Trio-based whole exome sequencing in patients with suspected sporadic inborn errors of immunity: A retrospective cohort study
- Department of Human Genetics, Radboud Institute of Molecular Life Sciences (RIMLS), Radboud University Medical Center, Netherlands
- Department of Laboratory Medicine, Laboratory for Medical Immunology, Radboud University Medical Center, Netherlands
- Department of Genetics, University of Groningen, University Medical Center Groningen, Netherlands
- Department of Pediatric Infectious Diseases and Immunology, Amalia Children’s Hospital, Radboud Center for Infectious Diseases (RCI), Radboud University Medical Center, Netherlands
- Department of Pediatric Rheumatology and Immunology, Amalia Children’s Hospital, Radboud University Medical Center, Netherlands
- Department of Clinical Genetics, Maastricht University Medical Center+, Netherlands
- Center for Human and Clinical Genetics, Leiden University Medical Center, Netherlands
- Department of Internal Medicine and Radboud Center for Infectious Diseases (RCI), Radboud University Medical Center, Netherlands
- Department for Immunology and Metabolism, Life and Medical Sciences Institute (LIMES), University of Bonn, Germany
- Department of Laboratory Medicine, Laboratory for Diagnostics, Radboud University Medical Center, Netherlands
Figures
Figure 1 with 1 supplement
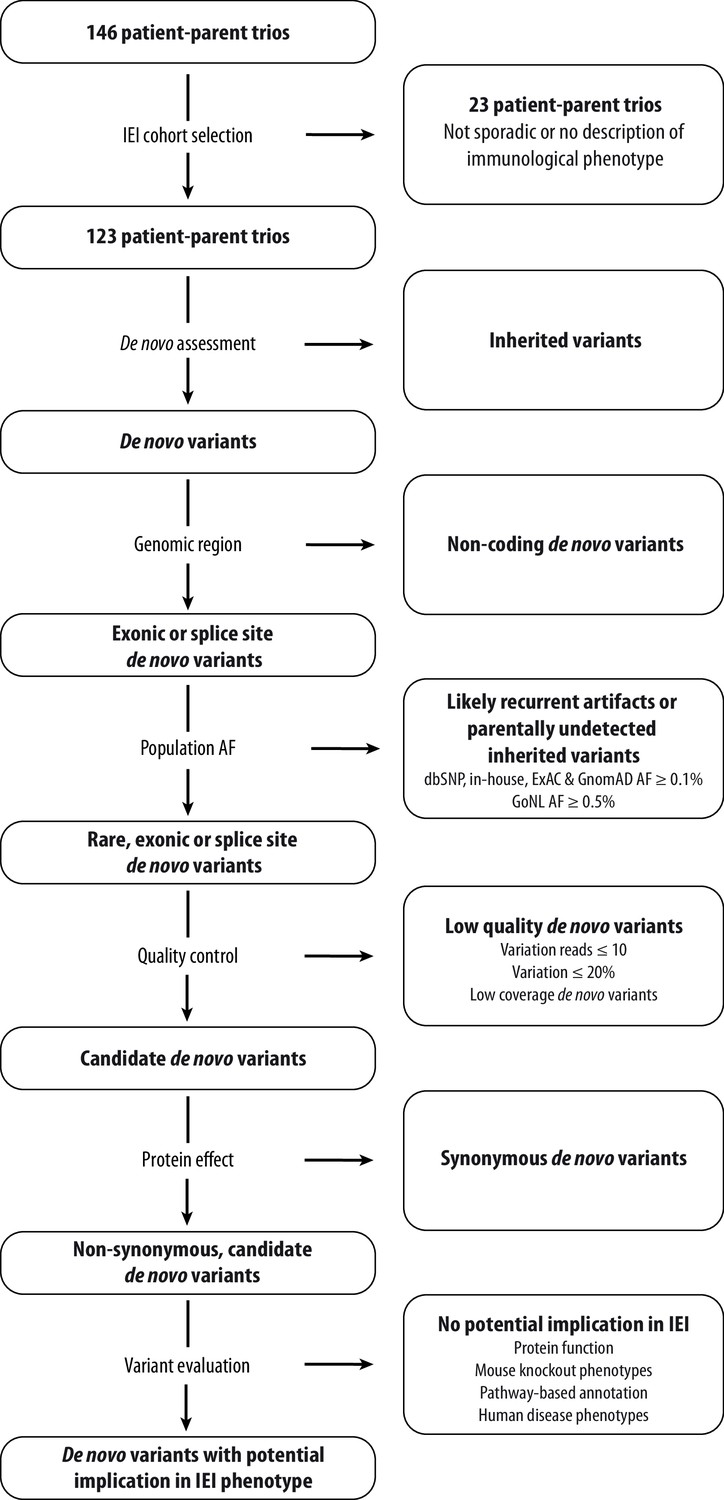
Schematic overview of patient inclusion, de novo variant filtering strategy and variant evaluation.
Of the 146 eligible patient-parent trios, 123 trios met the inclusion criteria for this IEI cohort study. Whole exome sequencing data from these patient-parent trios was filtered to retain rare, non-synonymous candidate de novo variants in coding regions. Subsequently, variants were systematically evaluated at variant and gene level for their potential involvement in the patient’s immunological phenotype. Abbreviations: IEI = inborn errors of immunity; dbSNP = Single Nucleotide Polymorphism Database; ExAC = Exome Aggregation Consortium; GnomAD = Genome Aggregation Database; AF = allele frequency; GoNL = Genome of the Netherlands.
-
Figure 1—source code 1
R script for de novo variant filtering.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig1-code1-v2.zip
-
Figure 1—source data 1
List of 123 patient-parent trios with patient characteristics and whole exome sequencing performance statistics.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig1-data1-v2.docx
-
Figure 1—source data 2
List of all candidate rare, coding de novo variants found in the cohort of IEI patients.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig1-data2-v2.docx
-
Figure 1—source data 3
De novo variant rate and distribution of de novo variant types across our IEI cohort in comparison to a reference cohort from Kaplanis et al., 2020.
