Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats
- Department of Pathology and Laboratory Medicine, University of British Columbia, Canada
- Public Health Laboratory, British Columbia Centre for Disease Control, Canada
- Department of Biology, Faculty of Science, University of Regina, Canada
- Institute for Microbial Systems and Society, Faculty of Science, University of Regina, Canada
- Metabiota Inc, Democratic Republic of the Congo
- Institut National de Recherche Biomédicale, Democratic Republic of the Congo
- Labyrinth Global Health Inc, United States
- Metabiota Inc, United States
- Development Alternatives, United States
- Mosaic, Cameroon
- Metabiota, Canada
- Southbridge Care, Canada
- One Health Institute, School of Veterinary Medicine, University of California, Davis, United States
- Nyati Health Consulting, Canada
- Institute for Global Health Sciences, University of California, San Francisco, United States
Figures
Figure 1

Custom hybridization probe panel provided broadly inclusive coverage of known bat coronavirus diversity in silico.
Bat coronavirus (CoV) sequences were obtained by downloading all available alphacoronavirus, betacoronavirus, and unclassified coronaviridae and coronavirinae sequences from GenBank on 4 October 2020 and searching for bat-related keywords in sequence headers. A custom panel of 20,000 probes was designed to target these sequences using the makeprobes module in the ProbeTools package. The ProbeTools capture and stats modules were used to assess probe coverage of bat CoV reference sequences. (A) Each bat CoV sequence is represented as a dot plotted according to its probe coverage, that is, the percentage of its nucleotide positions covered by at least one probe in the custom panel. (B) The same analysis was performed on the subset of sequences representing full-length genomes (>25 kb in length).
Figure 2
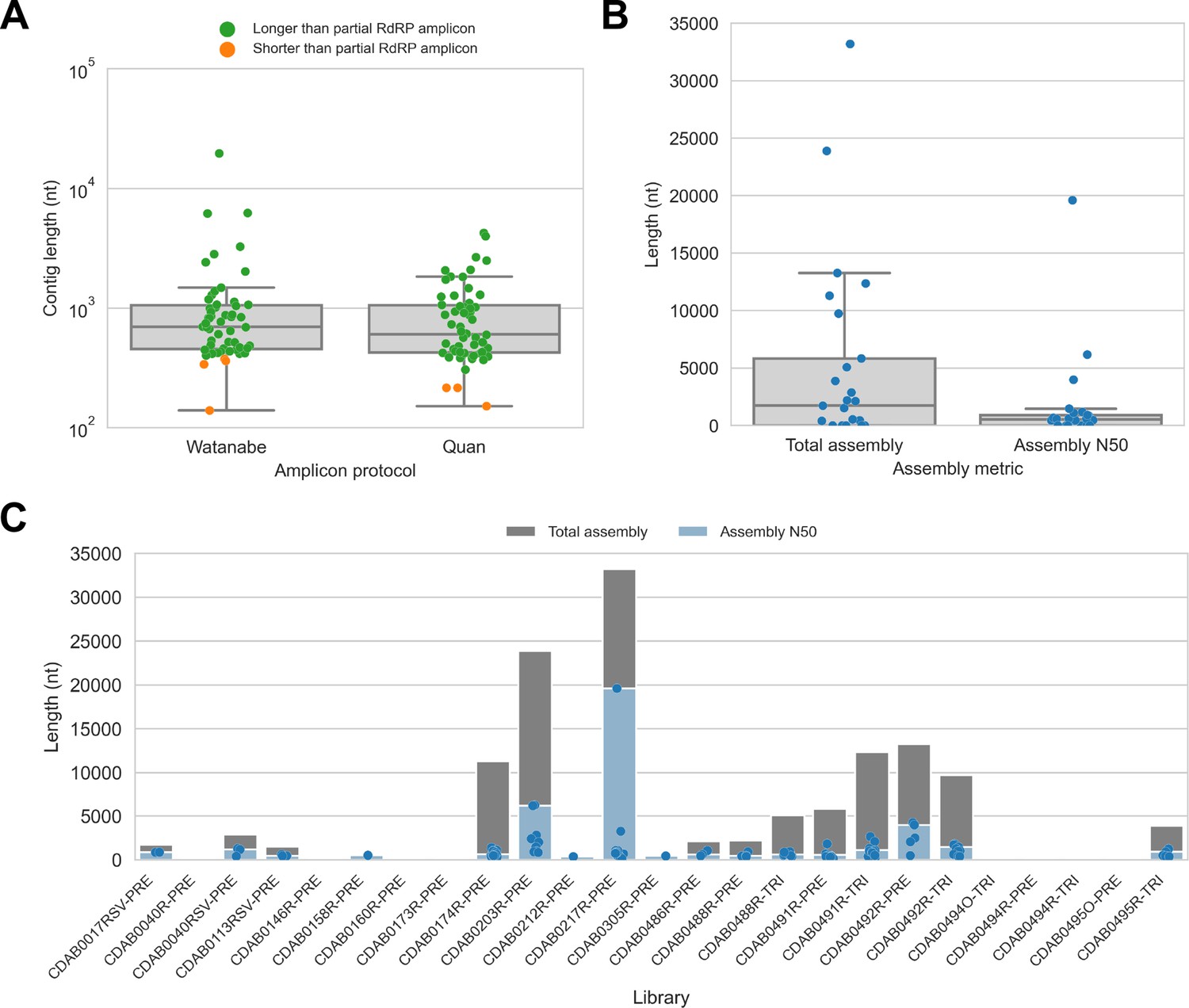
De novo assembly of probe captured libraries yielded more genome sequence than standard amplicon sequencing methods for most specimens.
