Admixture of evolutionary rates across a butterfly hybrid zone
- Department of Organismic and Evolutionary Biology, Harvard University, United States
- Kunming Institute of Zoology, Chinese Academy of Sciences, China
- The University Museum, The University of Tokyo, Japan
Figures
Figure 1
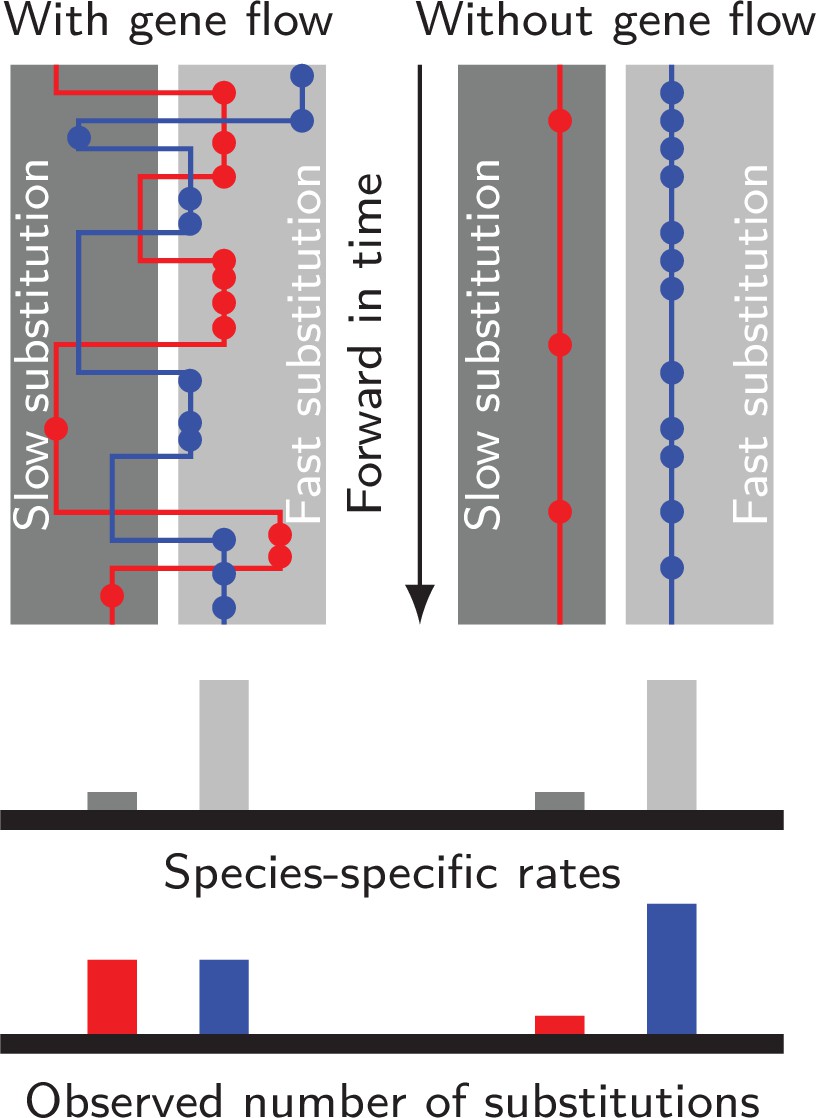
Gene flow interacts with divergent substitution rates and affects observed numbers of substitutions.
Each gray block represents a species with its species-specific substitution rate. Solid lines represent gene genealogies prior to coalescence, and horizontal jumps between species represent inter-specific gene flow. Dots are substitutions. When a gene sequence inherits mutations derived under multiple rates, the number of substitutions it carries will reflect a mixture of substitution rates among different species. If gene flow is strong, each lineage carries a similar number of substitutions; If gene flow is weak, genes evolve independently with species-specific rates, and the distribution of substitutions in each lineage will likely be skewed towards the distribution of species-specific substitution rates.
Figure 2 with 4 supplements
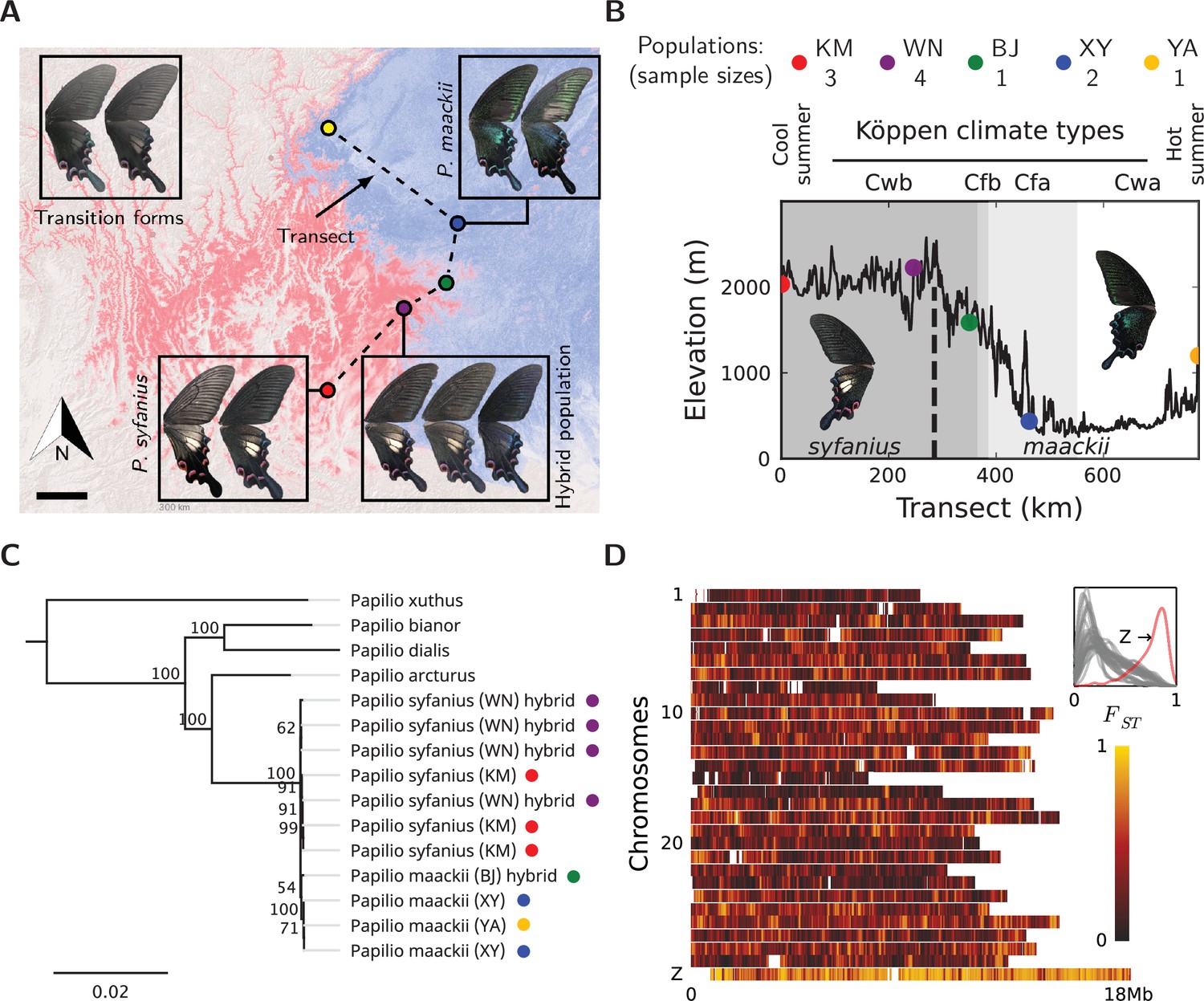
Overview of the study system.
(A) The geographic distribution of P. syfanius (red) and P. maackii (blue). Scale-bar: 100 km. Dashed line is the sampling transect covering five populations (colored circles). (B) Elevation, climate, and sample sizes along the transect. (C) Mitochondrial tree with four outgroups. (D) across chromosomes (50 kb windows with 10 kb increments). The inset shows the estimated density of on each chromosome.
-
Figure 2—source data 1