The total amount of candidate de novo variants was retrieved from our cohort, consisting of 123 individuals. All identified de novo variants with at least 20% variation reads of the Kaplanis et al., 2020. cohort were obtained. The amount of predicted loss-of-function, synonymous and non-synonymous de novo single nucleotide variants or small insertion-deletions were extracted from both cohorts. The total amount of all, predicted loss-of-function, synonymous and non-synonymous de novo variants were divided by the respective cohort size to obtain the average number of respective variants per individual for each cohort.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig1-data3-v2.docx
Figure 1—figure supplement 1
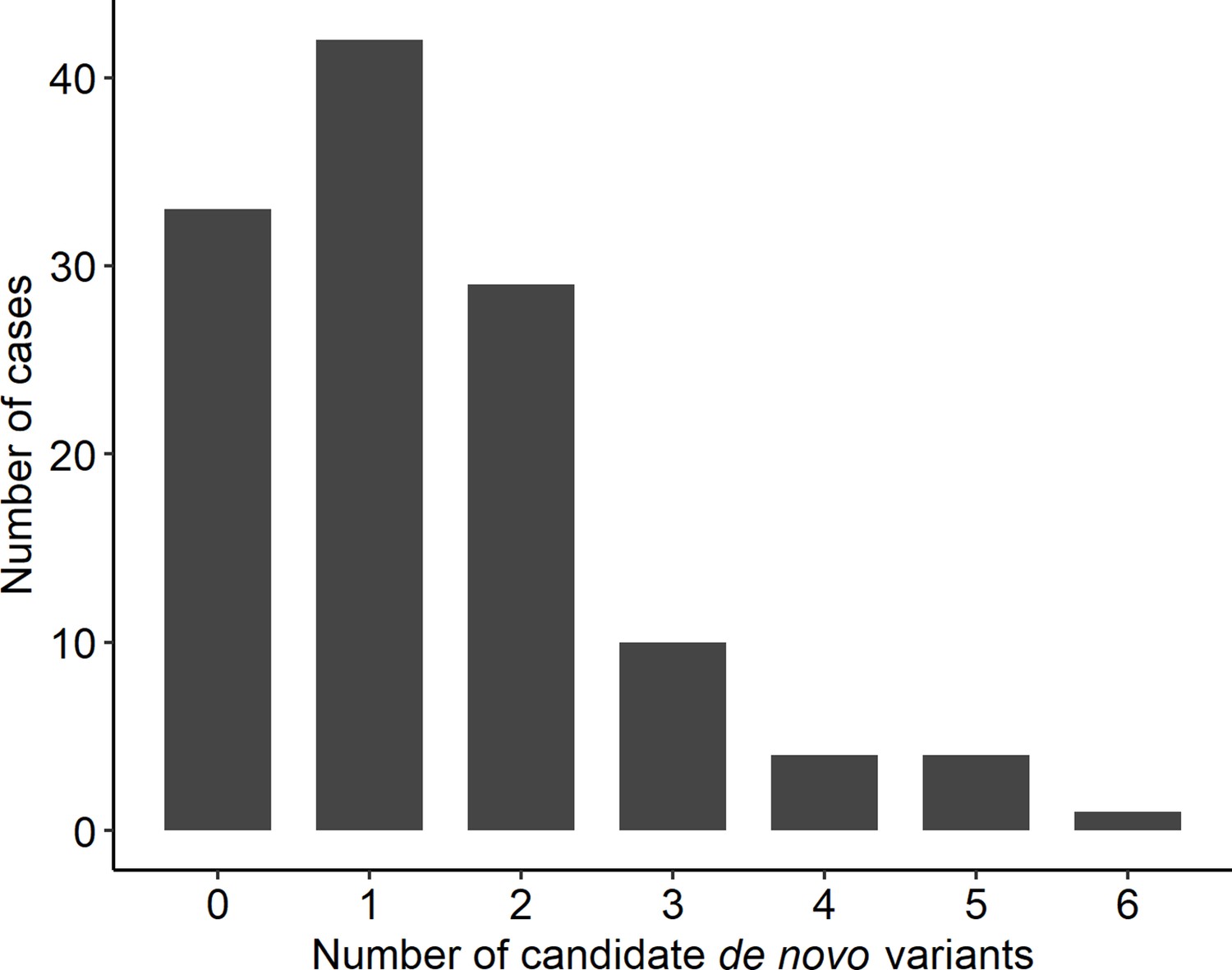
Distribution of rare, non-synonymous coding de novo variants among cases.
-
Figure 1—figure supplement 1—source data 1
Number of candidate de novo variants per case.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig1-figsupp1-data1-v2.xlsx
Figure 2 with 1 supplement
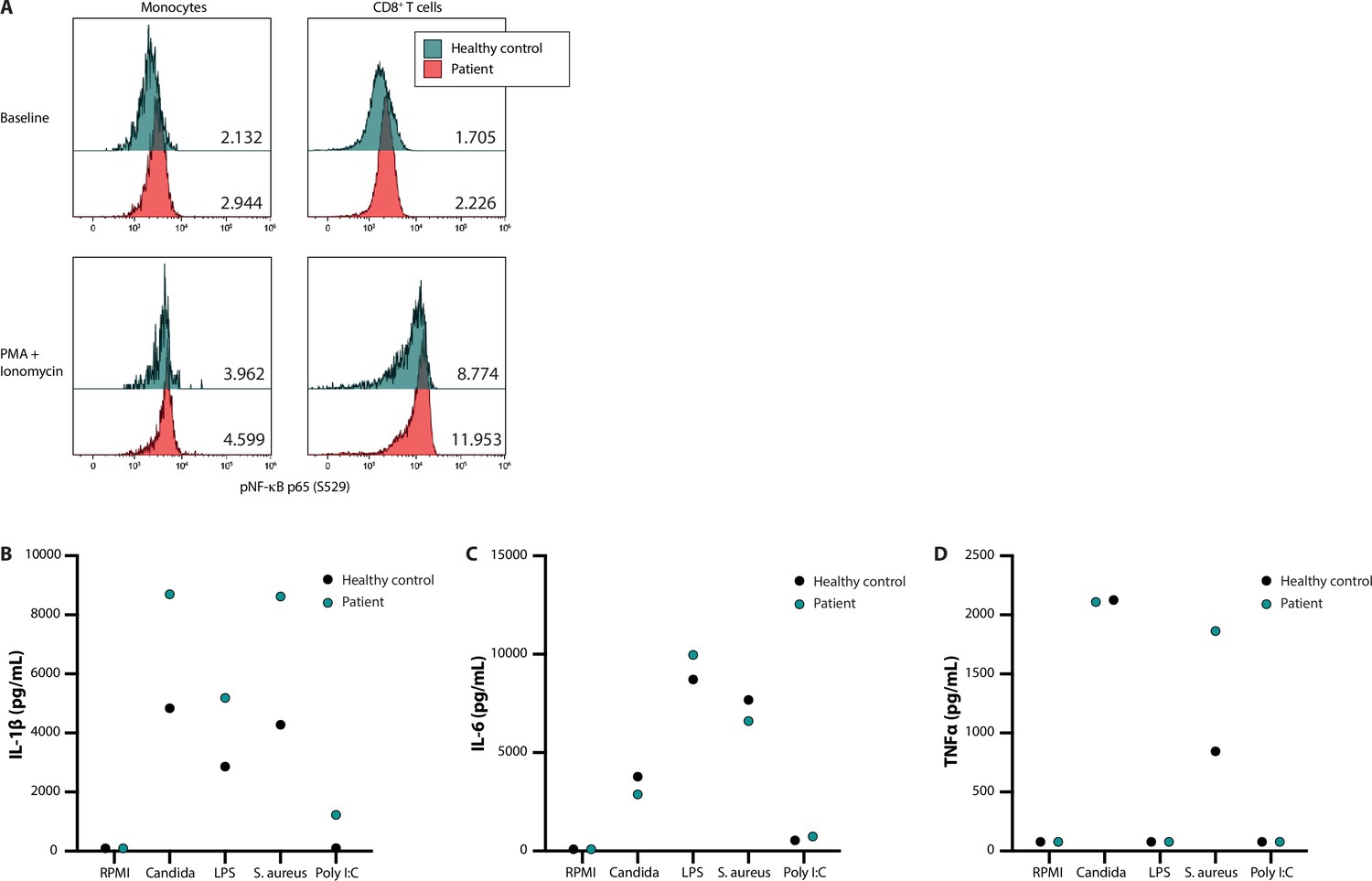
NF-κB signalling and production of innate cytokines upon ex vivo PBMC stimulation.
