The meningeal transcriptional response to traumatic brain injury and aging
- Department of Neuroscience, Center for Brain Immunology and Glia (BIG), University of Virginia School of Medicine, United States
- Department of Microbiology, Immunology and Cancer Biology, University of Virginia School of Medicine, United States
- Medical Scientist Training Program, University of Virginia School of Medicine, United States
- Immunology Training Program, Immunology Training Program, United States
- Department of Biochemistry and Molecular Genetics, University of Virginia School of Medicine, United States
- Center for Public Health Genomics, University of Virginia School of Medicine, United States
Figures
Figure 1 with 2 supplements
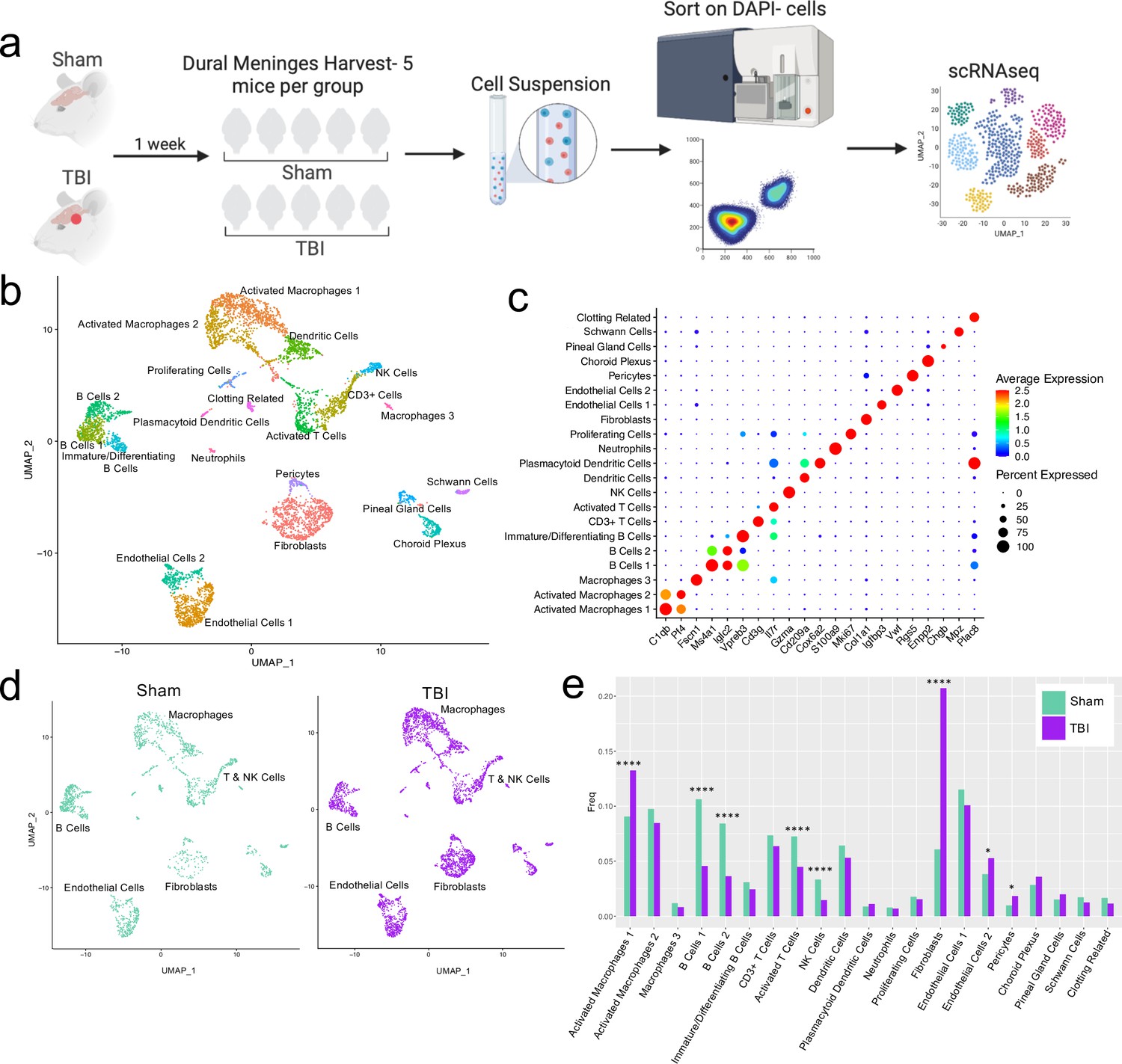
Alterations in the composition of meningeal cell populations following brain injury.
Male C57BL/6 J wild-type (WT) mice at 10 weeks of age were subjected to a mild closed-skull injury above the right inferior temporal lobe or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Schematic of scRNA-seq protocol. (b) Uniform Manifold Approximation and Projection (UMAP) representation of the cell populations present in the meninges where both Sham and TBI groups are included. Colors are randomly assigned to each cell population. (c) Dot plot representation of cluster defining genes for each cell population, where each gene represents the most significant cluster-defining marker for each population. The color and size of each dot represents the average expression and percent of cells expressing each gene, respectively. (d) UMAP representations of the cell populations present in the meninges separated by Sham (sage) and TBI (purple). (e) Bar graph depicting frequencies of cell populations in Sham vs. TBI samples. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality. Each data point in a UMAP plot represents a cell. p Values were calculated using a two sample z-test. ****p<0.0001, *p<.05, bar chart pairs without * were not statistically significant. Exact statistics are provided in the source data file.
-
Figure 1—source data 1
Cluster-defining genes for single cell populations.
