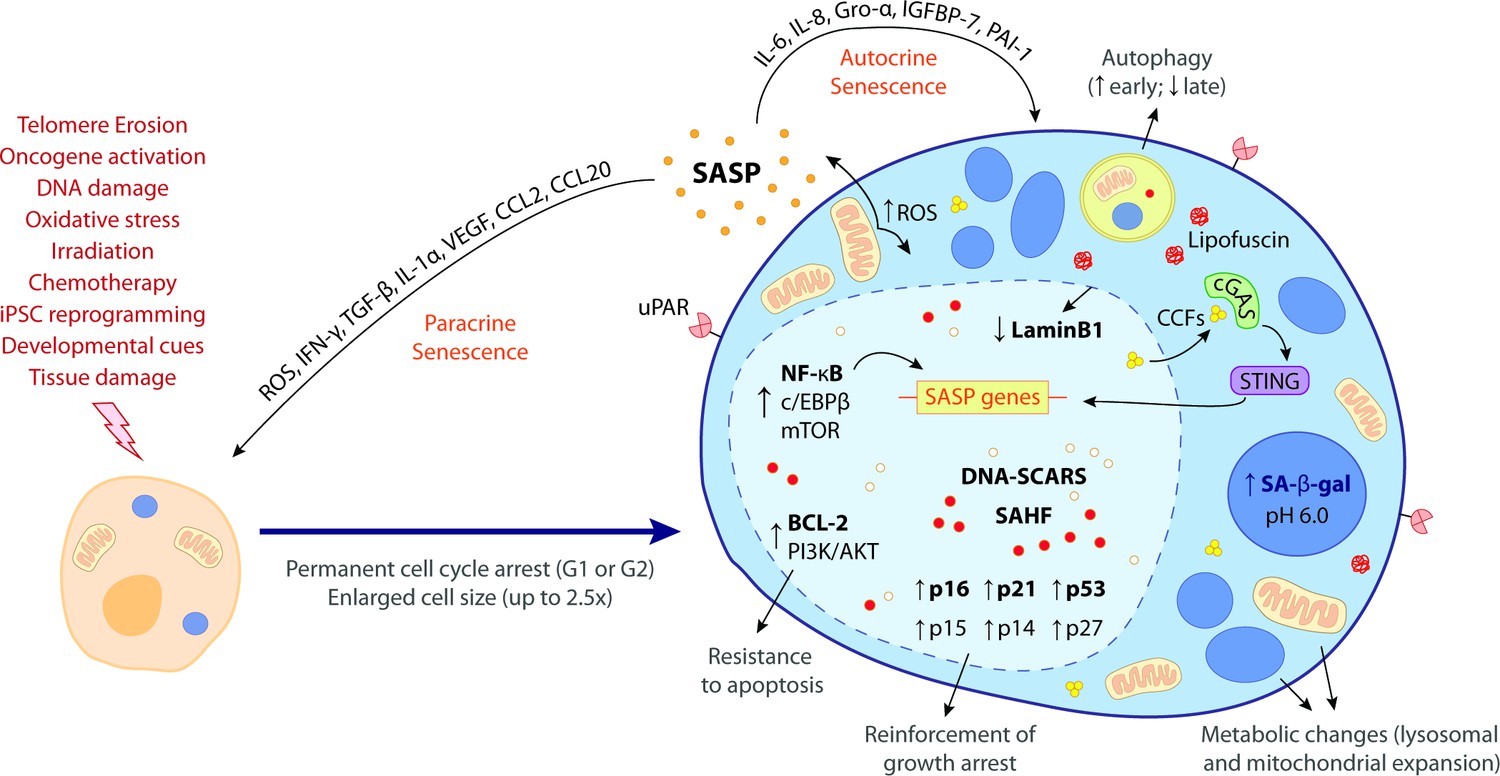
The hallmark features of cellular senescence.