Panel A shows the median fluorescence intensity expression levels of pNF-κB p65 (S529) in peripheral blood CD14 +monocytes and CD8 +T cells from a healthy control (blue) and patient 53 (red), in the absence (baseline) or presence of phorbol 12-myristate 13-acetate and ionomycin stimulation, with the absolute values indicated in the lower right corner. Panels B, C, and D display the production of IL-1β, IL-6, and TNFα, respectively, after ex vivo stimulation for 24 hr.
-
Figure 2—source data 1
Original vector file of Figure 2.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig2-data1-v2.eps
-
Figure 2—source data 2
Uncropped gel with labels.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig2-data2-v2.zip
-
Figure 2—source data 3
Raw data of cytokine measurements.
- https://cdn.elifesciences.org/articles/78469/elife-78469-fig2-data3-v2.xlsx
Figure 2—figure supplement 1
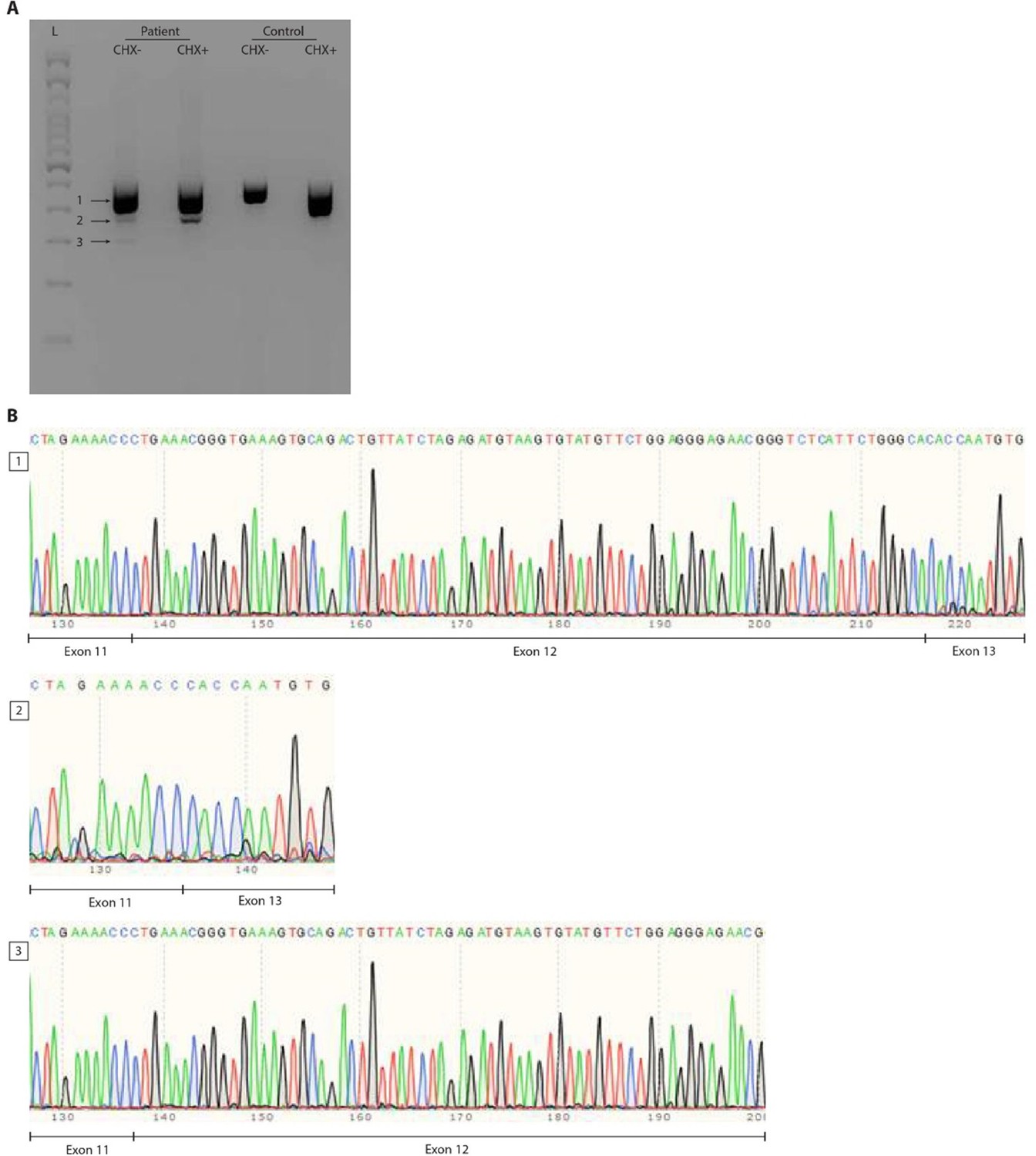
RNA splicing effect of the FBXW11 de novo splice site variant (c.1468-2A>G).
Panel A shows the agarose gel on cDNA PCR products of patient and control Epstein–Barr virus (EBV)-transformed lymphoblastoid cell lines (EBV-LCLs) treated with or without cycloheximide (CHX). Three distinct bands were identified and are indicated by arrows next to a 100bp ladder (L). Both the wildtype allele of the patient and the control show a smear, possibly indicating the presence of multiple FBXW11 isoforms. Panel B shows traces of the three bands from the agarose gel that were cut out and sent for Sanger sequencing. As the splice site variant in the patient was expected to lead to skipping of exon 12, the boundaries between exons 11, 12 and 13 were shown. The second band confirms skipping of exon 12 that results in a shorter transcript of the mutated FBXW11 allele. After CHX treatment, this band increased in size, indicating that the mutated allele undergoes nonsense mediated decay, but incomplete. Furthermore, a smaller transcript is formed in the patient, which is shown to contain part of exon 12, but not exon 13.