Reads from probe captured libraries were assembled de novo with coronaSPAdes, and coronavirus contigs were identified by local alignment against a database of all coronaviridae sequences in GenBank. (A) The size distribution of contigs from all libraries is shown. Dots are coloured to indicate whether the length of the contig exceeded partial RNA-dependent RNA polymerase (RdRP) gene amplicons previously sequenced from these specimens. (B) Total assembly size and assembly N50 distributions for all libraries. (C) Each contig is represented as a dot plotted according to its length. Assembly N50 sizes and total assembly sizes are indicated by the height of their bars.
Figure 3 with 4 supplements
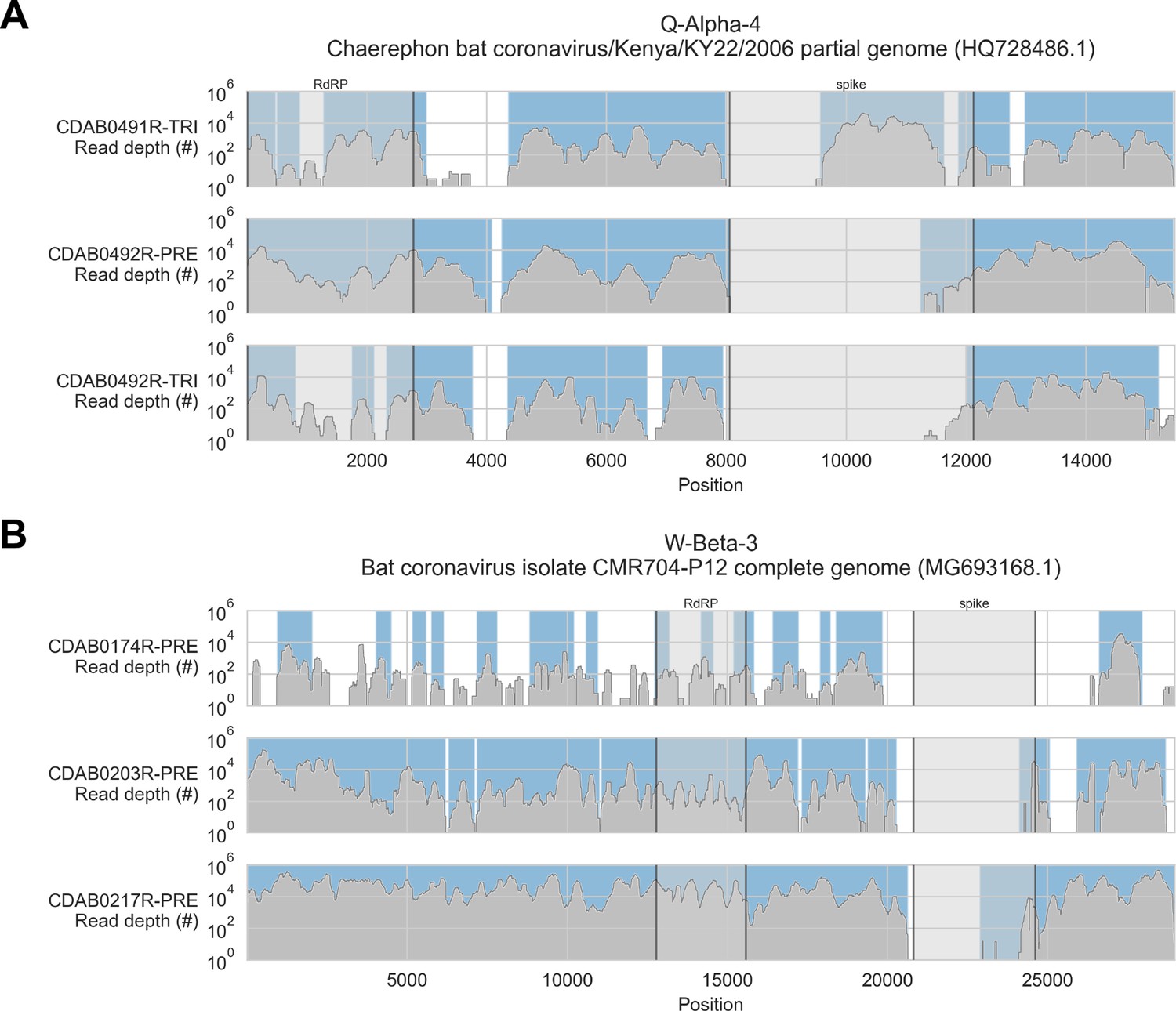
Coverage of reference sequences by probe captured libraries was used to assess extent and location of recovery.
Reference sequences were chosen for each previously identified phylogenetic group (indicated in panel titles). Coverage of these reference sequences was determined by mapping reads and aligning contigs from probe captured libraries. Dark grey profiles show depth of read coverage along reference sequences. Blue shading indicates spans where contigs aligned. The locations of spike and RNA-dependent RNA polymerase (RdRP) genes are indicated in each reference sequence and shaded light grey. This figure shows the six libraries with the most extensive reference sequence coverage. Similar plots are provided as figure supplements for all libraries where any coronavirus sequence was recovered (Figure 3—figure supplements 1–4) .