Tables depicting the top 20 most significant cluster-defining genes for clusters 1–21. Raw data for top cluster defining genes for each cell population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig1-data1-v1.xlsx
-
Figure 1—source data 2
Frequency, cell count, and p-value for each single cell population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig1-data2-v1.xlsx
Figure 1—figure supplement 1
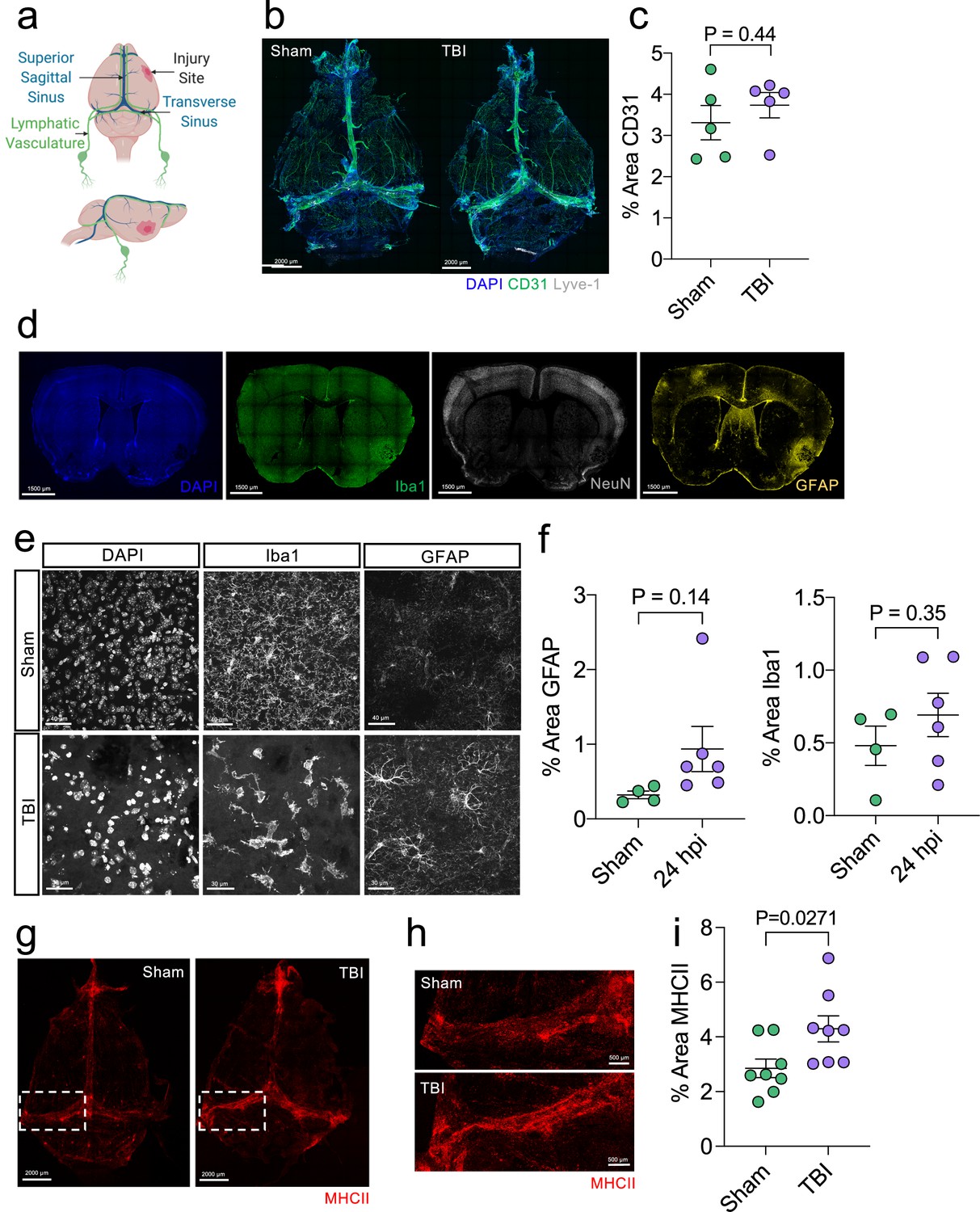
Initial brain and meningeal response following TBI.
Male C57BL/6 J wild-type (WT) mice at 10 weeks of age received a TBI or Sham procedure and then the brains or meninges were harvested for immunohistochemistry at 24 hours. (a) Schematic depicting the location of the TBI in relation to dorsal anatomical structures. (b) Representative images of meningeal whole mounts stained with DAPI (blue), CD31 (green) and Lyve-1 (grey). (c) Quantification of the percent area of CD31 in each meningeal whole mount. Each data point represents an individual mouse (Sham n=5, TBI n=5, rep = 1). (d) Representative images of brains with injury site (right) taken 24 hr following TBI stained with DAPI (blue), Iba1 (green), NeuN (grey) and GFAP (yellow). (e) Representative high-magnification images (63 x) of Iba1 and GFAP-positive cells (microglia/macrophages and reactive astrocytes respectively) and (f) quantification of the percent area of GFAP and Iba1-positive cells in the hemisphere ipsilateral to the injury 24 hr after TBI. Each data point represents an individual mouse (Sham n=4, TBI n=6, rep = 2). (g–i) Meningeal whole mounts stained with MHCII (red) taken 24 hr post TBI. Dashed boxes in (g) represent zoomed areas of transverse sinus shown in (h). (i) Quantification of the % area MHCII staining in each meningeal whole mount. Each dot represents one mouse (Sham n=8, TBI n=8, rep = 1). Error bars depict mean ± s.e.m. p Values were calculated using an unpaired students t-test.
-
Figure 1—figure supplement 1—source data 1
Table depicting the percent area of CD31 + immunofluorescence in the meninges 24 hr after TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig1-figsupp1-data1-v1.xlsx
-
Figure 1—figure supplement 1—source data 2
Tables depicting the percent area of GFAP + and Iba1 + immunofluorescence in the brain 24 hr after TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig1-figsupp1-data2-v1.xlsx
-
Figure 1—figure supplement 1—source data 3
Table depicting the percent area of MHCII + immunofluorescence in the meninges 24 hr after TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig1-figsupp1-data3-v1.xlsx
Figure 1—figure supplement 2
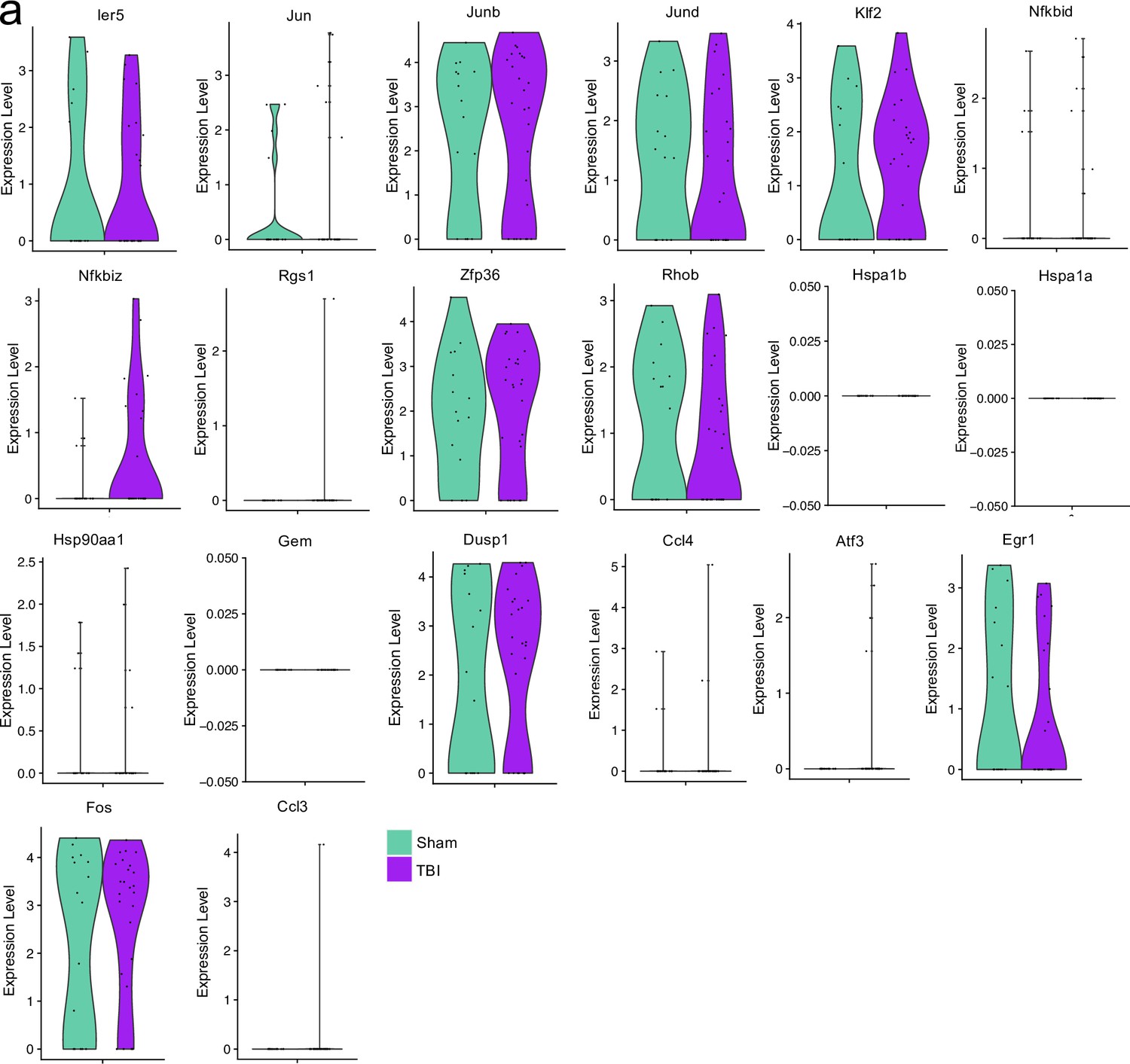