Author response image 1
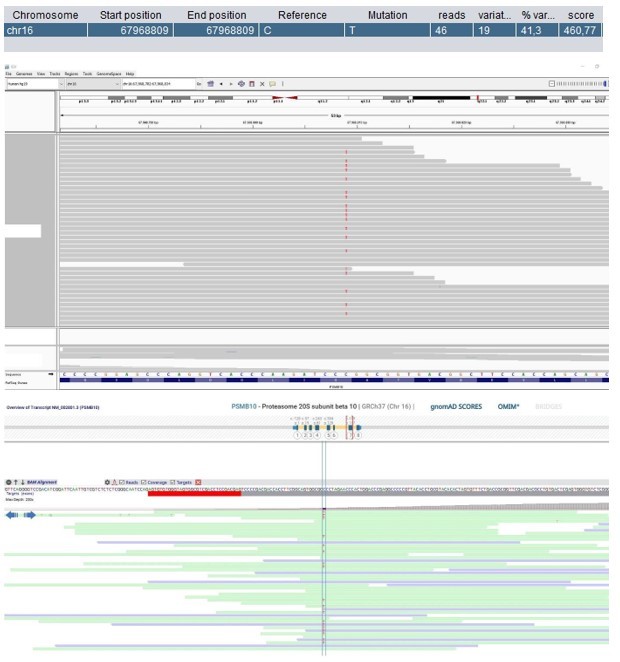
Patient 1: PSMB10 Chr16(GRCh37):g.67968809C>T NM_002801.3:c.601G>A p.(Gly201Arg).
Score <500, but validated by an independent test (Sanger sequencing).
Author response image 2
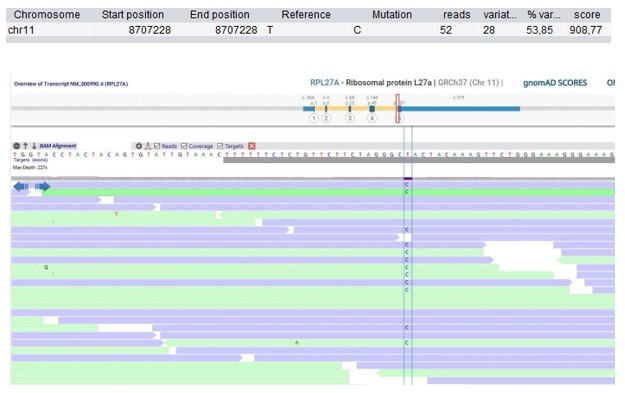
Patient 9: RPL27A Chr11(GRCh37):g.8707228T>C NM_000990.4:c.322T>C p.(Tyr108His).
Validated by an independent test (identified by single and trio-based WES).
Author response image 3
Patient 21: seq[GRCh37] del(17)(q25.3qter) NC_000017.10:g.(80544076_80544938)_qterdel.
Validated by independent test (identified by two different enrichment kits: Agilent Technologies and Twist Bioscience).
Author response image 4
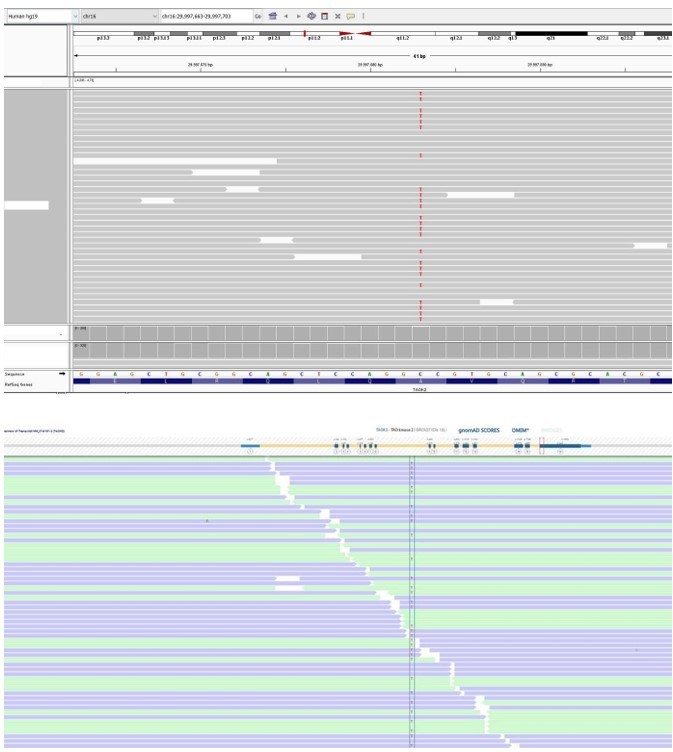
Patient 27: TAOK2 Chr16(GRCh37):g.29997683C>T NM_016151.3:c.2090C>T p.(Ala697Val).
Author response image 5
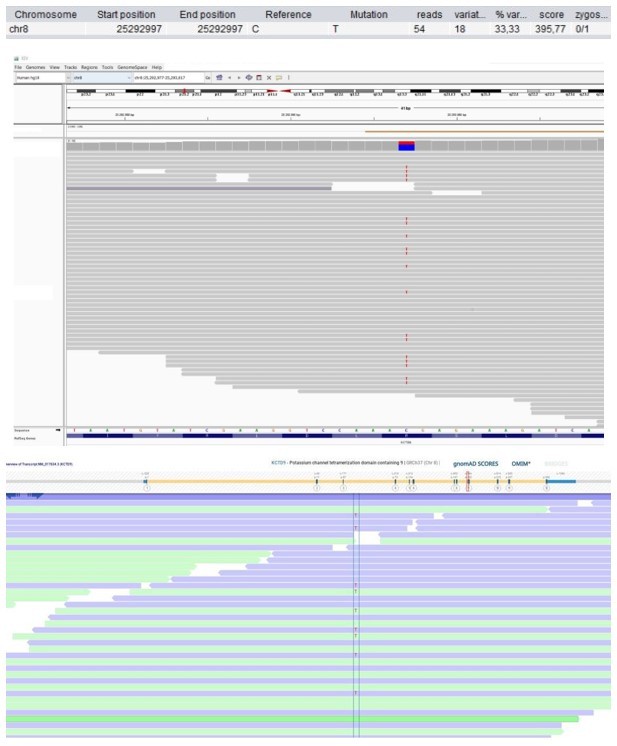
Patient 28: KCTD9 Chr8(GRCh37):g.25292997C>T NM_017634.3:c.695G>A p.(Arg232His).
Score <500, but validated by an independent test (Sanger sequencing).
Author response image 6

Patient 49: DDX1 Chr2(GRCh37):g.15769802dup NM_004939.2:c.1952dup p.(Trp652fs).