Figure 3—figure supplement 1
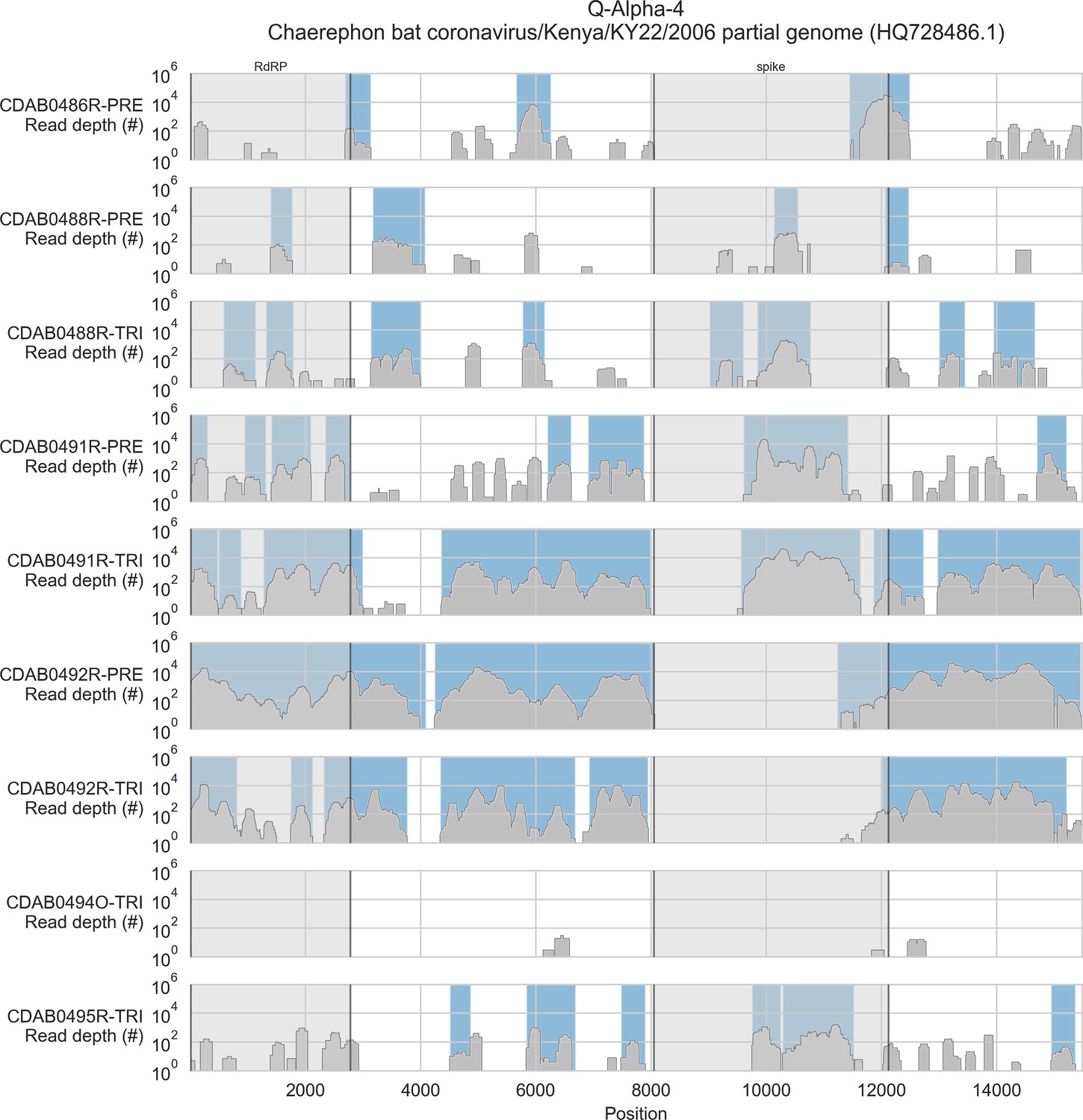
Coverage of reference sequence by probe captured libraries for specimens from phylogenetic group Q-Alpha-4.
Coverage of reference sequence was determined by mapping reads and aligning contigs from probe captured libraries. Dark grey profiles show depth of read coverage along reference sequence. Blue shading indicates spans where contigs aligned. The locations of spike and RNA-dependent RNA polymerase (RdRP) genes are indicated and shaded light grey.
Figure 3—figure supplement 2

Coverage of reference sequence by probe captured libraries for specimens from phylogenetic group W-Beta-2.
Coverage of reference sequence was determined by mapping reads and aligning contigs from probe captured libraries. Dark grey profiles show depth of read coverage along reference sequence. Blue shading indicates spans where contigs aligned. The locations of spike and RNA-dependent RNA polymerase (RdRP) genes are indicated and shaded light grey.