Cells may be induced to senesce by different stimuli, including telomere erosion, oncogene activation (oncogene-induced senescence [OIS]), DNA damage, oxidative stress, irradiation, chemotherapy, induced pluripotent stem cell (iPSC) reprogramming, developmental cues, and tissue damage. These stresses trigger the upregulation of key cell cycle inhibitors, namely p16INK4a, p21CIP1, p53, p15INK4b, p14ARF, and p27KIP1, leading to a permanent cell cycle arrest (either in G1 or G2 phase). BCL-2 and PI3K/AKT anti-apoptotic pathways also become upregulated, bestowing senescent cells (SCs) with resistance to apoptotic cues. Other phenotypic alterations include an enlarged cell size (up to 2.5 times the normal counterpart) and metabolic changes such as mitochondrial and lysosomal expansion, which increase the production of reactive oxygen species (ROS) as well as the activity of lysosomal senescence-associated β-galactosidase (SA-β-gal, detected at pH 6.0). ROS accumulation induces protein aggregates which crosslink with sugars and lipids and form insoluble lipofuscin aggresomes. Autophagy rate is increased during the early senescence programme but is highly compromised in later stages. SCs are characterized by a complex senescence-associated secretory phenotype (SASP) that comprises a plethora of different growth factors, cytokines, chemokines, proteases, and matrix components. Primary transcriptional activators of the SASP include nuclear factor kappa B (NF-κB), CCAAT/enhancer binding protein β (c/EBPβ), and mammalian target of rapamycin (mTOR). SASP factors, such as IL-6, IL-8, Gro-α, IGFBP-7, and PAI-1, reinforce the senescence programme in an autocrine manner, while others, like ROS, IFN-γ, TGF-β, IL-1α, VEGF, CCL2, and CCL20, induce paracrine senescence in neighbouring cells. Upon the initiation of the senescence programme, the chromatin suffers deep modifications towards the repression of proliferation-related genes and the stimulation of SASP-related genes. This results in the appearance of DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) as well as senescence-associated heterochromatic foci (SAHF). Chromatin alterations are accompanied by a downregulation of lamin B1, a major component of the nuclear lamina, compromising its integrity and leading to the extravasation of chromatin fragments into the cytosol. Cytosolic chromatin fragments (CCFs) are recognized by cyclic GMP-AMP synthase (cGAS) which, in turn, triggers the activation of stimulator of interferon genes (STING). The cGAS-STING pathway stimulates pro-inflammatory SASP responses through upregulation of NF-κB. Recent studies suggest that urokinase-type plasminogen activator receptor (uPAR), a cell-surface protein, is also broadly expressed during senescence. The biomarkers in bold represent the most common hallmark features of cellular senescence. None of these hallmarks is exclusively specific and their manifestation can diverge according to the nature of the senescence trigger, the cell/tissue type and time of the senescence programme.
Figure 2
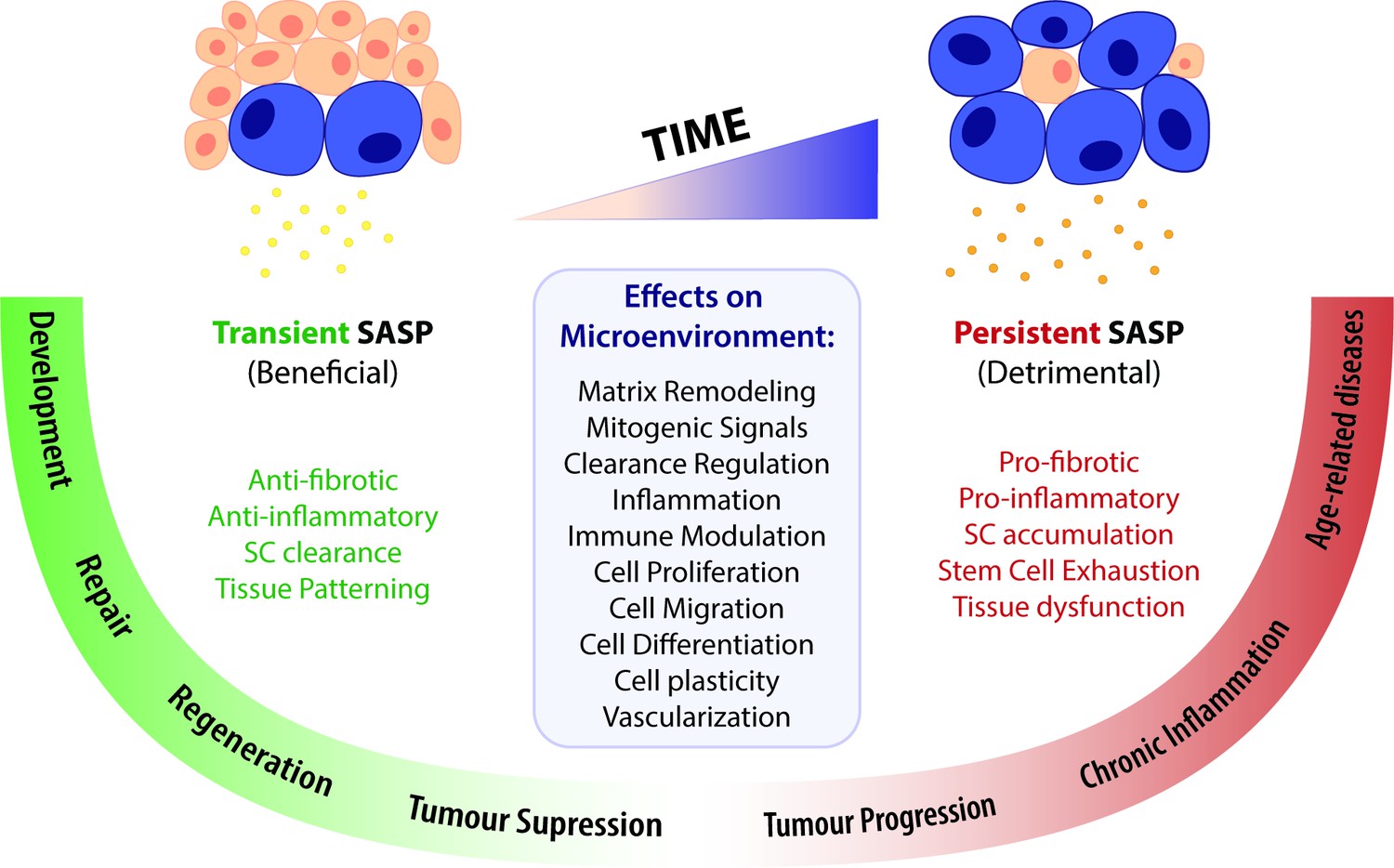
A time-gated model for the senescence-associated secretory phenotype (SASP)-mediated biological activities of cellular senescence.