Author response image 7
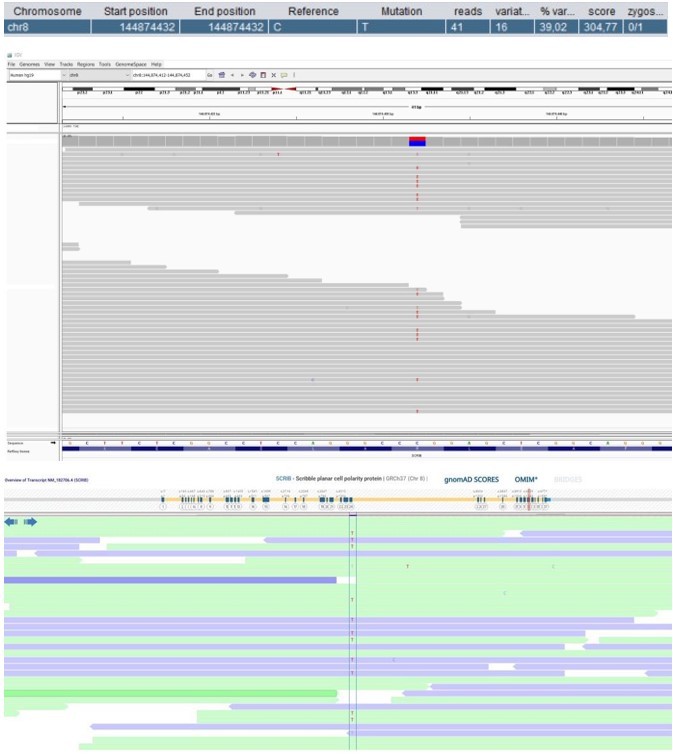
Patient 52: SCRIB Chr8(GRCh37):g.144874432C>T NM_182706.4:c.4472G>A p.(Arg1491Gln).
Score <500 and not validated by an independent test.
Author response image 8

Patient 53: FBXW11 Chr5(GRCh37):g.171295802T>C NM_012300.2:c.1468-2A>G p.?.
Author response image 9
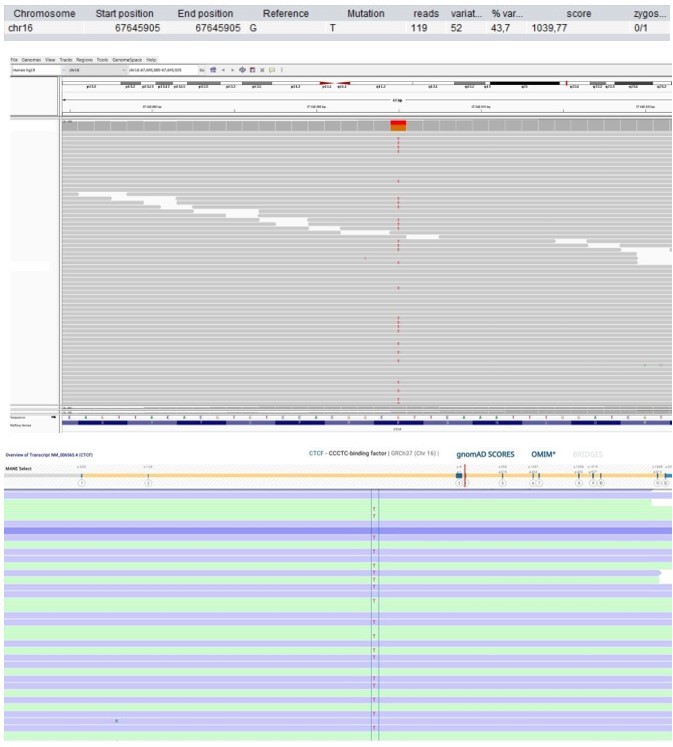
Patient 58: CTCF Chr16(GRCh37):g.67645905G>T NM_006565.4:c.833G>T p.(Arg278Leu).
Author response image 10
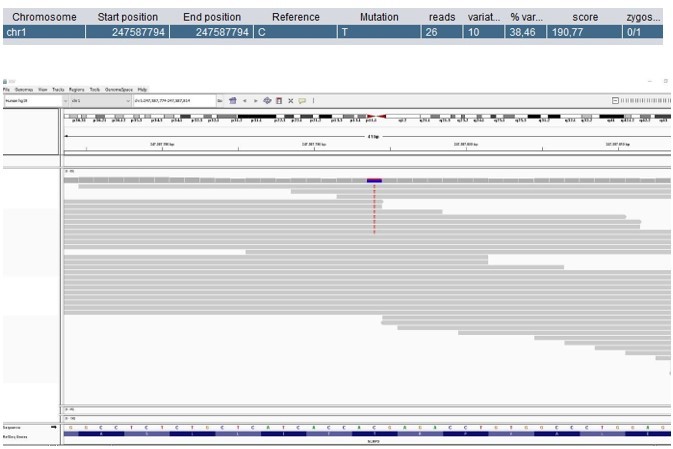
Patient 59: NLRP3 Chr1(GRCh37):g.247587794C>T NM_001079821.2:c.1049C>T p.(Thr350Met).
Score <500, but validated by an independent test (identified prior to trio-based WES in a different hospital).
Author response image 11
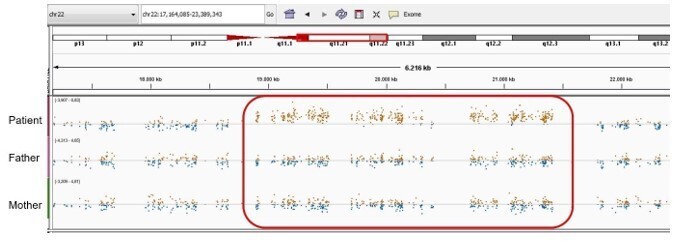
Patient 59: NLRP3 Chr1(GRCh37):g.Patient 69: seq[GRCh37] dup(22)(q11.21q11.21) NC_000022.10:g.(18775421_18893960)_(21414845_21576183)dup.
Author response image 12
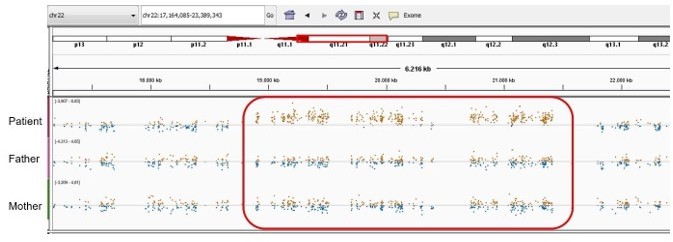
Patient 75: FUBP1 Chr1(GRCh37):g.78435621A>C NM_001303433.1:c.199T>G p.(Leu67Val).
Validated by an independent test (identified by single and trio-based WES).
Author response image 13

Patient 78: KMT2C Chr7(GRCh37):g.151860074del NM_170606.2:c.10588del p.(Ser3530Leufs*3).
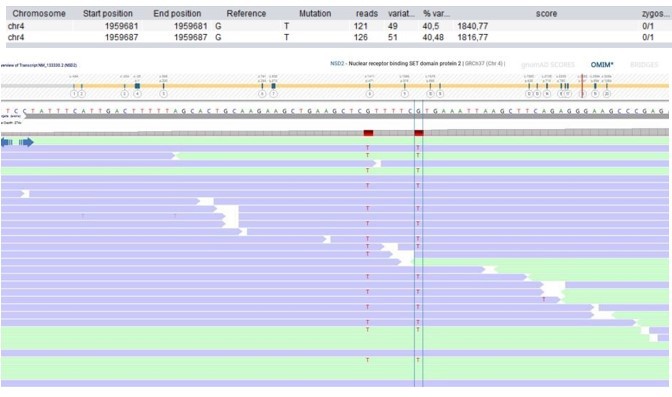
Author response image 14

Patient 108: NSD2 Chr4(GRCh37):g.1959681_1959687delinsTTTTTCT NM_133330.2:c.2903_2909delinsTTTTTCT p.(Arg968_Arg970delinsLeuPheLeu).