Estimated between populations KM and XY (50 kb windows with 10 kb increments).
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig2-data1-v3.zip
-
Figure 2—source data 2
and estimated for populations KM and XY (non-overlapping 10 kb windows).
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig2-data2-v3.zip
-
Figure 2—source data 3
The phylogeny and alignment among mitochondrial genomes.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig2-data3-v3.zip
-
Figure 2—source data 4
Results and data for MaxEnt species distribution models.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig2-data4-v3.zip
-
Figure 2—source data 5
Sample information.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig2-data5-v3.zip
Figure 2—figure supplement 1

Polymorphic mimetic color patterns in P. syfanius.
The mimicry polymorphism in P. syfanius. The occurrence of spotted and spotless forms matches local forms of poisonous swallowtails in the genus Byasa.
Figure 2—figure supplement 2
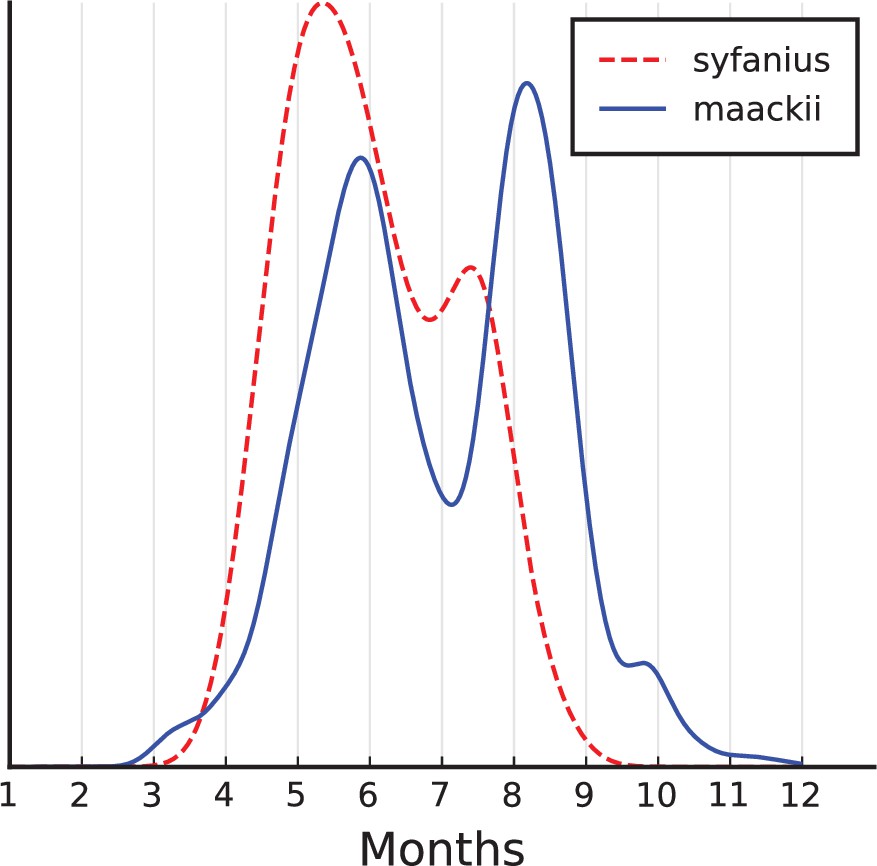
Estimated seasonal occurrence times of P. syfanius and P. maackii adults.
Seasonal distributions of adults. Records with verifiable time information were used (must have months, but we also kept records with missing days or years). Records explicitly mentioning larvae, pupae or eggs were removed, and records from December to February were also removed since adults do not occur in winter. Together, there were 62 P. syfanius records and 3138 P. maackii records passing all filters. Then we estimated kernel density of variable (month +day/30). If day is missing, it was randomly picked among 1–30. Results show the average kernel density from 1000 repeated estimations.