Stress and processing related genes after single cell RNA-sequencing.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. Violin plots depicting various genes split by experimental group: Sham (sage) and TBI (purple). Each dot represents an individual cell. The width of the violin plot represents the frequency of observations at that given y-value. Therefore, the wider the violin plot, the higher the frequency of observations. Plots without sage or purple coloring did not have enough cells expressing the gene to create the plot. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality.
Figure 2 with 2 supplements
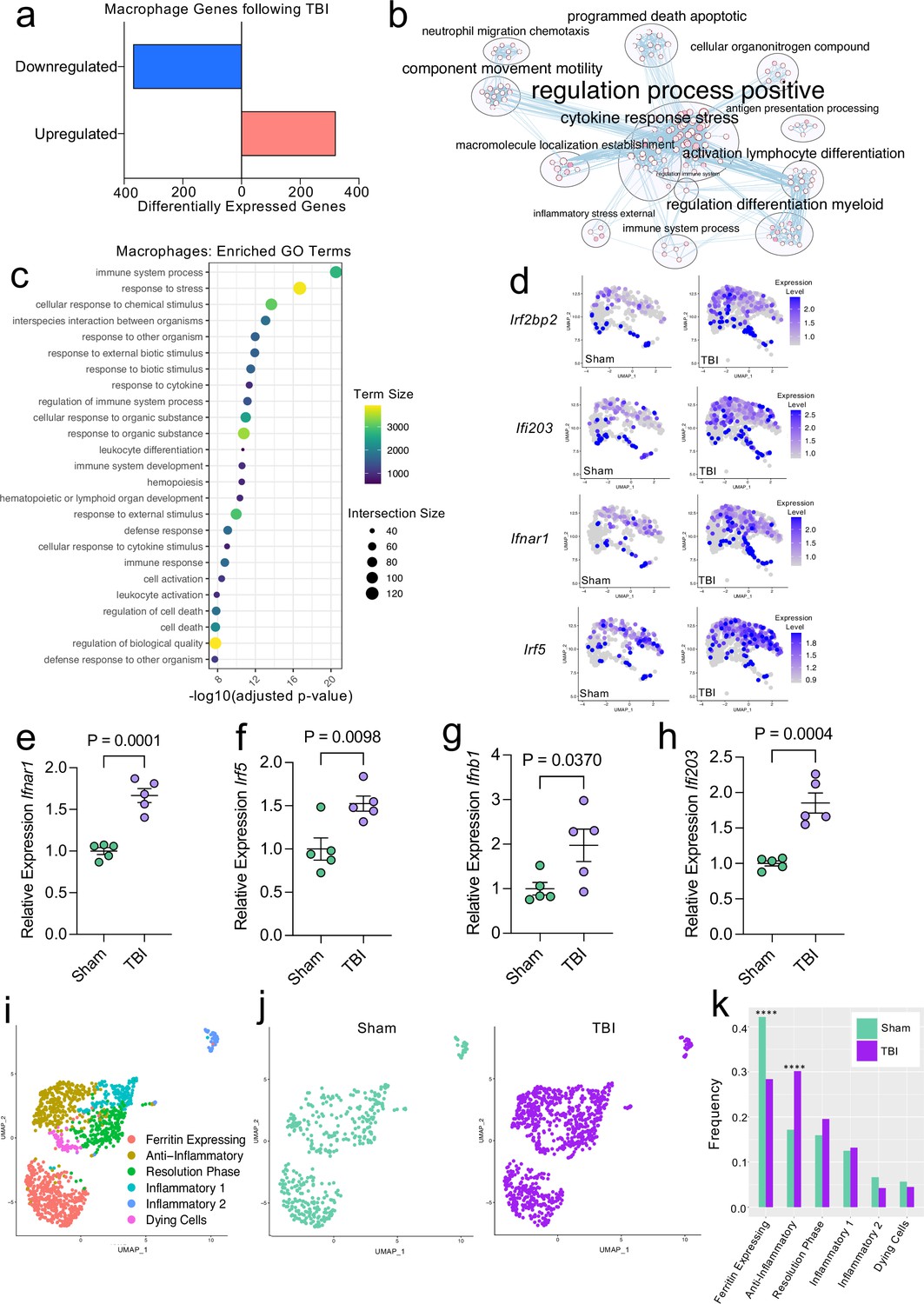
Transcriptional response of meningeal macrophages to mild TBI.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Quantification of the number of upregulated and downregulated macrophage genes following injury (FDR < 0.1). (b) Network analysis of significantly upregulated genes in meningeal macrophages following injury. Text size is proportional to the number of genes enriched in that cluster. Node size is roughly proportional to the number of GO terms in that cluster (node size was manually adjusted so may not be exactly proportional to GO terms included). Dot size is proportional to the number of genes contributing to each GO term. Dot color is proportional to p-value, where colors closer to white have lower p-values. Connecting lines represent GO terms with shared genes, more lines represents a higher number of shared genes between nodes. (c) Dot plot showing the 25 most enriched GO terms with significantly upregulated genes following TBI in the meningeal macrophage population. The color and size of each dot represents the size of the GO term and the number of upregulated genes that contribute to each term, respectively. (d) Feature plots depicting several significantly upregulated genes following injury (FDR < 0.1). The color of each data point represents the expression level of the indicated gene within that cell. Quantitative PCR relative expression of (e) Ifnar1, (f) Irf5, (g) Ifnb1, and (h) Ifi203 within the dural meninges one week after TBI (Sham n=5, TBI n=5, rep = 1). (i) UMAP representation showing re-clustering of the meningeal macrophage populations. (j) UMAP representation of the macrophages present in the meninges separated by Sham (sage) and TBI (purple). (k) Frequencies of meningeal macrophage populations in Sham vs. TBI samples represented as a gradient bar chart. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality. Differential gene expression was calculated using the ZINB-WaVE function for zero-enriched datasets and DESeq2. Each data point in a UMAP plot represents a cell. Error bars in (e–h) depict mean ± s.e.m. p Values for (e–h) were calculated using unpaired two-sample students t-tests and p values for (k) were calculated using a two sample z-test. ****p<0.0001. Bar chart pairings without * were not statistically significant, exact statistics are provided in the source data file. FDR; false discovery rate.