The SASP can have a wide range of effects in the surrounding microenvironment, including matrix remodelling, mitogenic signalling, clearance regulation, inflammation, immune modulation, cell proliferation, migration, differentiation and plasticity, as well as vascularization. Depending on the time duration of the senescence programme and the associated SASP response, effects can be beneficial or detrimental. Anti-fibrotic and anti-inflammatory effects are normally correlated to a transient SASP and favour tissue repair and regeneration. A short-term SASP also favours immune-mediated clearance of SCs in order to avoid their accumulation and persistence. Likewise, a transient senescence profile is fundamental for tissue patterning during development. In contrast, long-lasting SASP responses have detrimental pro-fibrotic and pro-inflammatory effects on the microenvironment. Therefore, the persistent accumulation of SCs leads to tissue dysfunction and is associated with chronic inflammation and a broad spectrum of aging-related diseases. Persistent senescence responses can also deplete stem cell progenitor pools, impairing the repair/regenerative capability of affected tissues. In turn, the role of the SASP in cancer is more ambiguous than the rest, as SCs can both promote tumour suppression and tumour progression/invasiveness. However, current knowledge suggests that the SASP suppresses tumour growth in early stages, while supplying pro-tumourigenic chronic inflammatory environments in later stages.
Tables
Table 1
Senescence in tissue repair and regeneration.
| Species | Model (tissue/type of injury) | Role (beneficial vs. detrimental) | Profile (transient vs. persistent) | Main findings | Biomarkers | SASP factors (identified) | Reference |
|---|---|---|---|---|---|---|---|
| Mouse | Cutaneous wound healing | BeneficialAnti-fibrotic. | N/E | - CCN1 → DNA damage/p53 activation + ROS-dependent p16INK4a/pRB activation → fibroblast senescence → ↑ expression of antifibrotic genes- CCN1 mutant mice → ↓ senescence → exacerbated fibrosis | SA-β-Gal, p16INK4a, p53 | IL-6, IL-8, IL-11, MMP1, MMP3 | Jun and Lau, 2010 |
| Mouse | Cutaneous wound healing | BeneficialAnti-fibrotic.Promote differentiation. | Transient | - Fibroblast and endothelial cell senescence at the wound site → PDGF-AA secretion → myofibroblast differentiation- Elimination of p16INK4a+ cells → delayed wound healing + ↑ fibrosis | SA-β-Gal, p16INK4a, p21CIP1,↓ laminB1 | PDGF-AA, VEGF, PAI-1, CCL5, and CCL2 | Demaria et al., 2014 |
| Mouse | Chronic liver damage | BeneficialAnti-fibrotic.Recruitment of immune system. | N/E | - CCl4 administration → activation of CCN1/p53/p16INK4a pathway → hepatic stellate cell senescence → secretion of MMPs → fibrotic resolution → recruitment of NK cells to promote their own clearance- p53;INK4A/ARF null mice → ↓ senescence upon CCl4 treatment → ↑ cirrhosis | SA-β-Gal, p21CIP1, p53, HMGA1 | MICA, IL-8, ULBP2, CD58,MMP1, MMP3 | Krizhanovsky et al., 2008 |
| Mouse | Heart infarction | BeneficialAnti-fibrotic. | Transient | - CCN1-dependent cardiac myofibroblast senescence → ↓ fibrosis → improved heart function- p53 deficiency → ↑ collagen deposition | SA-β-Gal, p16INK4a, p21CIP1, p53, p19ARF | MMP2, MMP9, IL-6, IL-11, CXCL1 | Zhu et al., 2013 |
| Mouse | Cardiac hypertrophy | BeneficialAnti-fibrotic. | N/E | - CCN1-dependent cardiac myofibroblast senescence → ↓ fibrosis → improved heart function- SC genetic ablation → ↑ fibrosis | SA-β-Gal, p16INK4a, p21CIP1, | N/E | Meyer et al., 2016; |