Figure 3—figure supplement 3
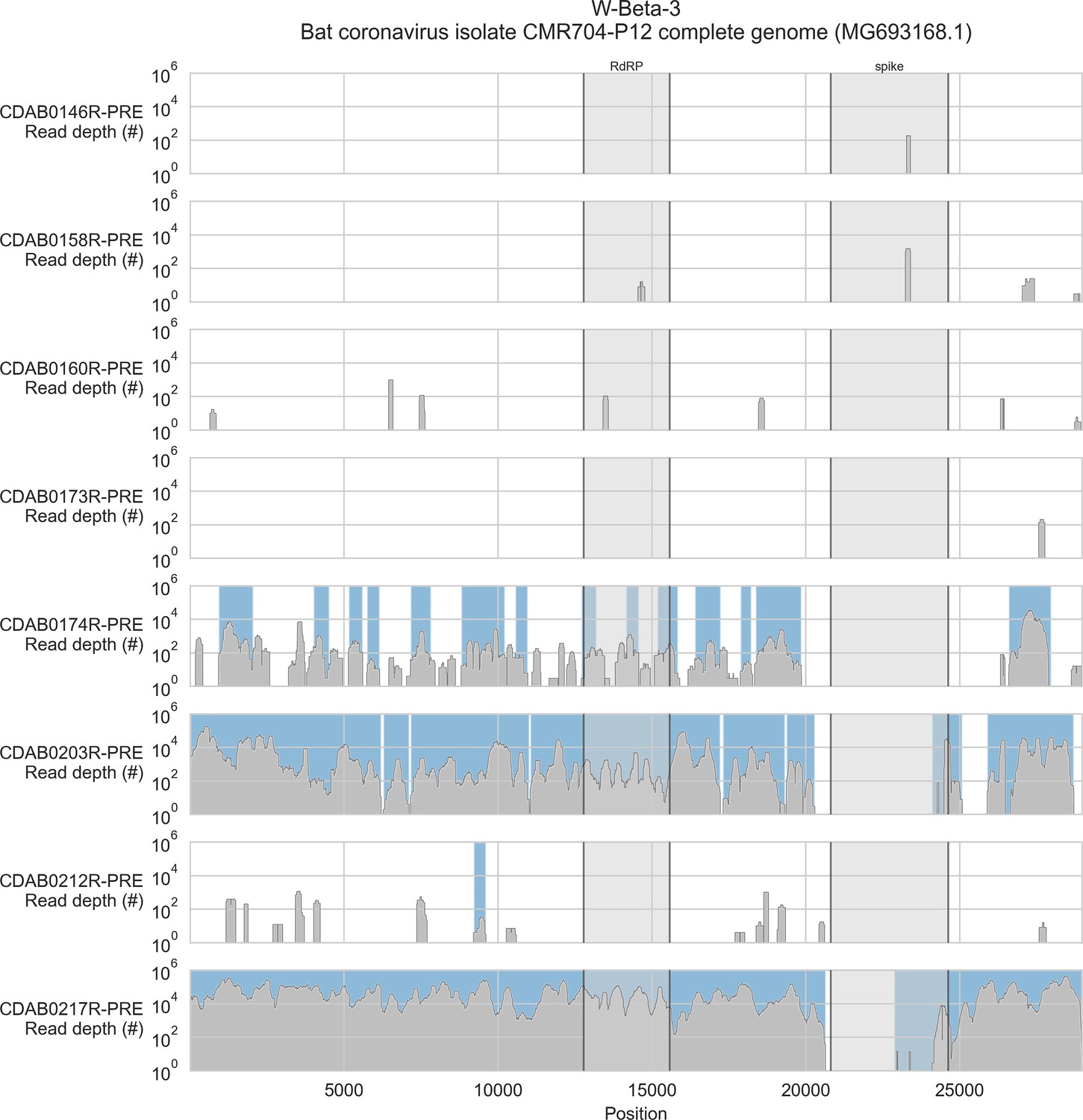
Coverage of reference sequence by probe captured libraries for specimens from phylogenetic group W-Beta-3.
Coverage of reference sequence was determined by mapping reads and aligning contigs from probe captured libraries. Dark grey profiles show depth of read coverage along reference sequence. Blue shading indicates spans where contigs aligned. The locations of spike and RNA-dependent RNA polymerase (RdRP) genes are indicated and shaded light grey.
Figure 3—figure supplement 4
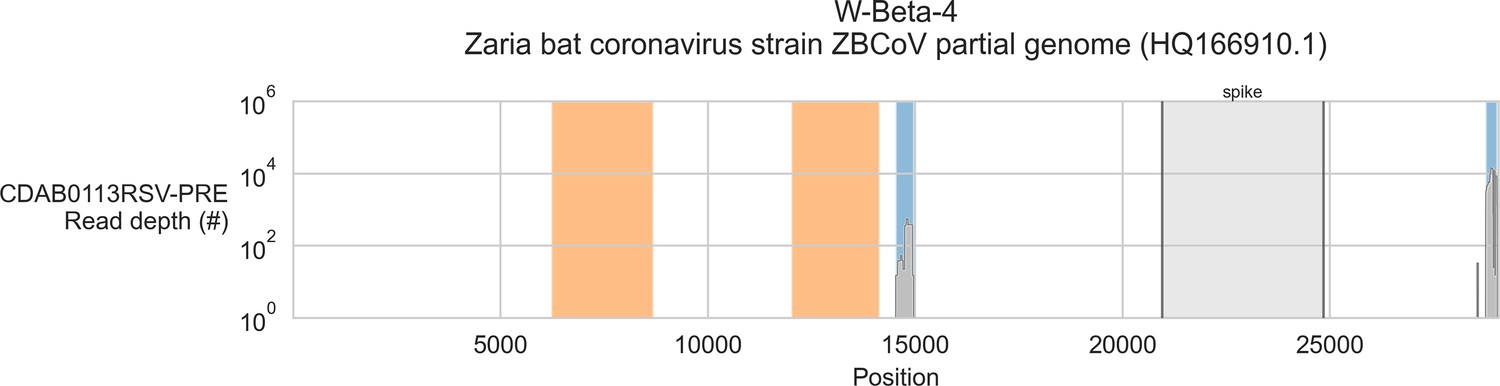
Coverage of reference sequence by probe captured libraries for specimens from phylogenetic group W-Beta-4.
Coverage of reference sequence was determined by mapping reads and aligning contigs from probe captured libraries. Dark grey profiles show depth of read coverage along reference sequence. Blue shading indicates spans where contigs aligned. The locations of spike gene are indicated and shaded light grey. Ambiguous bases (Ns) are shaded orange.