-
Figure 2—source data 1
Number of up- and downregulated macrophage genes 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data1-v1.xlsx
-
Figure 2—source data 2
Raw data showing enriched GO-terms and contributory differentially expressed macrophage genes.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data2-v1.xlsx
-
Figure 2—source data 3
Raw data for the differential expression analysis in the macrophage population 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data3-v1.xlsx
-
Figure 2—source data 4
Relative expression of interferon-related genes by qPCR in whole meninges 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data4-v1.xlsx
-
Figure 2—source data 5
Cluster-defining genes for macrophage populations.
Tables depicting the top 10 most significant cluster-defining genes for macrophage populations. Raw data for top cluster defining genes for each cell population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data5-v1.xlsx
-
Figure 2—source data 6
Frequency, cell count, and p-value for each macrophage population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-data6-v1.xlsx
Figure 2—figure supplement 1
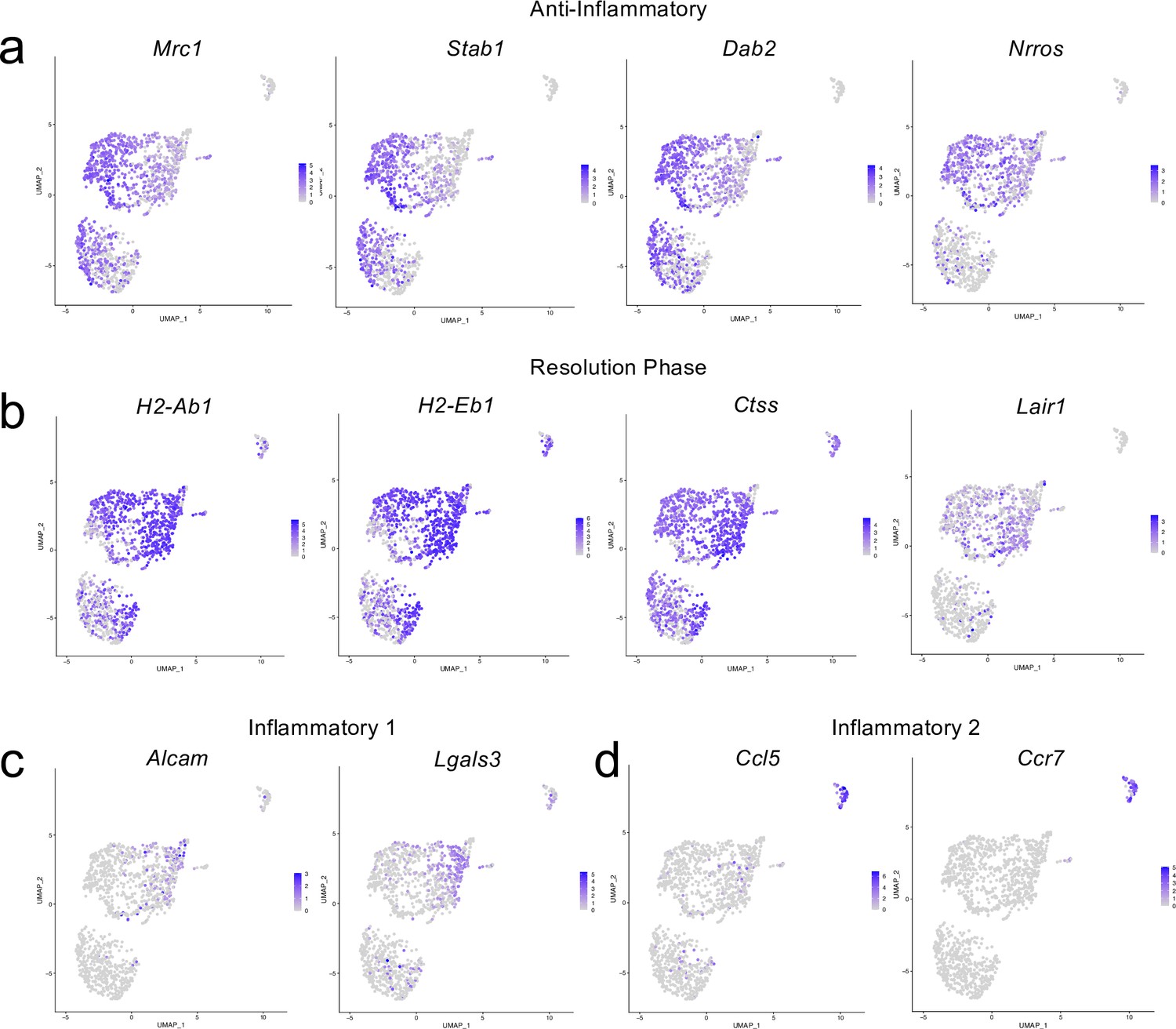
Cluster-defining genes for macrophage subpopulations.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a–d) Feature plots showing expression patterns of (a) Anti-inflammatory, (b) Resolution phase, (c) Inflammatory 1, and (d) Inflammatory 2 macrophage cluster-defining genes. The color of each data point represents the expression level of the indicated gene within that cell. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality.