Validated by independent test (WES on different tissue (skin biopsy)).
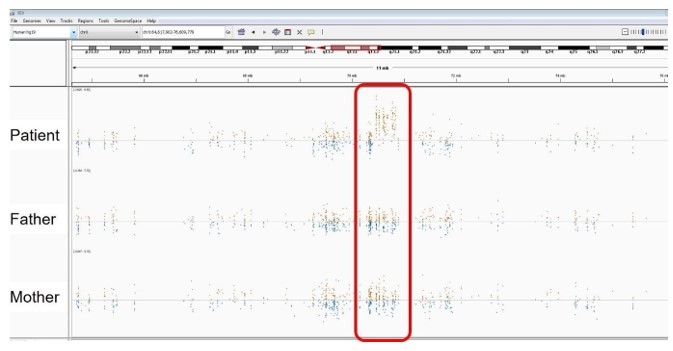
Author response image 15
Patient 115: seq[GRCh37] dup(X)(q13).
Author response image 16
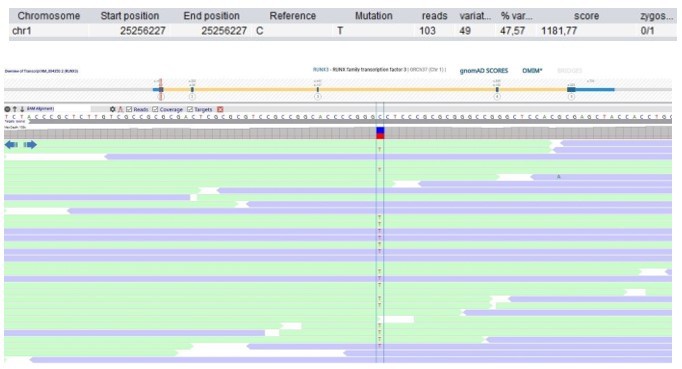
Patient 118: RUNX3 Chr1(GRCh37):g.25256227C>T NM_004350.2:c.133G>A p.(Gly45Arg).
Author response image 17
Patient 119: RELA Chr11(GRCh37):g.65423234C>T NM_021975.3:c.959-1G>A p.?.
Tables
Table 1
Patient cohort characteristics.
| Characteristic | Total N=123 |
|---|---|
| Demographics | |
| Age*, median (IQR) y | 9 (2-17) |
| <18 y, % | 67.4 |
| >18 y, % | 33.6 |
| Sex ratio, M:F | 50.4:49.6 |
| Distribution of clinical phenotypes † | |
| Severe combined immunodeficiency, n (%) | 9 (7.3) |
| Suspected SCID (low TRECs), n | 5 |
| Other, n | 4 |
| Combined immunodeficiency, n (%) | 22 (17.9) |
| Syndromal, n | 20 |
| Non-syndromal, n | 2 |
| Primary antibody deficiency, n (%) | 14 (11.4) |
| CVID, n | 14 |
| Agammaglobulinemia, n | 0 |
| Other, n | 0 |
| Immune dysregulation, n (%) | 20 (16.3) |
| HLH/EBV, n | 5 |
| Autoimmunity, n | 15 |
| Autoinflammatory syndrome, n (%) | 22 (17.9) |
| Periodic fever syndrome, n | 19 |
| Interferonopathy, n | 0 |
| Other, n | 3 |
| Phagocyte defect, n (%) | 5 (4.1) |
| Functional defect, n | 1 |
| Neutropenia/other, n | 4 |
| Innate/intrinsic immune defect, n (%) | 16 (13.0) |
| Bacterial/parasitic, n | 2 |
| MSMD/Viral, n | 7 |
| Other, n | 7 |
| Complement deficiencies, n (%) | 0 (0.0) |
| Bone marrow failure, n (%) | 10 (8.1) |
| Phenocopies of PIDs, n (%) | 0 (0.0) |
| Unclassified, n (%) | 5 (4.1) |
-
Abbreviations: IQR = interquartile range; SCID = severe combined immunodeficiency; TREC = T cell receptor excision circle; CVID = common variable immunodeficiency; HLH = haemophagocytic lymphohistiocytosis; EBV = Epstein-Barr virus; MSMD = Mendelian susceptibility to mycobacterial disease; PID = primary immunodeficiency.
-
*
The age at the time of genetic testing is indicated, since the age of onset has not been documented for all cases.
-
†
Categorization of phenotypes is based on the IUIS classification of 2019 (14).
-
Table 1—source data 1
List of patient-parent trios with variants identified in genes outside the diagnostic IEI gene panel, or classified as risk factors, carriership or variants of uncertain significance.
The table displays information on inherited single nucleotide variants and small insertion-deletions or copy number variants that were identified after diagnostic in silico gene panel and/or exome-wide analysis prior to the systematic DNV analysis in this study.
- https://cdn.elifesciences.org/articles/78469/elife-78469-table1-data1-v2.zip
Table 2
Genetic findings after routine diagnostic panel analysis.
Genetic variants reported after routine diagnostic whole exome sequencing analysis of the 123 patients included in this cohort of inborn errors of immunity.
| Total cases in which a genetic variant was reported, n (%) | 36 (29.3) | Patient nr. |
|---|---|---|
| (Likely) pathogenic mutation, n (%) | 18 (14.6) | |
| Within IEI gene panel, n (%) | 12 (9.8) | All patients listed in Table 3 |
| Beyond IEI gene panel, n (%) | 6 (4.9) | 1, 3, 40, 69, 85, 103 (Table 1—source data 1) |
| Other variants, n (%) | 19 (15.4) | Table 1—source data 1 |
| Risk factor, n (%) | 6 (4.9) | 21, 44, 55, 56, 68, 112 |
| Carriership recessive allele, n (%) | 7 (5.7) | 3, 7, 16, 23, 32, 44, 76 |
| Variant of unknown significance, n (%) | 9 (7.3) | 6, 21, 23, 45, 54, 80, 100, 101, 115 |
Table 3
Patients with previously reported single nucleotide variants, small insertion-deletions, or copy number variants that may (partially) explain the patient’s immunological phenotype.