Figure 2—figure supplement 3
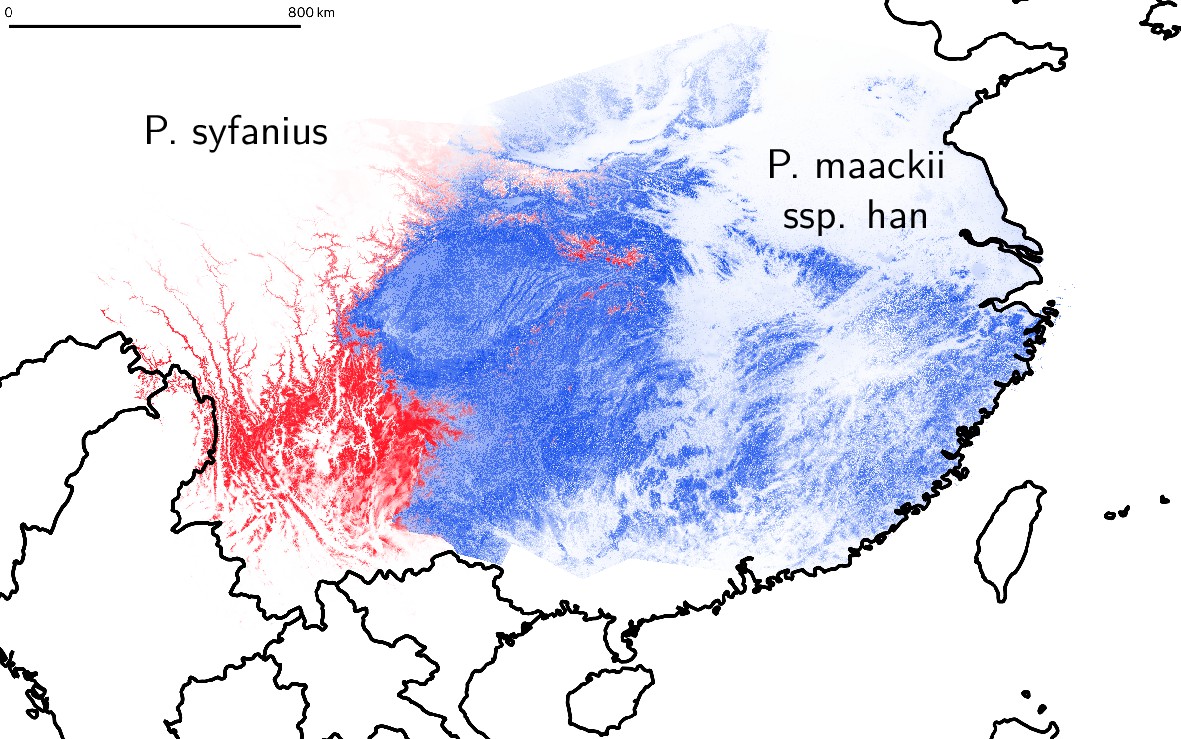
Estimated geographic distribution of P. syfanius and P. maackii ssp. han.
Estimated geographic distribution of P. syfanius (red) and P. maackii ssp. han (blue) predicted by MaxEnt. Scale-bar represents 800 km.
Figure 2—figure supplement 4

Window-based and estimated for populations KM and XY.
and on chromosomes. This series of plots include genetic diversity (red: population KM; blue: population XY) and absolute sequence divergence (gray: between populations XY and KM). and are calculated on 10 kb non-overlapping windows. The horizontal axis has range (0 bp, bp), and the vertial axis has range (0, 0.03). Empty regions are masked due to abnormal coverage.
Figure 3 with 1 supplement
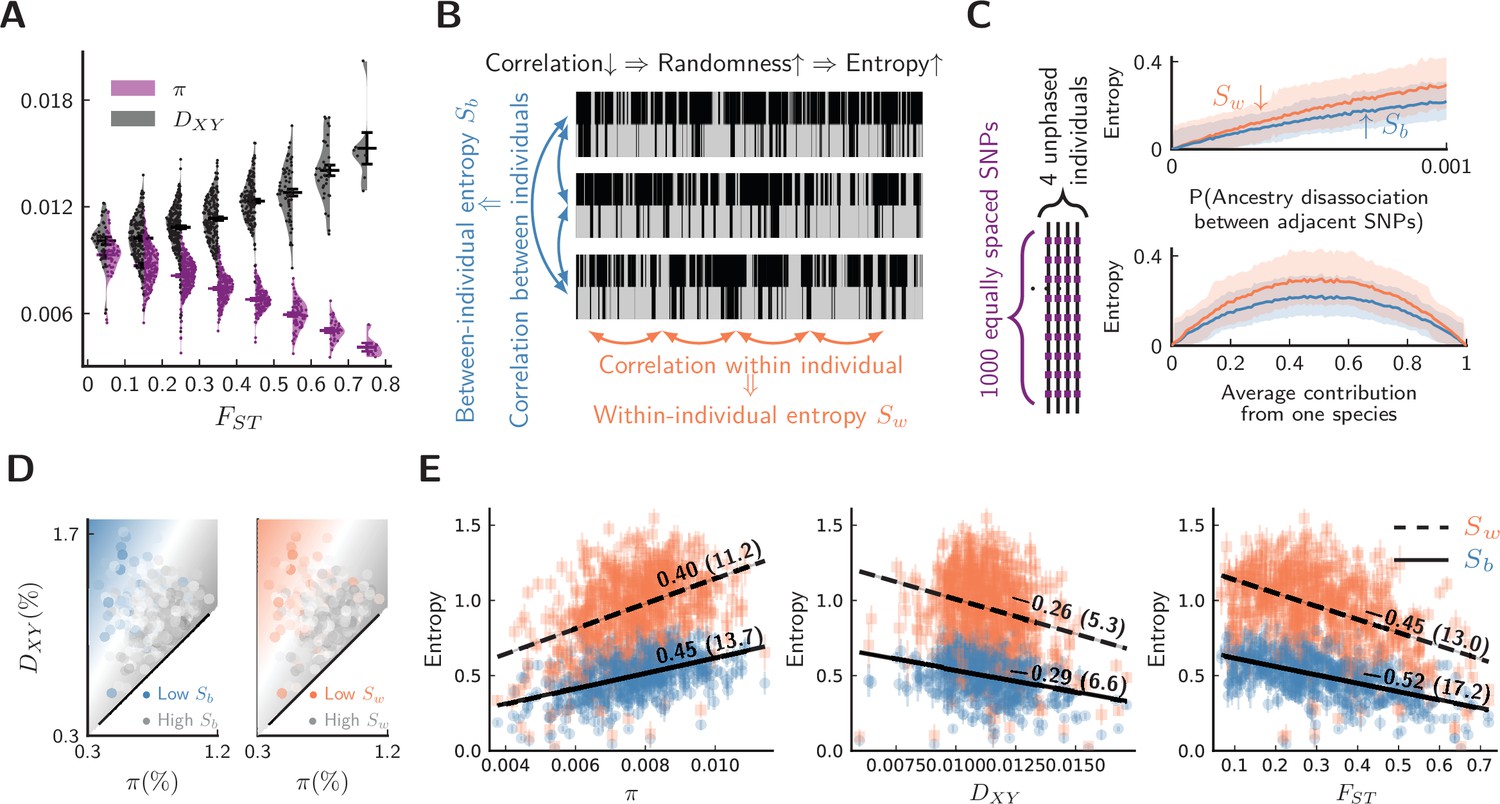
Evidence for barrier loci on autosomes.