Figure 2—figure supplement 2
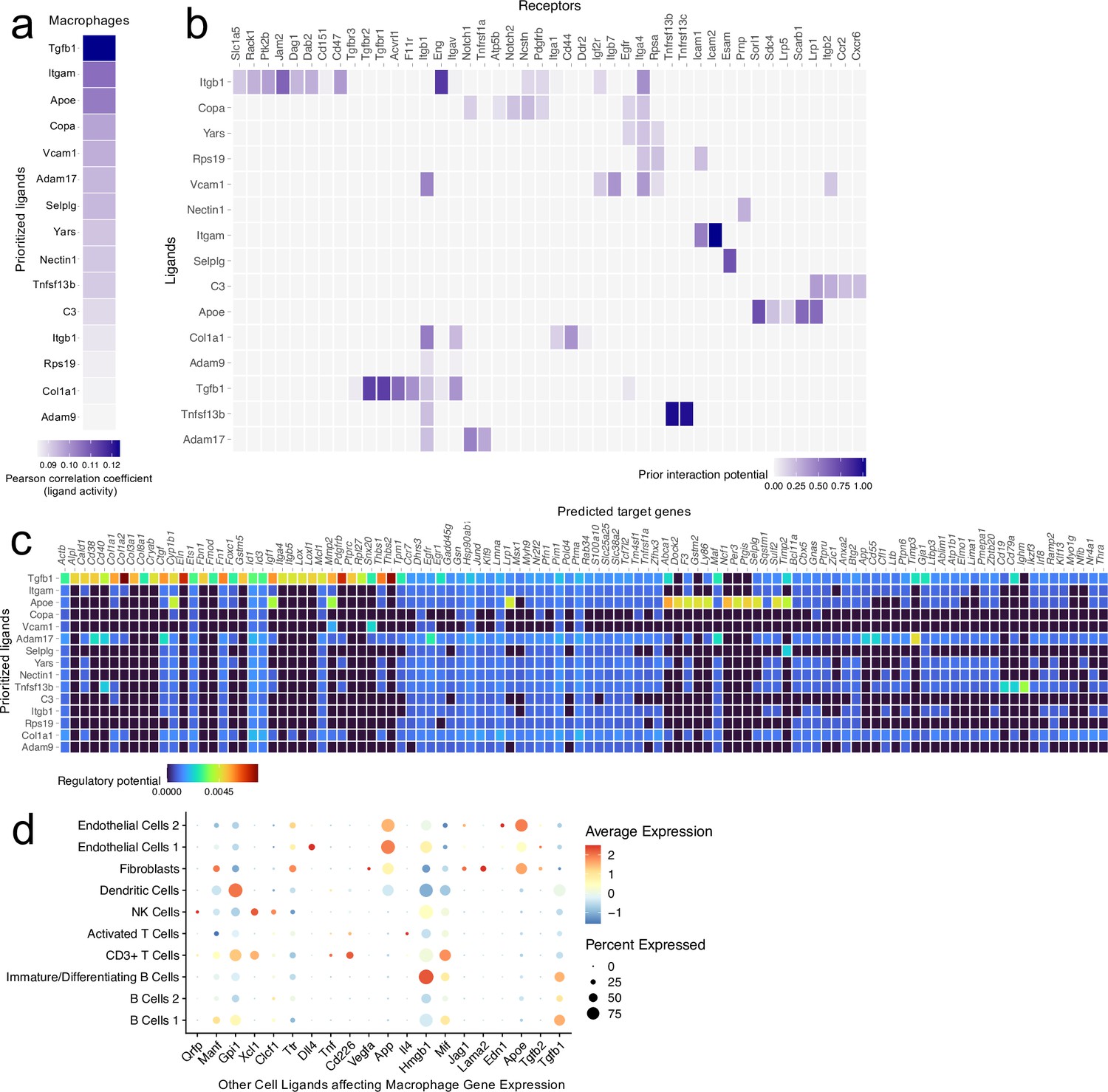
Ligand-target interactions between macrophages and other meningeal cells.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Heatmap showing the top prioritized macrophage ligands where the darkest shades represent the highest Pearson correlation coefficient, which corresponds to the ligands that best predict the TBI target genes. (b) Heatmap showing the prioritized ligands and their predicted receptors. The darkest shades represent the ligand-receptor pairs that interact most robustly. (c) Heatmap depicting the prioritized ligands and their predicted target genes. Warmer colors represent genes that are more likely regulated by the shown ligand whereas cooler colors are less likely to be regulated by the shown ligand. (d) Dotplot depicting which other cell populations are most likely to affect gene expression in the macrophage population through ligand-target interactions. The warmer colors represent higher average expression of the shown ligand within a cell population and the size of the dot represents the frequency at which the ligand is expressed within a certain cell population. Graphs were produced with NicheNet using the same Seurat object generated by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality.