Figure 4
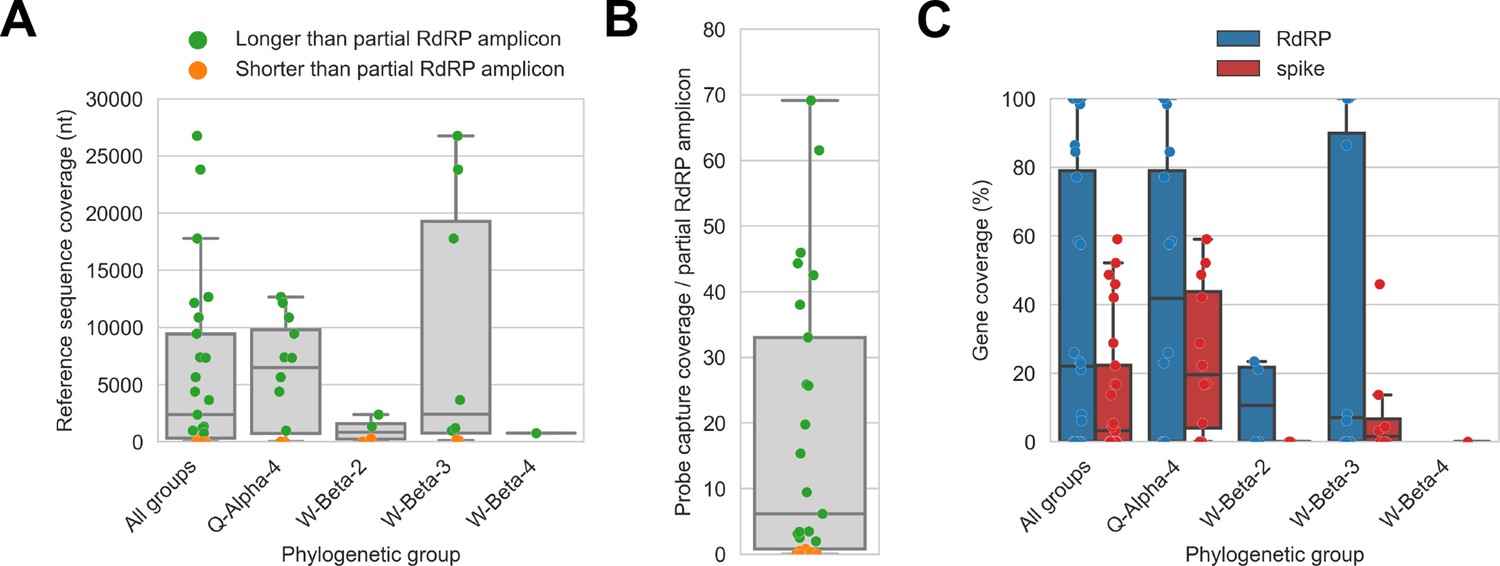
Probe captured libraries provided more extensive coverage of reference genomes than standard amplicon sequencing protocols for most specimens.
Reference sequences were selected for the previously identified phylogenetic groups to which these specimens had been assigned by Kumakamba et al., 2021. (A) Coverage of these reference sequences was determined by mapping reads and aligning contigs from probe captured libraries. Each library is represented as a dot, and dots are coloured according to whether reference sequence coverage exceeded the length of the partial RNA-dependent RNA polymerase (RdRP) gene sequence that had been previously generated by amplicon sequencing. (B) The number of reference sequence positions covered by probe captured libraries was divided by the length of the partial RdRP amplicon sequences from these specimens. This provided the fold-difference in recovery between probe capture and standard amplicon sequencing methods. (C) Percent coverage of the spike and RdRP genes were calculated for each specimen.
Figure 5
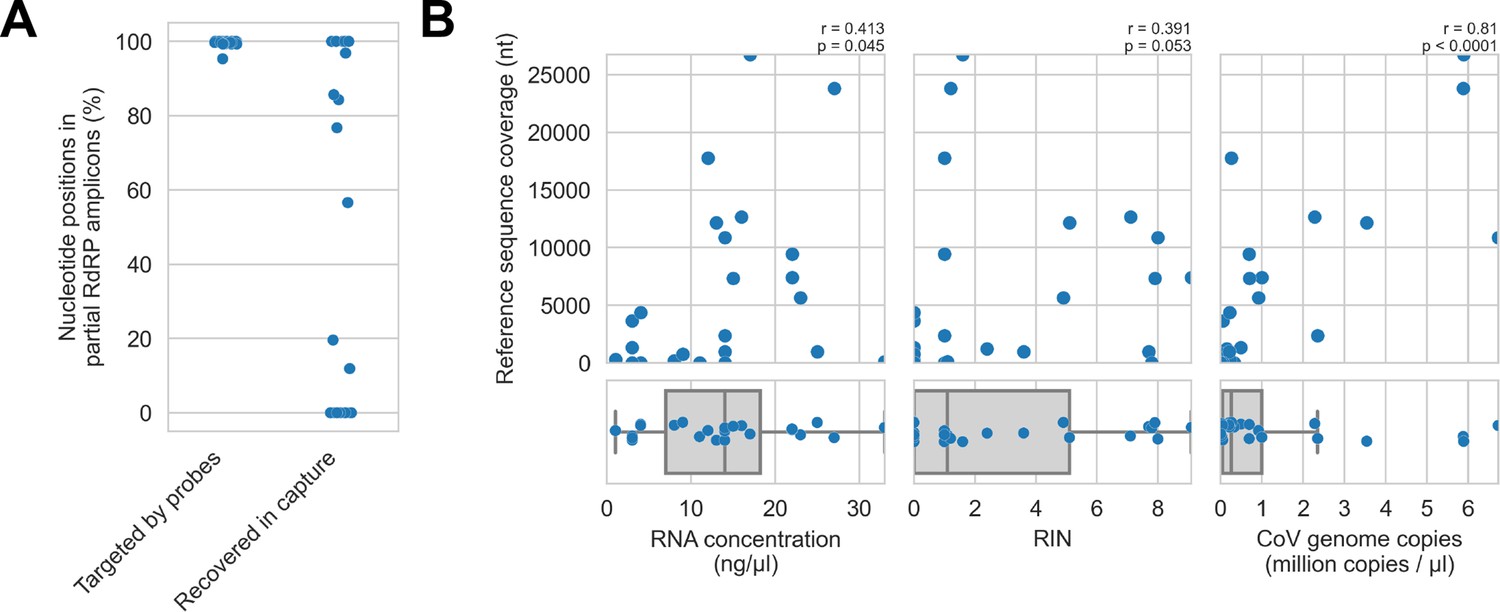
Recovery of coronavirus (CoV) genomic material was limited in vitro by method sensitivity.