For all plots, pure populations refer to XY & KM, and hybrid population refers to WN. (A) Between pure populations, reduced diversity (, showing values averaged between pure populations) is associated with increased divergence () across autosomes (30 segments per chromosome). Error bars are standard errors. (B) The conceptual picture of entropy metrics on diploid, unphased ancestry signals. In a genomic window, between-individual entropy () measures local ancestry randomness among individuals, while within-individual entropy () measures ancestry randomness along a chromosomal interval. (C) Simulated behaviors of entropy in a simplified model of biparental local ancestry. Chromosomes are assumed to be spatially homogeneous, thus recombination rate is uniform among 1000 equally spaced SNPs, and adjacent SNPs have a single probability of ancestry disassociation. For each haplotype block with linked ancestry, its ancestry is randomly assigned according to the average contribution from each species. Each pair of haploid chromosomes are combined into an unphased ancestry signal before calculating entropy. The top plot assumes equal contribution from both species, and the bottom plot assumes ancestry disassociation probability = 0.001. Solid lines are average entropies across 1000 repeated simulations, and shaded areas represent averaged upper and lower deviations from the mean. (D) The joint range among entropy, , and across autosomes (20 segments per chromosome). Color range is normalized by the range of entropy in each plot. Gray represents higher entropy, and colored regions are associated with lower entropy. Heatmaps represent linear fits to