-
Figure 2—figure supplement 2—source data 1
List of top macrophage ligands likely to influence the gene expression patterns of the other major cell populations in the scRNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig2-figsupp2-data1-v1.xlsx
Figure 3 with 1 supplement
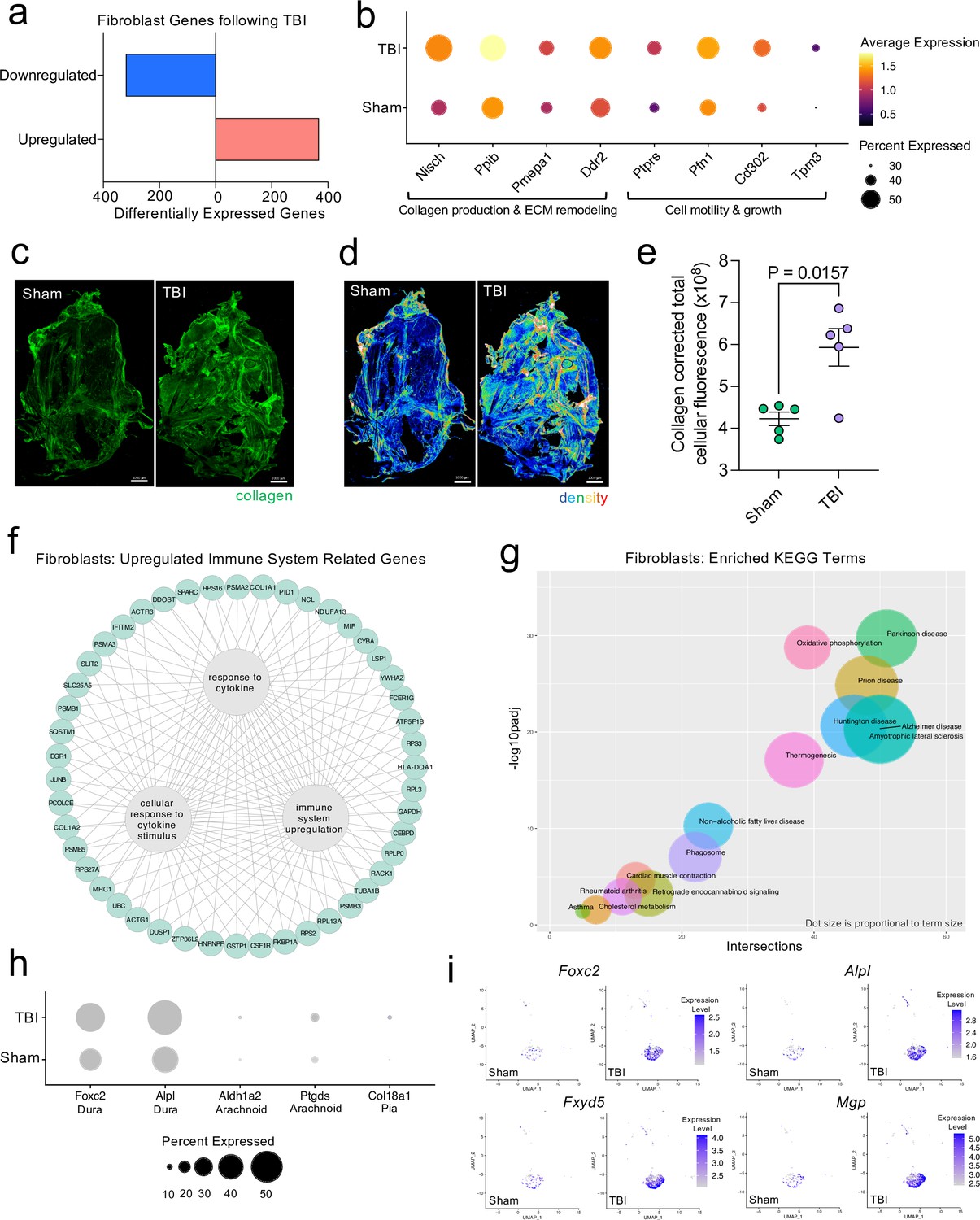
Dural fibroblasts express genes involved in tissue remodeling, cell migration, and immune activation in TBI.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Quantification of the number of upregulated and downregulated fibroblast genes following injury (FDR < 0.1). (b) Dot plot representation of highlighted fibroblast genes that were significantly upregulated following injury (FDR < 0.1). The color and size of each dot represents the average expression and percent of cells expressing each gene, respectively. (c–d) Representative images of meningeal whole mounts stained for collagen (green) (c) and a 16 color heatmap of the collagen staining intensity (d), where red is most intense and blue is least intense. (e) Quantification of collagen staining intensity using corrected total cellular fluorescence (CTCF). CTCF is calculated as mean fluorescence of meningeal whole mounts - (Area of meningeal whole mount x Mean fluorescence of background). Each data point represents an individual mouse (Sham n=5, TBI n=5, rep = 1). (f) Network map depicting significantly upregulated genes that enriched immune system-related GO terms (FDR < 0.1). The lines within the circle indicate which genes contribute to each GO term. (g) Scatter plot representation of the top enriched KEGG terms with significantly upregulated genes in the fibroblast population (FDR < 0.1). Dot size is proportional to term size. Genes contributing to one KEGG term may also contribute to other KEGG terms. (h) Dot plot depicting dural, arachnoid, and pial fibroblasts markers where the size of the circles represents the percent of cells expressing each gene. (i) Feature plots of genes characteristic of dural fibroblasts in both Sham and TBI conditions. The color of each data point represents the expression level of the indicated gene within that cell. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality. Differential gene expression was calculated using the ZINB-WaVE function for zero-enriched datasets and DESeq2. Each data point in a UMAP plot represents a cell. Error bars depict mean ± s.e.m. p value for (e) was calculated using an unpaired two-tailed t-test assuming unequal variances. FDR; false discovery rate, p.adj; adjusted p-value.
-
Figure 3—source data 1
Number of up- and downregulated fibroblast genes one week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig3-data1-v1.xlsx
-
Figure 3—source data 2
Raw data for the differential expression analysis in the fibroblast population 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig3-data2-v1.xlsx
-
Figure 3—source data 3
Table depicting the corrected total cellular fluorescence for collagen in meningeal whole mounts.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig3-data3-v1.xlsx
-
Figure 3—source data 4
Raw data showing enriched KEGG disease processes and contributory differentially expressed fibroblast genes.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig3-data4-v1.xlsx
Figure 3—figure supplement 1
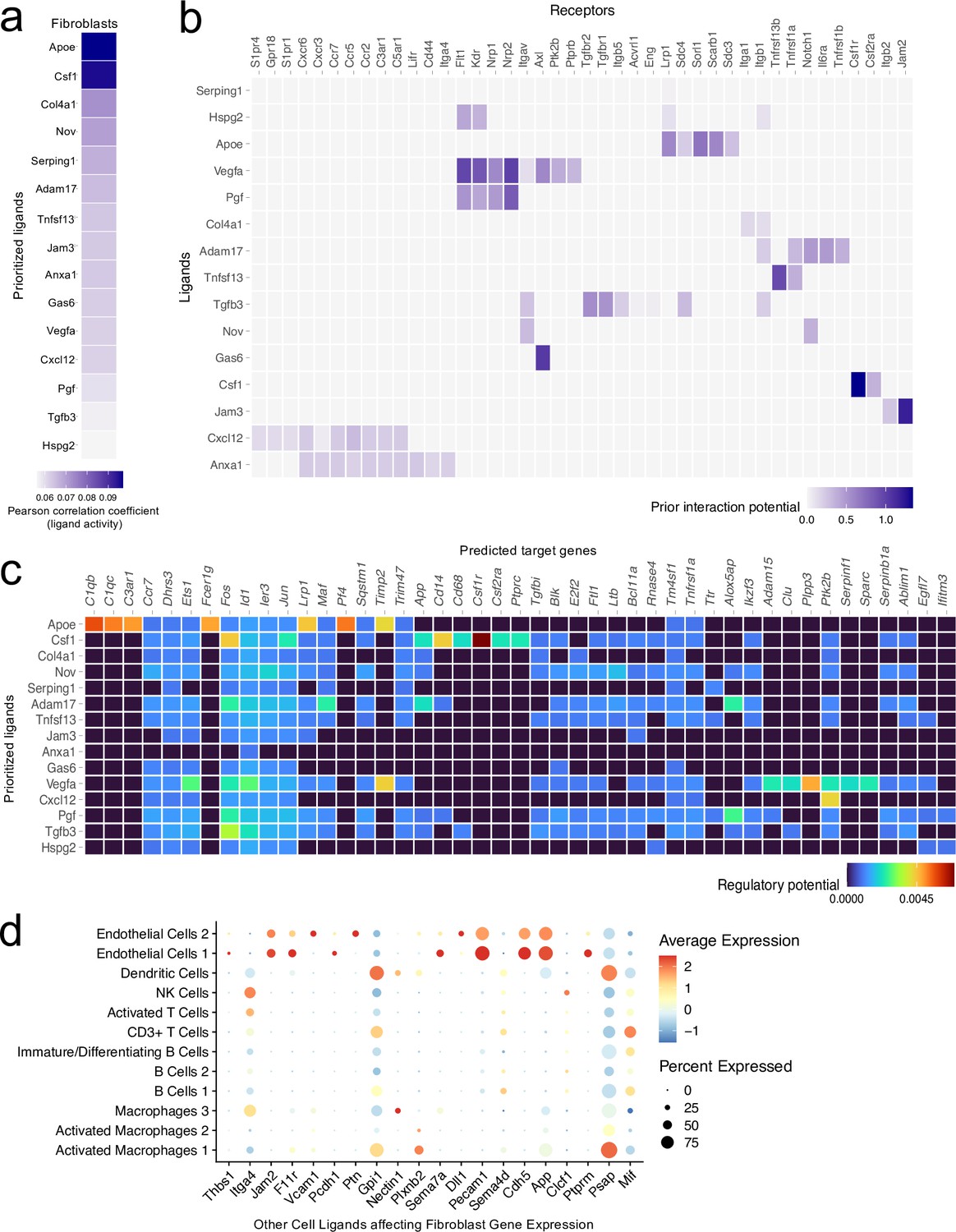