| Mouse | Cardiac ischaemia-reperfusion injury | Detrimental.Pro-fibrotic.Pro-inflammatory. | Persistent | - Oxidative stress → Cardiomyocyte and interstitial cell senescence → ↑ fibrosis and inflammation → impaired heart function- SC clearance → ↓ scar size and inflammation + ↑ myocardial vascularization → improved heart function | SA-β-Gal, p16INK4a, p21CIP1, | IP-10, TGF-β3, IL-6, IL-11, IL-16, CCL22, MIP-3β, CX3CL1 | Dookun et al., 2020 |
| Human/mouse | Ischemic retinopathy | BeneficialRecruitment of immune system.Pro-regenerative. | Transient | - Rapid proliferation of vascular cells → ↑ endothelial cell senescence → cytokine secretion+ neutrophil recruitment → formation of neutrophil extracellular traps (NETs) → removal of SCs → regression of pathological angiogenesis + regeneration of functional vessels- Inhibition of NETosis / neutrophil inactivation → impaired SC clearance → no regression of pathological angiogenesis | SA-β-Gal, PML | CXCL1, IL-1β | Binet et al., 2020 |
| Mouse | Idiopathic pulmonary fibrosis (IPF) | Detrimental.Promote differentiation.Pro-fibrotic (indirectly). | Persistent | - Accumulation of senescent epithelial cells and fibroblasts → myofibroblast differentiation → ↑ fibrosis- SC elimination (genetic ablation and pharmacological) → improved pulmonary function and physical health | P16INK4a, p53, γH2AX | PAPPA, IGFBP2, IGFBP4, COL1A1, MMP10, MMP12, VCAM1, MCP1, PAI1, TNF-α | Schafer et al., 2017 |
| Salamander | Limb amputation | N/E | Transient | - Turnover of SCs (unidentified) at injury site during regeneration | SA-β-Gal(in vivo) | N/E(in vivo) | Yun et al., 2015 |
| Zebrafish/mouse (P1) | Heart injury | N/E | Transient | - Turnover of senescent cardiac fibroblasts during regeneration | SA-β-Gal, p53 | N/E | Sarig et al., 2019 |
| Zebrafish | Caudal fin amputation | Beneficial. | Transient | - Turnover of SCs (unidentified) at injury site during regeneration.SC removal → impaired regeneration | SA-β-Gal, p21CIP1 | N/E | Da Silva‐Álvarez et al., 2019 |
| Mouse | Acute muscle injury | Beneficial.Anti-inflammatory.Pro-regenerative. | Transient | - Senescence of fibro-adipogenic progenitors (FAPs) → cytokine secretion → anti-inflammatory environment → muscle regeneration | SA-β-Gal, γH2AX, p16INK4a, p53 | TSG-6IL-33 | Saito et al., 2020 |
| Mouse | Biliary injury | Detrimental.Pro-fibrotic.Pro-inflammatory.Paracrine senescence. | Persistent | - Senescent cholangiocytes → TGF-β secretion → paracrine senescence of cholangiocytes and hepatocytes → ↑ fibrosis and inflammation → impaired liver regeneration- Inhibition of TGF-β-signaling → disruption of paracrine senescence → restoration of liver function | p16INK4a, p21CIP1, DCR2,γH2AX, p27, p53 | TGF-β, IL1α | Ferreira-Gonzalez et al., 2018 |
| Zebrafish | Spinal cord injury | N/E | Transient | - Turnover of senescent neurons at lesion periphery during regeneration | SA-β-Gal, p21CIP1,No BrdU | N/E | Paramos-de-Carvalho et al., 2021 |
| Mouse | Spinal cord injury | Detrimental.Pro-inflammatory.Pro-fibrotic. | Persistent | - Continuous accumulation of senescent neurons at lesion periphery.- SC pharmacological depletion → ↑ myelin and axonal sparing + ↓ fibrosis and inflammation → improved motor and sensory functions | SA-β-Gal, p16INK4a,γH2AX | Amphiregulin, PDGF-BB, IGFBP-3, Serpin E1, IL-15, TNF- α, M-CSF, I-TAC, CCL11, ICAM-1, CCL20 | Paramos-de-Carvalho et al., 2021 |
-
N/E - not explored.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The right time for senescence
eLife 10:e72449.
https://doi.org/10.7554/eLife.72449




{kind=link}
{kind=link}