(A) Sensitivity was assessed by evaluating recovery of partial RNA-dependent RNA polymerase (RdRp) gene regions that had been previously sequenced in these specimens by amplicon sequencing. Probe coverage of partial RdRp sequences was assessed in silico to exclude insufficient probe design as an alternate explanation for incomplete recovery of these targets. (B) Input RNA concentration, RNA integrity numbers (RINs), and CoV genome abundance were measured for each specimen. The impact of these specimen characteristics on recovery by probe capture (as measured by reference sequence coverage) was assessed using Spearman’s rank correlation (test results stated in plots). An outlier was omitted from this analysis: RNA concentration for specimen CDAB0160R was recorded as 190 ng/μl, a value 4.7 SDs from the mean of the distribution.
Figure 6

In silico assessment of probe panel coverage for reference genomes.
Reference sequences were chosen for each previously identified phylogenetic group (indicated in panel titles). Blue profiles show the number of probes covering each nucleotide position along the reference sequence. Probe coverage, that is, the percentage of nucleotide positions covered by at least one probe, is stated in panel titles. Ambiguity nucleotides (Ns) are shaded in orange, and these positions were excluded from the probe coverage calculations. The locations of spike and RNA-dependent RNA polymerase (RdRP) genes are indicated in each reference sequence (where available) and shaded grey.
Figure 7

Coronavirus (CoV) genomic material was low abundance in swab specimens but effectively enriched by probe capture.
(A) Reads from uncaptured, deep metagenomic sequenced libraries were mapped to complete genomes recovered from these specimens to assess abundance of CoV genomic material. On-target rate was calculated as the percentage of total reads mapping that mapped to the CoV genome sequence. (B) Reads from probe captured libraries were also mapped to assess enrichment and removal of background material. Most libraries used for probe capture (-PRE and -TRI) had insufficient volume remaining for deep metagenomic sequencing, so new libraries were prepared (-DEEP) from the same specimens.
Figure 8

Phylogenetic tree of translated spike gene sequences from alphacoronaviruses.
Spike sequences are coloured according to whether they were from study specimens (blue), human CoVs (red), RefSeq (black), or GenBank (grey). Only the 25 closest-matching spike sequences from GenBank were included, as determined by blastp bitscores. GenBank and RefSeq accession numbers are provided in parentheses. The scale bar measures amino acid substitutions per site.
Figure 9

Phylogenetic tree of translated spike gene sequences from betacoronaviruses.
Spike sequences are coloured according to whether they were from study specimens (blue), human coronaviruses (CoVs) (red), RefSeq (black), or GenBank (grey). Only the 25 closest-matching spike sequences from GenBank were included, as determined by blastp bitscores. GenBank and RefSeq accession numbers are provided in parentheses. The scale bar measures amino acid substitutions per site.
Tables
Table 1
Bat specimens and sequencing libraries analysed in this study.
Rectal and oral swabs collected for a previous study were used to evaluate hybridization probe capture (Kumakamba et al., 2021). For 19 swabs, archived RNA extracted during the previous study was assayed. For 6 swabs, freshly extracted RNA (using a conventional Trizol method) was assayed. As part of the previous study, Kumakamba et al. generated partial sequences from the RNA-dependent RNA polymerase gene, which were used to assign alpha- and betacoronaviruses in these specimens to four novel phylogenetic groups.