Listed variants were identified prior to the research-based systematic re-analysis of the current study following diagnostic gene panel analysis for inborn errors of immunity.
| Patient nr. | Sex | Age range at sampling | Phenotype (IUIS classification) | Variant | Mutational mechanism | ACMG classification | ClinVar accession | Comments |
|---|---|---|---|---|---|---|---|---|
| 10 | F | 0–5 | Immune dysregulation, HLH/EBV | AP3B1 Chr5(GRCh37):g.77563371del NM_003664.4:c.177del p.(Lys59fs) | AR (ch) LoF | Pathogenic | VCV000224763 | Hermansky-Pudlak syndrome 2 (OMIM #608233) |
| AP3B1 Chr5(GRCh37):g.77423980_77423983del NM_003664.4:c.1839_1842del p.(Asp613fs) | Pathogenic | VCV000224764 | ||||||
| 12 | F | 11–15 | CID, syndromal | FAS Chr10(GRCh37):g.90774167_90774186dup NM_000043.6:c.968_987dup p.(Glu330fs) | AD (htz) LoF | Pathogenic | VCV000016509 | Autoimmune lymphoproliferative syndrome, type IA (OMIM #601859) |
| seq[GRCh37] del(16)(p11.2p11.2) NC_000016.9:g.(29469093_29624260)_(30199846_30208282)del | AD (htz) LoF | Pathogenic | - | 16 p11.2 deletion syndrome (OMIM #611913) | ||||
| 26 | F | 0–5 | Bone marrow failure | DHFR Chr5(GRCh37):g.79950248C>T NM_000791.3:c.61G>A p.(Gly21Arg) | AR (hmz) LoF | Likely pathogenic | - | Megaloblastic anaemia due to dihydrofolate reductase deficiency (OMIM #613839) Affected sibling carries equal variant |
| 59 | M | 6–10 | Autoinflammatory disorder | NLRP3 Chr1(GRCh37):g.247587794C>T NM_001079821.2:c.1049C>T p.(Thr350Met) | AD (htz) LoF | Pathogenic | - | Muckle-Wells syndrome (OMIM #191900) De novo SNV |
| 61 | M | 0–5 | CID, syndromal | MKL1 Chr22(GRCh37):g.40815086dup NM_020831.4:c.1356dup p.(Val453Argfs) | AR (hmz) LoF | Likely pathogenic | - | Immunodeficiency 66 (OMIM #618847) Affected sibling carries equal variant |
| 77 | F | 0–5 | CID, syndromal | ALOXE3 Chr17(GRCh37):g.8006708G>A NM_021628.2:c.1889C>T p.(Pro630Leu) | AR (hmz) LoF | Pathogenic | - | Congenital ichthyosis 3 (OMIM #606545) |
| 91 | F | 0–5 | Suspected SCID (low TRECs) | FOXN1 Chr17(GRCh37):g.26857765A>G NM_003593.2:c.831–2A>G p.? | AD (htz) LoF | Likely pathogenic | - | T-cell lymphopenia, infantile, with or without nail dystrophy (OMIM #618806) |
| 102 | F | 11–15 | Immune dysregulation, autoimmunity and others | CD55 Chr1(GRCh37):g.207497984dup NM_001300902.1:c.367dup p.(Thr123fs) | AR (hmz) LoF | Pathogenic | - | Complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (OMIM #226300) |
| PET117 Chr20(GRCh37):g.18122927C>T NM_001164811.1:c.172C>T p.(Gln58*) | AR (hmz) LoF | Likely pathogenic | VCV000981504 | Mitochondrial complex IV deficiency, nuclear type 19 (OMIM #619063) | ||||
| 105 | M | 31–35 | Defects in intrinsic and innate immunity, MSMD and viral infection | TLR7 ChrX(GRCh37):g.12905756_12905759del NM_016562.3:c.2129_2132del p.(Gln710fs) | XLR (hemi) LoF | Pathogenic | VCV000977232 | Immunodeficiency 74, COVID19-related (OMIM #301051) Affected sibling carries equal variant |
| 114 | M | 6–10 | Immune dysregulation, autoimmunity and others | LRBA Chr4(GRCh37):g.151835415del NM_006726.4:c.1093del p.(Tyr365fs) | AR (hmz) LoF | Pathogenic | - | Common variable immunodeficiency 8 (OMIM #614700) |
| 120 | M | 11–15 | Congenital defect of phagocyte, functional defects | NCF1 Chr7(GRCh37):g.74191615_74191616del NM_000265.5:c.75_76del p.(Tyr26fs) | AR (hmz) LoF | Pathogenic | VCV000002249 | Chronic granulomatous disease 1 (OMIM #233700) |
| 122 | M | 0–5 | Suspected SCID (low TRECs) | FOXN1 Chr17(GRCh37):g.26851540del NM_003593.2.1:c.143del p.(Cys48fs) | AD (htz) LoF | Pathogenic | - | T-cell lymphopenia, infantile, with or without nail dystrophy (OMIM #618806) |
-
Abbreviations: IUIS = International Union of Immunological societies; ACMG = American College of Medical Genetics and Genomics; HLH = haemophagocytic lymphohistiocytosis; EBV = Epstein-Barr virus; OMIM = Online Mendelian Inheritance in Man; (S)CID = (severe) combined immunodeficiency; TREC = T cell receptor excision circle; MSMD = Mendelian susceptibility to mycobacterial disease; AR = autosomal recessive; AD = autosomal dominant; XLR = X-linked recessive; ch = compound heterozygous; htz = heterozygous; hmz = homozygous; hemi = hemizygous; LoF = loss-of-function; SNV = single nucleotide variant.
Table 4
Identification of 13 heterozygous, rare and non-synonymous candidate de novo variants.
The 124 non-synonymous candidate de novo variants were systematically evaluated based on the potential to be damaging to gene and protein function and the possible involvement in the patient’s immunological phenotype.