Ligand-target interactions between fibroblasts and other meningeal cells.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Heatmap showing the top prioritized fibroblast ligands where the darkest shades represent the highest Pearson correlation coefficient, which corresponds to the ligands that best predict the TBI target genes. (b) Heatmap showing the prioritized ligands and their predicted receptors. The darkest shades represent the ligand-receptor pairs that interact most robustly. (c) Heatmap depicting the prioritized ligands and their predicted target genes. Warmer colors represent genes that are more likely regulated by the shown ligand whereas cooler colors are less likely to be regulated by the shown ligand. (d) Dotplot depicting which other cell populations are most likely to affect gene expression in the fibroblast population through ligand-target interactions. The warmer colors represent higher average expression of the shown ligand within a cell population and the size of the dot represents the frequency at which the ligand is expressed within a certain cell population. Graphs were produced with NicheNet using the same Seurat object generated by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality.
-
Figure 3—figure supplement 1—source data 1
List of top fibroblast ligands likely to influence the gene expression patterns of the other major cell populations in the scRNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig3-figsupp1-data1-v1.xlsx
Figure 4 with 2 supplements
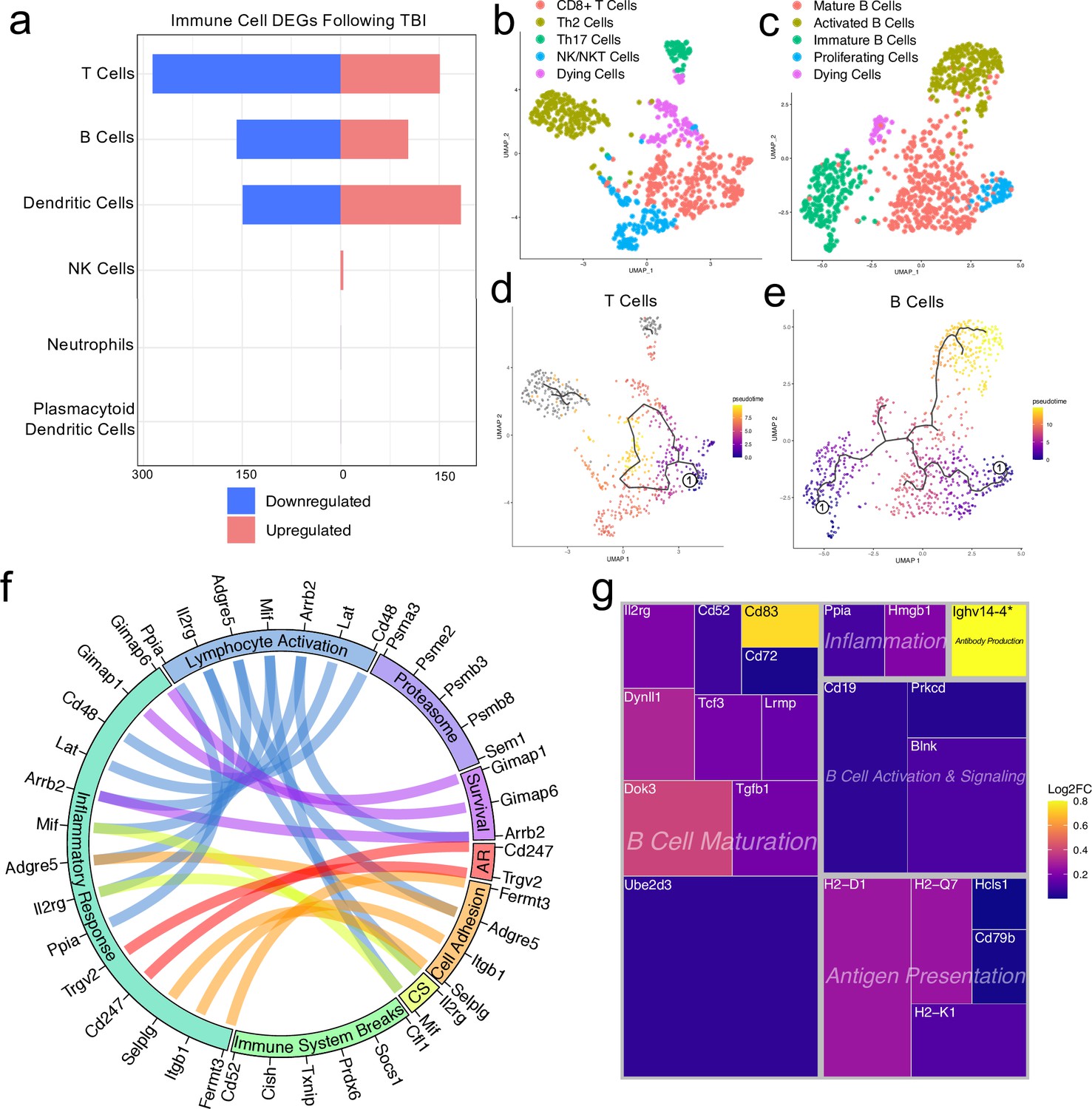
Transcriptional response of meningeal lymphocytes to mild TBI.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a) Quantification of the number of upregulated and downregulated genes in different immune cell populations following injury (FDR <0.1). (b–c) UMAP representation showing re-clustering of the (b) T cell and (c) B cell populations present within the meninges. (d-e) UMAP representation of pseudotime cellular trajectory profiles showing (d) T cell and (e) B cell maturation trajectories. The circle with the number ‘1’ represents the root node. The color of each data point represents advancement in pseudotime, with dark purple representing ‘early’ pseudotime and yellow representing ‘late’ pseudotime. The line represents the ‘path’ of pseudotime with intersections representing possible different differentiation events. Grey data points represent cell populations that were not connected in pseudotime with the selected node. (f) Circos plot depicting differentially expressed genes in the T cell populations within the TBI meninges (FDR <0.1) associated with different cellular processes. The proportion of the circle’s circumference allocated to each cellular process represents the number of T cell genes associated with that process that are differentially expressed in the TBI meninges. The lines connecting genes within the circle indicate which genes were shared amongst cellular processes. Colors were randomly assigned. (g) Treemap depicting significantly upregulated genes in the B cell population and the cellular process to which each gene contributes. The size of the square around each gene represents the Wald statistic, which is used to calculate the overall significance of the change in gene expression (a larger square indicates a larger Wald statistic, which leads to a lower adjusted p-value). The color of the boxes represents log2FC, where purple represents a lower log2FC and yellow represents a higher log2FC. An asterisk (*) indicates that the log2FC of the gene was higher than the scale (Ighv14-4 had a log2FC of 18.08). Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality. Each data point in a UMAP plot represents a cell. Differential gene expression was calculated using the ZINB-WaVE function for zero-enriched datasets and DESeq2. Pseudotime graphs were created using Monocle. AR, antigen recognition; CS, cytokine signaling; FDR; false discovery rate, log2FC; log 2 fold change.