the ensembles of points. (E) The correlation on autosomes between entropy in hybrid populations and {, , } within and between pure populations. is shown above each regression line. Error bars are standard errors of entropy from 50 repeated estimates of local ancestry using software ELAI (parameters are in Materials and methods). The significance of is estimated using block-jackknifing among all segments: -scores are shown in parentheses.
-
Figure 3—source data 1
Estimated entropy (, ), , , on 20 segments per chromosome (including the Z chromosome).
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig3-data1-v3.zip
Figure 3—figure supplement 1

An example run of the local ancestry estimation software ELAI on chromosomes 1–30.
An example run of the local ancestry estimation software ELAI on chromosomes 1–30 of four diploid individuals from the hybrid population WN (generation parameter: 5000). Gray represents the ancestry contribution from P. maackii.
Figure 4 with 1 supplement
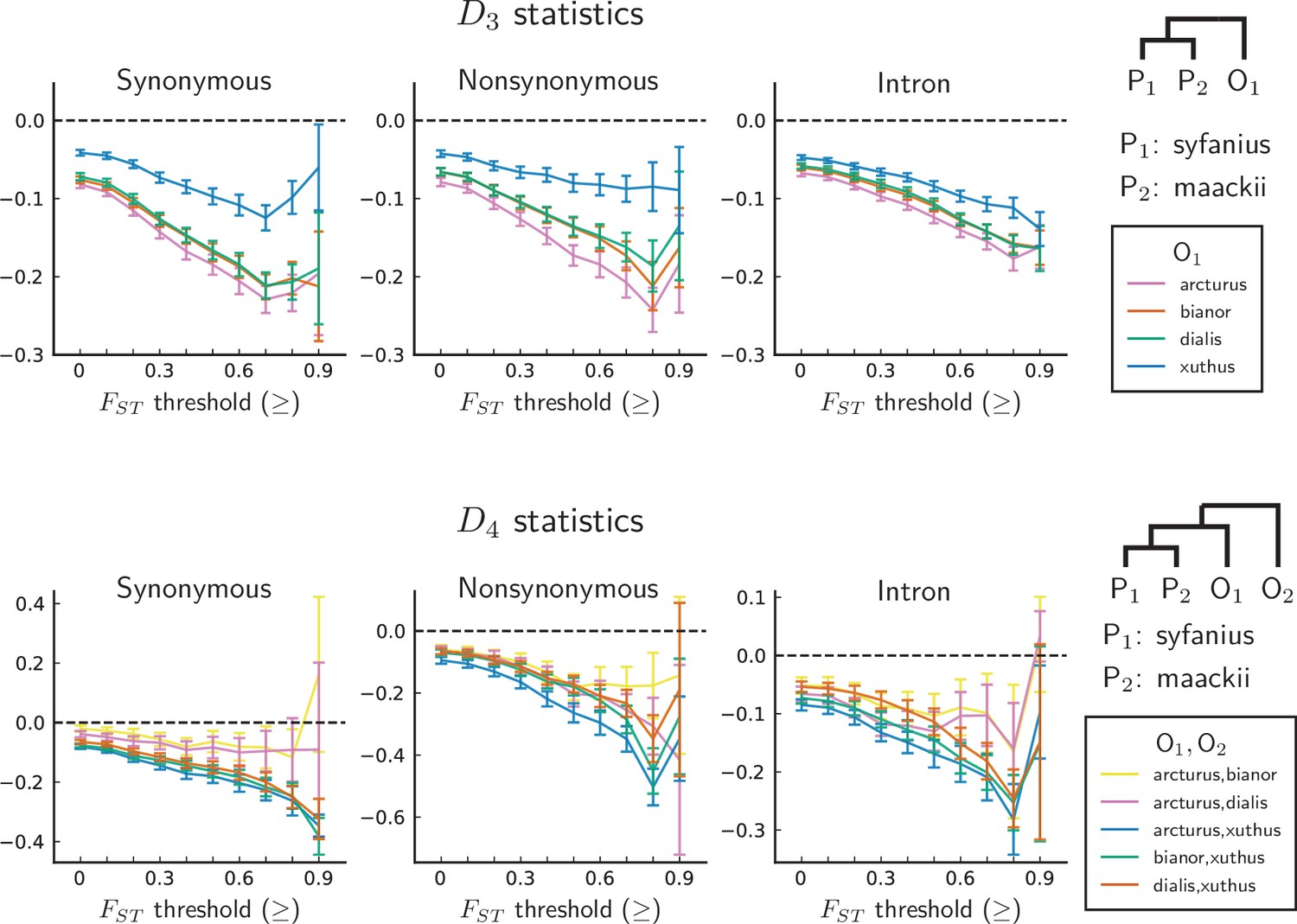
statistics are unanimously negative.
For each data point, we choose an threshold (x-axis) and report statistics on SNPs with a background (50 kb windows and 10 kb increments) no less than the given threshold. Error bars are standard errors estimated using block-jackknife with 1 Mb blocks. The aberrant behavior for the highest bins is we believe mostly due to low sample sizes for these bins.
-
Figure 4—source data 1
, statistics with their -scores.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig4-data1-v3.zip
Figure 4—figure supplement 1
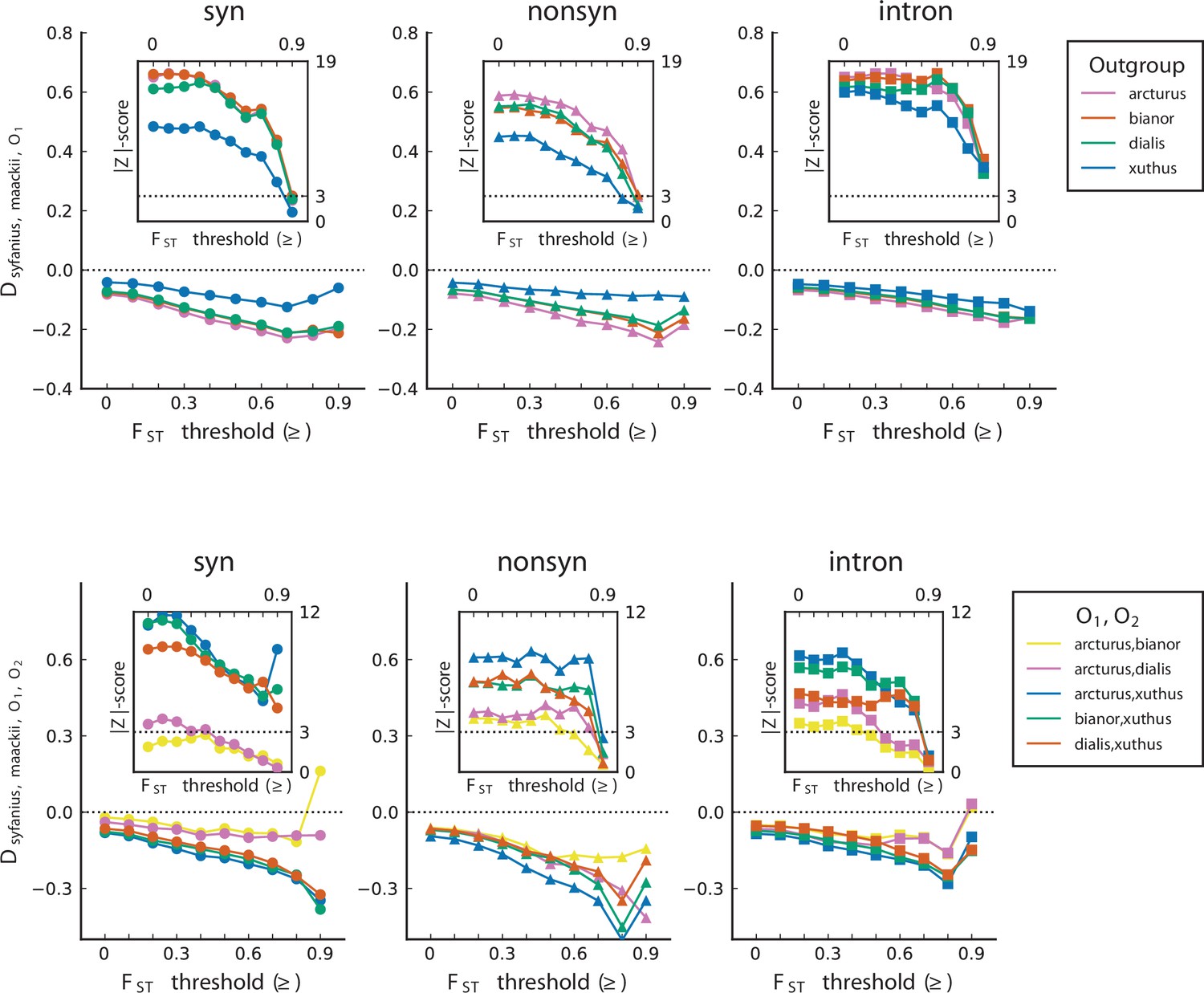
, statistics plotted with their -scores.