| Patient nr. | Sex | Age range at sampling | Phenotype (IUIS classification) | De novo variant | GnomAD AF in % | in-house AF in % | PhyloP | CADD | VarMap | MetaDome | Coding DNV in denovo-db (protein effect) | LOEUF | Function | Literature | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Missense SNVs | |||||||||||||||
| 1 | M | 11–15 | SCID | PSMB10 Chr16(GRCh37): g.67968809C>T NM_002801.3: c.601G>A p.(Gly201Arg) | 0 | 0 | 5 | 32 | Likely deleterious | Neutral | - | 1.37 | Immuno- and thymoproteasome subunit | Homozygous Psmb10 variant in mice causes SCID and systemic autoinflammation (Treise et al., 2018). Homozygous PSMB10 variant in humans cause PRAAS, no immunodeficiency (Sarrabay et al., 2020). | Revertant somatic mosaicism (VAF: 39.7%). Additional inherited SNV and partial somatic UPD16 (Table 1—source data 1). |
| 9 | M | 6–10 | Predominantly antibody deficiency, hypogamma-globulinemia | RPL27A Chr11(GRCh37): g.8707228T>C NM_000990.4: c.322T>C p.(Tyr108His) | 0.0032 | 0.0041 | 7.4 | 27.4 | Likely deleterious | Intolerant | - | 0.39 | Ribosomal subunit | Ribosomopathies may include immunological defects (Khan et al., 2011). | |
| 27 | M | 11–15 | Autoinflammatory disorder | TAOK2 Chr16(GRCh37): g.29997683C>T NM_016151.3: c.2090C>T p.(Ala697Val) | 0 | 0 | 4.8 | 22.5 | Possibly deleterious | Slightly intolerant | 6 (4 mis) | 0.24 | Serine/threonine-protein kinase (p38 MAPK pathway) | Homozygous TAOK2 variant causes abnormal T cell activation in two patients with inflammatory bowel disease (Molho-Pessach et al., 2017). | |
| 28 | F | 16–20 | Predominantly antibody deficiency, hypogamma-globulinemia | KCTD9 Chr8(GRCh37): g.25292997C>T NM_017634.3: c.695G>A p.(Arg232His) | 0 | 0.0082 | 5.8 | 32 | Likely deleterious | Intolerant | - | 0.52 | Substrate-specific adapter | Involved in NK cell activation (Chen et al., 2013). | |
| 52 | M | 11–15 | Predominantly antibody deficiency, hypogamma-globulinemia | SCRIB Chr8(GRCh37): g.144874432C>T NM_182706.4: c.4472G>A p.(Arg1491Gln) | 0.0032 | 0 | 4.2 | 29.9 | Possibly deleterious | Intolerant | 5 (4 mis) | 0.31 | Scaffold protein | Involved in uropod and immunological synapse formation, and ROS production by antigen-presenting cells (Barreda et al., 2020). | |
| 58 | F | 21–25 | Unclassified | CTCF Chr16(GRCh37): g.67645905G>T NM_006565.4: c.833G>T p.(Arg278Leu) | 0 | 0 | 9.7 | 24.7 | Possibly deleterious | Highly intolerant | 12 (11 mis) | 0.15 | Transcriptional insulator | CTCF variants cause neurodevelopmental disorders, sometimes associated with recurrent infections and minor facial dysmorphisms (Konrad et al., 2019). | Published (Konrad et al., 2019). |
| 75 | F | 6–10 | Bone marrow failure | FUBP1 Chr1(GRCh37): g.78435621A>C NM_001303433.1: c.199T>G p.(Leu67Val) | 0 | 0 | 2.6 | 24.8 | Possibly deleterious | Intolerant | 1 (0 mis) | 0.12 | Transcriptional regulator that binds FUSE upstream of the c-myc promoter | Essential for long-term repopulating hematopoietic stem cell renewal (Rabenhorst et al., 2015). Fubp1 KO mice show cerebral hyperplasia, pulmonary hypoplasia, pale livers, hypoplastic spleen, thymus, and bone marrow, cardiac hypertrophy, placental distress, and small size (Zhou et al., 2016). | |
| 118 | F | 0–5 | Immune dysregulation, autoimmunity and others | RUNX3 Chr1(GRCh37): g.25256227C>T NM_004350.2: c.133G>A p.(Gly45Arg) | 0 | 0 | 2.4 | 18 | Possibly deleterious | Slightly tolerant | 1 (1 mis) | 0.42 | Transcriptional regulator | RUNX3 regulates CD8+T cell thymocyte development, maturation of cytotoxic CD8+T cells and the function of innate lymphoid cells 3 via stimulation of RORγt (Ebihara et al., 2015). Runx3 KO mice spontaneously develop inflammatory bowel disease and gastric lesions (Brenner et al., 2004). | |
| Frameshift SNVs | |||||||||||||||
| 49 | M | 26–30 | Predominantly antibody deficiency, hypogamma-globulinemia | DDX1 Chr2(GRCh37): g.15769802dup NM_004939.2: c.1952dup p.(Trp652fs) | 0 | 0 | - | - | - | - | 4 (0 fs) | 0.28 | RNA helicase | Part of a dsRNA sensor that activates the NF-κB pathway and type I interferon responses (Zhang et al., 2011). | |
| 78 | F | 6–10 | CID, syndromal | KMT2C Chr7(GRCh37): g.151860074del NM_170606.2: c.10588del p.(Ser3530Leufs*3) | 0 | 0 | - | - | - | - | 19 (4 fs) | 0.12 | Histone methyltransferase | KMT2C de novo variant causes Kleefstra syndrome 2, sometimes associated with recurrent respiratory infections (Koemans et al., 2017). | |
| Small in-frame indel | |||||||||||||||
| 108 | M | 21–25 | Bone marrow failure | NSD2 Chr4(GRCh37): g.1959681_1959687delins TTTTTCT NM_133330.2: c.2903_2909delins TTTTTCT p. (Arg968_Arg970delinsLeuPheLeu) | - | - | - | - | - | - | 0.12 | Histone methyltransferase | NSD2 de novo LoF variant causes mild Wolf-Hirschhorn syndrome (Barrie et al., 2019). Unclear role in immunity. | Postzygotic mosaicism (VAF 29%). | |
| Patient nr. | Sex | Age range at sampling | Phenotype (IUIS classification) | De novo variant | GnomAD AF in % | in-house AF in % | PhyloP | CADD | SpliceAI Acceptor Gain | SpliceAI Acceptor Loss | Coding DNV in denovo-db (protein effect) | LOEUF | Function | Literature | Comments |
| Splice site SNVs | |||||||||||||||
| 53 | F | 11–15 | Autoinflammatory disorder | FBXW11 Chr5(GRCh37): g.171295802T>C NM_012300.2: c.1468–2A>G p.? | 0 | 0 | 7.9 | 34 | 0.0134 | 0.9862 | 2 (0 ss) | 0.31 | Component of SCF (SKP1-CUL1-F-box) E3 ubiquitin ligase complex | Involved in the regulation of NF-κB signalling (Wang et al., 2018). | |
| 119 | F | 11–15 | Autoinflammatory disorder | RELA Chr11(GRCh37): g.65423234C>T NM_021975.3: c.959–1G>A p.? | 0 | 0 | 3.5 | 34 | 0.7968 | 0.9991 | - | 0.18 | Transcription factor p65 (NF-κB subunit) | Heterozygous RELA variant causes chronic mucocutaneous ulceration (Badran et al., 2017). | |
-
Abbreviations: IUIS = International Union of Immunological Societies; GnomAD = Genome Aggregation Database; AF = allele frequency; CADD = Combined Annotation Dependent Depletion; DNV = de novo variant; LOEUF = loss-of-function observed/expected upper bound fraction; SNV = single nucleotide variant; indel = insertion-deletion; (S)CID = severe combined immunodeficiency; NA = not applicable; mis = missense; fs = frameshift; ss = splice site; MAPK = mitogen-activated protein kinase; FUSE = far upstream element; PRAAS = proteasome-associated autoinflammatory syndrome; NK = natural killer; ROS = reactive oxygen species; KO = knockout; dsRNA = double-stranded RNA; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells; LoF = loss-of-function; VAF = variant allele fraction; UPD16 = uniparental disomy of chromosome 16.
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Trio-based whole exome sequencing in patients with suspected sporadic inborn errors of immunity: A retrospective cohort study
eLife 11:e78469.
https://doi.org/10.7554/eLife.78469




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}