| Specimen ID | Library ID | Host | Swab type | RNA extraction method | Phylogenetic group |
|---|---|---|---|---|---|
| CDAB0017RSV | CDAB0017RSV-PRE | Micropteropus pusillus | Rectal | Previously extracted | W-Beta-2 |
| CDAB0040R | CDAB0040R-PRE | Myonycteris sp. | Rectal | Previously extracted | W-Beta-2 |
| CDAB0040RSV | CDAB0040RSV-PRE | Myonycteris sp. | Rectal | Previously extracted | W-Beta-2 |
| CDAB0305R | CDAB0305R-PRE | Micropteropus pusillus | Rectal | Previously extracted | W-Beta-2 |
| CDAB0146R | CDAB0146R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0158R | CDAB0158R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0160R | CDAB0160R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0173R | CDAB0173R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0174R | CDAB0174R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0203R | CDAB0203R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0212R | CDAB0212R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0217R | CDAB0217R-PRE | Eidolon helvum | Rectal | Previously extracted | W-Beta-3 |
| CDAB0113RSV | CDAB0113RSV-PRE | Hipposideros cf. ruber | Rectal | Previously extracted | W-Beta-4 |
| CDAB0486R | CDAB0486R-PRE | Chaerephon sp. | Rectal | Previously extracted | Q-Alpha-4 |
| CDAB0488R | CDAB0488R-PRE | Mops condylurus | Rectal | Previously extracted | Q-Alpha-4 |
| CDAB0488R | CDAB0488R-TRI | Mops condylurus | Rectal | Trizol re-extraction | Q-Alpha-4 |
| CDAB0491R | CDAB0491R-PRE | Mops condylurus | Rectal | Previously extracted | Q-Alpha-4 |
| CDAB0491R | CDAB0491R-TRI | Mops condylurus | Rectal | Trizol re-extraction | Q-Alpha-4 |
| CDAB0492R | CDAB0492R-PRE | Mops condylurus | Rectal | Previously extracted | Q-Alpha-4 |
| CDAB0492R | CDAB0492R-TRI | Mops condylurus | Rectal | Trizol re-extraction | Q-Alpha-4 |
| CDAB0494O | CDAB0494O-TRI | Mops condylurus | Oral | Trizol re-extraction | Q-Alpha-4 |
| CDAB0494R | CDAB0494R-PRE | Mops condylurus | Rectal | Previously extracted | Q-Alpha-4 |
| CDAB0494R | CDAB0494R-TRI | Mops condylurus | Rectal | Trizol re-extraction | Q-Alpha-4 |
| CDAB0495O | CDAB0495O-PRE | Mops condylurus | Oral | Previously extracted | Q-Alpha-4 |
| CDAB0495R | CDAB0495R-TRI | Mops condylurus | Rectal | Trizol re-extraction | Q-Alpha-4 |
Table 2
Sequencing metrics for probe captured libraries.
Total reads and sequencing output were measured for each library. Raw metrics describe unprocessed FASTQ files directly from the sequencer. Valid metrics describe FASTQ files following pre-processing to trim adapters, trim trailing low-quality bases, remove index hops, and remove PCR chimeras. On-target metrics were estimated by mapping valid data to the coronavirus reference sequence selected for each specimen and to the contigs assembled from each specimen.
| Library ID | Raw reads(#) | Raw output (kb) | Valid reads(#) | Valid output(kb) | Mapped reads(#) | Mapped size(kb) |
|---|---|---|---|---|---|---|
| CDAB0017RSV-PRE | 115,280 | 14,225.8 | 47,609 | 7362 | 36,716 | 4919 |
| CDAB0040R-PRE | 37,950 | 4708.9 | 695 | 121.1 | 0 | 0 |
| CDAB0040RSV-PRE | 373,254 | 45,333.7 | 176,783 | 26,783.3 | 48,136 | 7068.4 |
| CDAB0113RSV-PRE | 31,394 | 4261 | 16,861 | 2875 | 16,302 | 2772.2 |
| CDAB0146R-PRE | 11,870 | 1520 | 193 | 23.7 | 186 | 22.6 |
| CDAB0158R-PRE | 48,014 | 6422.5 | 4189 | 706.9 | 1548 | 239.8 |
| CDAB0160R-PRE | 83,524 | 9948.5 | 2513 | 376.6 | 1363 | 191.2 |
| CDAB0173R-PRE | 10,628 | 1403 | 206 | 34.4 | 203 | 33.7 |
| CDAB0174R-PRE | 900,578 | 118,821 | 107,979 | 17,525.4 | 82,679 | 13,290.9 |
| CDAB0203R-PRE | 6,832,218 | 849,284.9 | 1,186,186 | 188,188.9 | 456,474 | 68,384.7 |
| CDAB0212R-PRE | 60,838 | 7526 | 4158 | 681.4 | 4152 | 678.3 |
| CDAB0217R-PRE | 20,078,142 | 2,617,427.3 | 8,946,935 | 1,467,955.3 | 5,173,448 | 81,5381.3 |
| CDAB0305R-PRE | 27,054 | 3182.7 | 3971 | 594.9 | 1787 | 250 |
| CDAB0486R-PRE | 442,456 | 5,8326.7 | 56,838 | 9377 | 20,687 | 3385.1 |
| CDAB0488R-PRE | 188,294 | 24,679 | 2913 | 506.2 | 2867 | 493.1 |
| CDAB0488R-TRI | 343,916 | 45,867.1 | 8415 | 1381.2 | 8225 | 1346.2 |
| CDAB0491R-PRE | 791,120 | 96,136.3 | 46,509 | 7081.8 | 45,289 | 6854.6 |
| CDAB0491R-TRI | 1,561,144 | 204,995.4 | 173,533 | 29,280.5 | 157,889 | 26,421.2 |
| CDAB0492R-PRE | 3,453,456 | 448,217.9 | 277,176 | 48,023.8 | 185,665 | 31,641.3 |
| CDAB0492R-TRI | 4,200,520 | 518,837.7 | 139,804 | 20,074.7 | 93,442 | 13,294.3 |
| CDAB0494O-TRI | 141,494 | 18,980.9 | 290 | 49.7 | 60 | 11.3 |
| CDAB0494R-PRE | 82,360 | 11,162.7 | 22 | 4 | 0 | 0 |
| CDAB0494R-TRI | 95,924 | 12,762.1 | 9 | 2.1 | 0 | 0 |
| CDAB0495O-PRE | 27,074 | 3776 | 0 | 0 | 0 | 0 |
| CDAB0495R-TRI | 470,850 | 63,267 | 8896 | 1440.9 | 8672 | 1399.2 |
Table 3
Alignments between translated spike sequences from study specimens and phylogenetically proximate entries from GenBank and RefSeq.