Figure 5
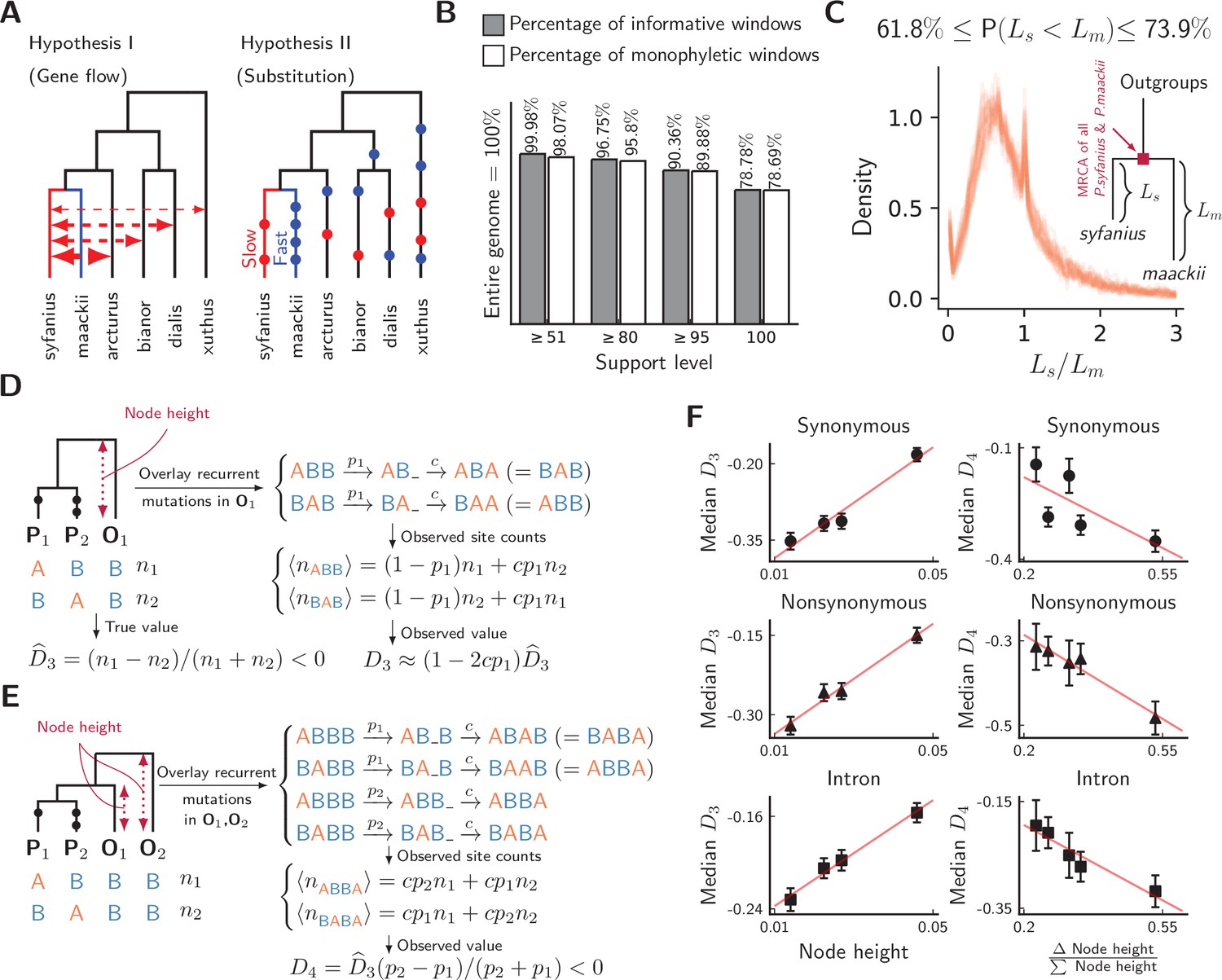
Unequal substitution rates between pure population of P. syfanius and P. maackii.
(A) Two hypotheses to explain negative statistics. (B) The percentage of local gene trees (50 kb non-overlapping windows) where P. syfanius and P. maackii are together monophyletic. For each level of support, we filter out windows with Support(monophyly) and Support(paraphyly) below a given level, and report both the percentage of windows passing the filter (informative windows) and the percentage of monophyletic windows. (C) The distribution of P. syfanius branch lengths () relative to those of P. maackii () among highly supported monophyletic trees (Support ≥95). Each curve corresponds to a pairwise comparison between a syfanius individual and a maackii individual. Branch lengths are distances from tips to the most-recent common ancestor of all syfanius +maackii individuals. (D) Behavior of under divergent substitution rates and recurrent mutations in outgroups (O1). (E) Behavior of under divergent substitution rates and recurrent mutations in outgroups (O1 & O2). (F) Left: Median is positively correlated with node height; Right: Median is negatively correlated with . Node height is used as a proxy for the probability of recurrent mutation ().
-
Figure 5—source data 1
Concatenated gene trees based on 50 kb windows and the underlying support for each binary split.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig5-data1-v3.zip
-
Figure 5—source data 2
Results of the signed-rank test on branch elongation in P. maackii.