Alignments were conducted with blastp. Reference sequence host and collection location were obtained from GenBank entry summaries.
| Specimen | Specimen host | Reference sequence GenBank accession number | Reference sequence host | Reference sequence collection location | Alignment query coverage(%) | Alignment identity(%) | Alignment positivity(%) |
|---|---|---|---|---|---|---|---|
| CDAB0492R | Mops condylurus | HQ728486.1 | Chaerephon sp. | Kenya | 100 | 71.2 | 80.1 |
| CDAB0492R | Mops condylurus | MZ081383.1 | Chaerephon plicatus | Yunnan, China | 100 | 65.8 | 77.5 |
| CDAB0017RSV | Micropteropus pusillus | HQ728482.1 | Eidolon helvum | Kenya | 99 | 76.5 | 85.7 |
| CDAB0017RSV | Micropteropus pusillus | MG693168.1 | Eidolon helvum | Cameroon | 99 | 63.7 | 77.7 |
| CDAB0040RSV | Myonycteris sp. | HQ728482.1 | Eidolon helvum | Kenya | 99 | 75.9 | 84.7 |
| CDAB0040RSV | Myonycteris sp. | MG693168.1 | Eidolon helvum | Cameroon | 99 | 64.4 | 77.7 |
| CDAB0203R | Eidolon helvum | HQ728482.1 | Eidolon helvum | Kenya | 100 | 73.7 | 85.3 |
| CDAB0203R | Eidolon helvum | MG693168.1 | Eidolon helvum | Cameroon | 100 | 65.6 | 78.8 |
| CDAB0217R | Eidolon helvum | HQ728482.1 | Eidolon helvum | Kenya | 100 | 73.5 | 85.1 |
| CDAB0217R | Eidolon helvum | MG693168.1 | Eidolon helvum | Cameroon | 100 | 65.2 | 79.0 |
Table 4
Nucleotide alignments between novel spike genes from study specimens and phylogenetically related sequences from GenBank and RefSeq.
Alignments were conducted with blastn. Discontinuous alignments are represented as multiple lines in the table, for example, CDAB0217R vs. MG693168.1.
| Specimen | Reference sequence GenBank accession number | Alignment query coverage(%) | Alignment identity(%) |
|---|---|---|---|
| CDAB0492R | HQ728486.1 | 60 | 81.0 |
| CDAB0492R | MZ081383.1 | 18 | 71.5 |
| CDAB0040RSV | HQ728482.1 | 83 | 75.4 |
| CDAB0203R | HQ728482.1 | 78 | 75.5 |
| CDAB0203R | MG693168.1 | 45 | 76.6 |
| CDAB0217R | HQ728482.1 | 71 | 76.0 |
| CDAB0217R | MG693168.1 | 47 | 75.7 |
| CDAB0217R | MG693168.1 | 47 | 84.6 |
Table 5
RT-qPCR primer sequences.
| Specimen | Primers | Primer sequences | Standard curve R2 |
|---|---|---|---|
| CDAB0146R-PRE CDAB0158R-PRE CDAB0160R-PRE CDAB0173R-PRE CDAB0174R-PRE CDAB0203R-PRE CDAB0212R-PRE CDAB0217R-PRE | Beta-3_rdrp_FWD Beta-3_rdrp_REV | ATA TAT GTC AGG CCG TTA GTG C CCA TAT AGA GGC GAT GTT GC | 0.995 |
| CDAB0486R-PRE CDAB0488R-PRE CDAB0488R-TRI CDAB0491R-PRE CDAB0491R-TRI CDAB0492R-PRE CDAB0492R-TRI CDAB0494O-TRI-PRE CDAB0494R-PRE CDAB0494R-TRI CDAB0495O-PRE CDAB0495R-TRI | Alpha_4_rdrp_FWD Alpha_4_rdrp_REV | GCG ACT ACC TGG TAA ACC TAT C CTT TGC CGC ACT CAC AAA C | 0.989 |
| CDAB0017R-PRE CDAB0040R-PRE CDAB0040RSV-PRE | Beta-2_rdrp_FWD Beta-2_rdrp_REV | CAC TAC TTG TAC CAC CAG GTT T TTG TAG TGG TTC TGA TCG GTT T | 0.998 |
| CDAB0305R-PRE | D0305_rdrp_FWD D0305_rdrp_REV | GAC GGC AAT AAG GTG CAT AAC AGT CAG AAA CCA AGT CCT CAT C | 0.999 |
| CDAB0113RSV-PRE | D0113_rdrp_FWD D0113_rdrp_REV | GTA CGT TGA GTG AGC GGT ATT GAT GAA GTT CCA CCT GGC TTA | 0.998 |
Additional files
-
Supplementary file 1
Probe sequences, fasta format.
- https://cdn.elifesciences.org/articles/79777/elife-79777-supp1-v1.txt
-
Supplementary file 2
gBlock sequences, fasta format.
Two gBlocks were generated for this study. ‘Gblck_Beta2_17_40_B3_Alpha’ is a composite of RdRp nucleotide sequences from W-Beta-2 (samples CDAB0017 and CDAB0040), W-Beta-3, and Q-Alpha-4 (Table 1). ‘Gblck_Beta2_0305_B4_Alpha’ is a composite of RdRp nucleotide sequences from W-Beta-2 (samples CDAB0305), W-Beta-4, and Q-Alpha-4. The Q-Alpha-4 sequence is identical in both gBlocks.
- https://cdn.elifesciences.org/articles/79777/elife-79777-supp2-v1.txt
-
MDAR checklist
- https://cdn.elifesciences.org/articles/79777/elife-79777-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats
eLife 11:e79777.
https://doi.org/10.7554/eLife.79777




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}