- https://cdn.elifesciences.org/articles/78135/elife-78135-fig5-data2-v3.zip
Figure 6 with 3 supplements
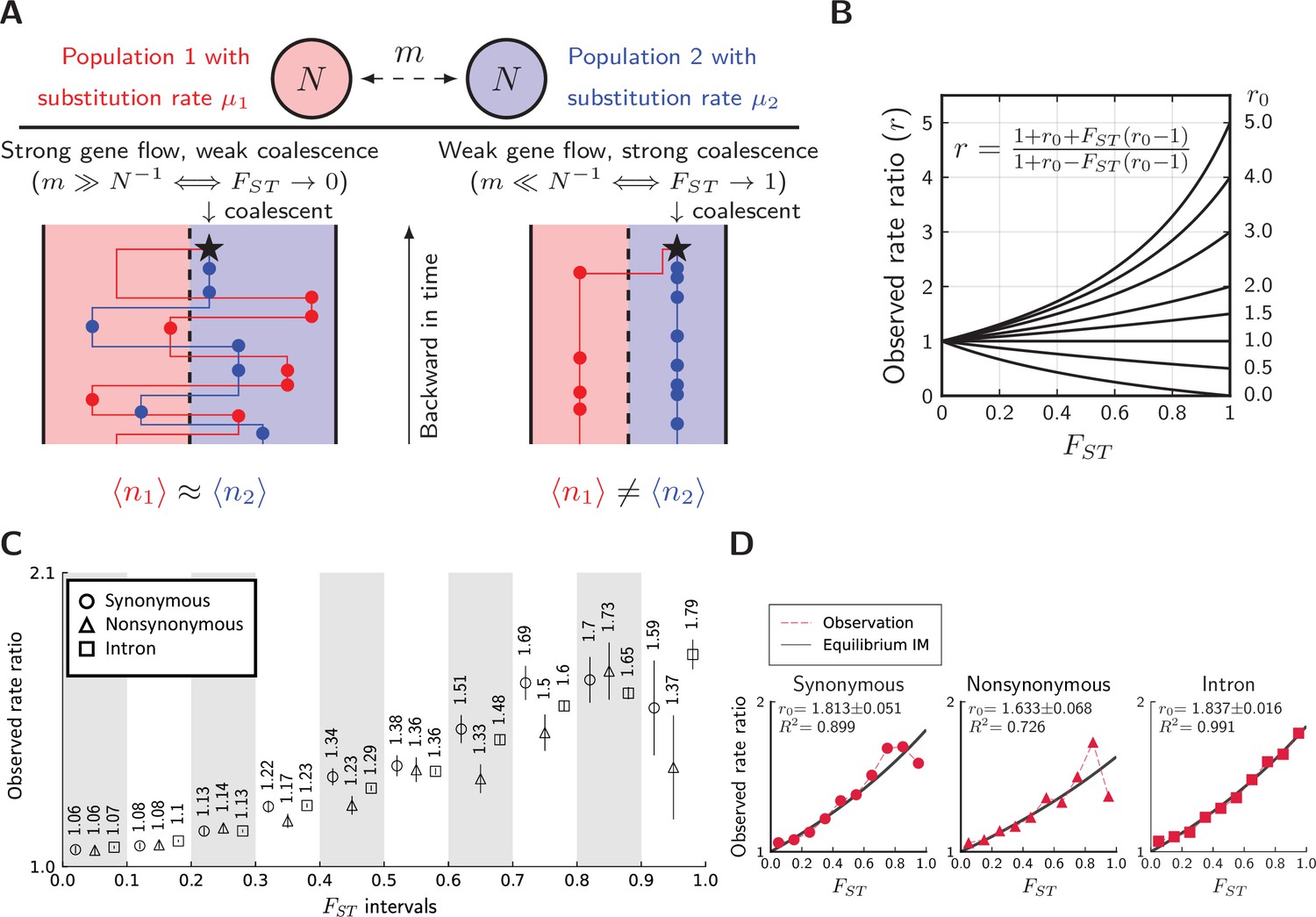
Divergence is correlated with increased differences in the relative number of substitutions.
(A) Behavior of the equilibrium isolation-with-migration model with divergent substitution rates. If coalescence is weaker than gene flow, each lineage has a similar number of derived alleles. If coalescence is stronger than gene flow, lineages sampled from the population with a faster substitution rate will also inherit more derived alleles. (B) Theoretical relationship between observed rate ratio () and relative divergence (), parameterized by the true ratio r0) of substitution rates. (C) Observed rate ratios between P. maackii (population XY) and P. syfanius (Population KM), partitioned by ten intervals. Error bars are standard errors calculated using 1 Mb block-jackknifing. (D) The theoretical relationship between and is a good fit to observation.
Figure 6—figure supplement 1
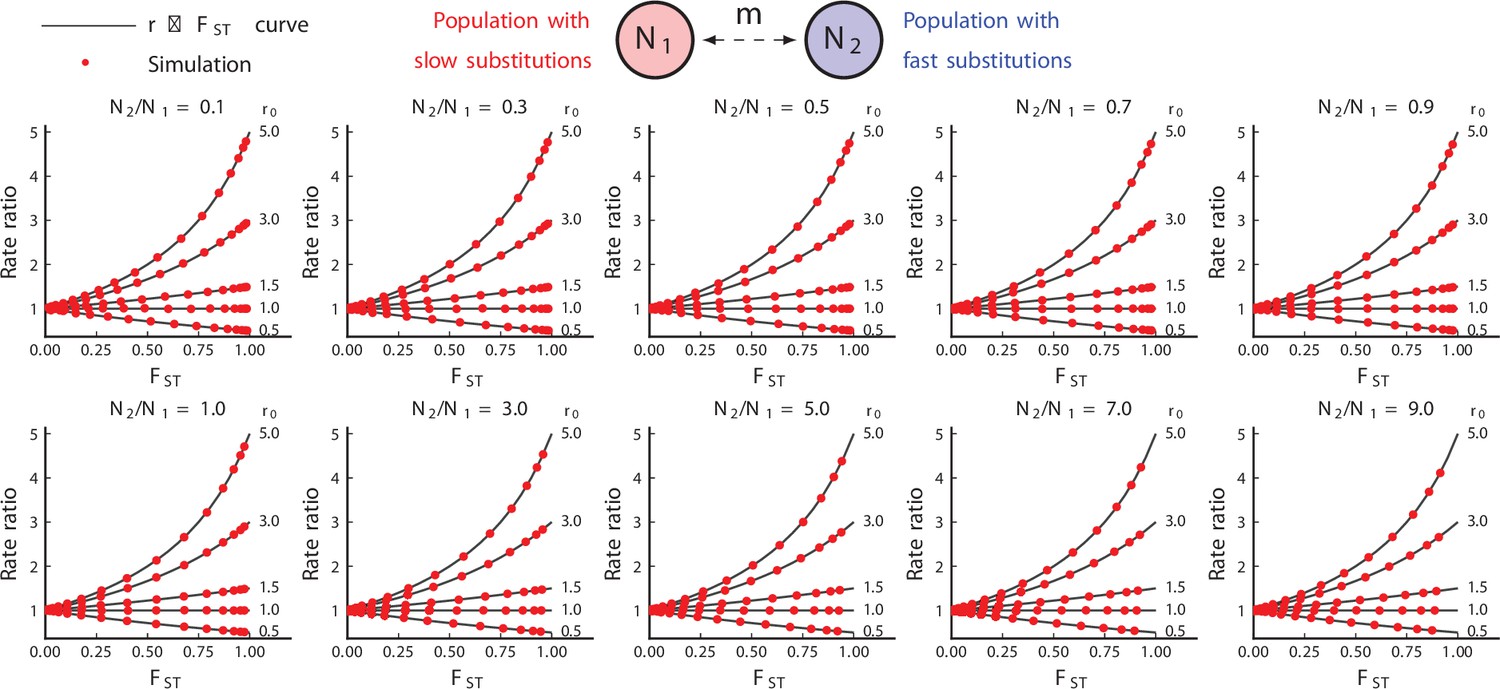
Simulated results from equilibrium symmetric migration models of rate-mixing.
Symmetric migration models. Parameters: , (substitution rate in the red species), . Substitution rate in the blue species is determined by . Theoretical curves appear to be exact. (106 repetitions).
Figure 6—figure supplement 2

Simulated results from equilibrium conservative migration models of rate-mixing.
Conservative migration models. Parameters: , (substitution rate in the red species), . Substitution rate in the blue species is determined by . Theoretical curves are still robust, although less accurate (106 repetitions).
Figure 6—figure supplement 3
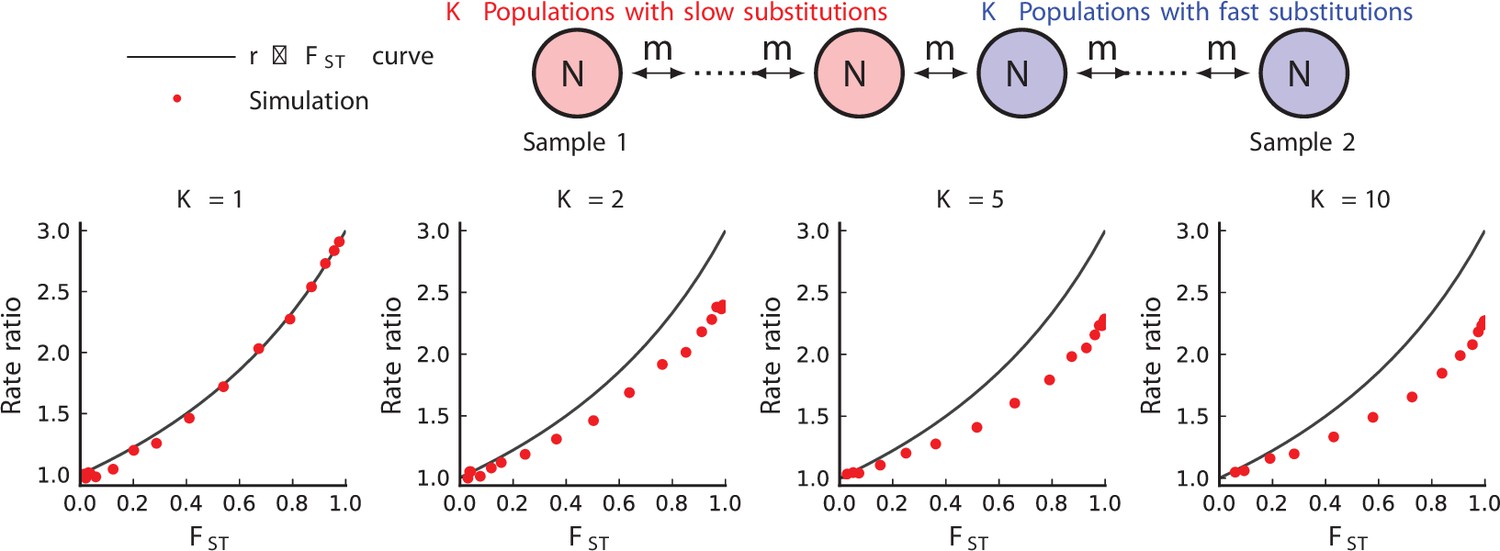
Simulated results from equilibrium stepping-stone models of rate-mixing.
Stepping-stone models. Parameters: , (substitution rate in the red species), . The blue species is three times slower in substitution. Within-species population structure dampens observed relationship below the theoretical curve (104 repetitions).
Author response image 1
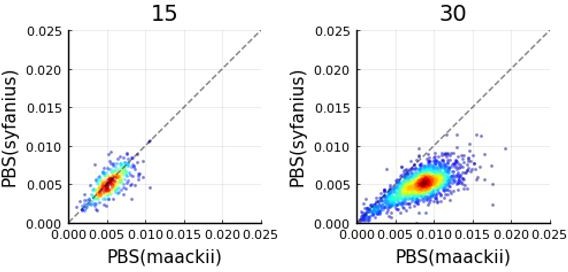
Tables
Table 1
Coverage abnormality in SNPs from coding sequences (CDSs).
Abnormal coverage is inferred if the average coverage of a CDS exceeds twice of the median coverage of all CDSs in the genome.
| Number of SNPs from CDSs with abnormal coverage | 1993 |
| Total number of SNPs from all CDSs | 1060525 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Admixture of evolutionary rates across a butterfly hybrid zone
eLife 11:e78135.
https://doi.org/10.7554/eLife.78135




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}