-
Figure 4—source data 1
Number of up- and downregulated immune-cell genes 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig4-data1-v1.xlsx
-
Figure 4—source data 2
Cluster-defining genes for T cell populations.
Tables depicting the top 10 most significant cluster-defining genes for T cell populations. Raw data for top cluster defining genes for each cell population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig4-data2-v1.xlsx
-
Figure 4—source data 3
Cluster-defining genes for B cell populations.
Tables depicting the top 10 most significant cluster-defining genes for B cell populations. Raw data for top cluster defining genes for each cell population.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig4-data3-v1.xlsx
-
Figure 4—source data 4
Differentially expressed T cell genes contributing to immune-related cellular functions.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig4-data4-v1.xlsx
-
Figure 4—source data 5
Raw data for the differential expression analysis in B cells 1 week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig4-data5-v1.xlsx
Figure 4—figure supplement 1
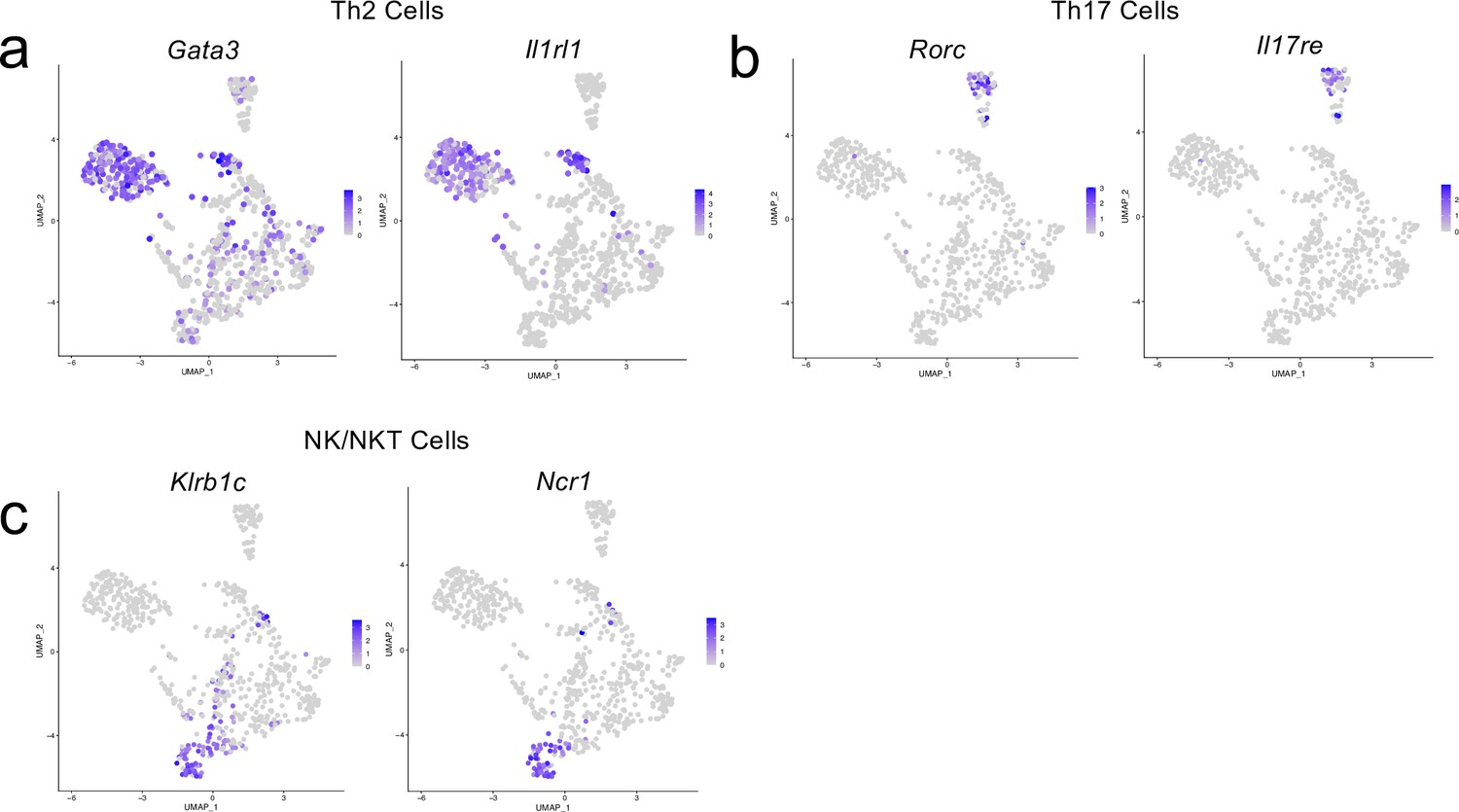
Cluster-defining genes for T cell subpopulations.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a–c) Feature plots showing expression patterns of (a) Th2, (b) Th17 and (c) NK/NKT T cell subset cluster-defining genes. The color of each data point represents the expression level of the indicated gene within that cell. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and reducing the dimensionality.
Figure 4—figure supplement 2
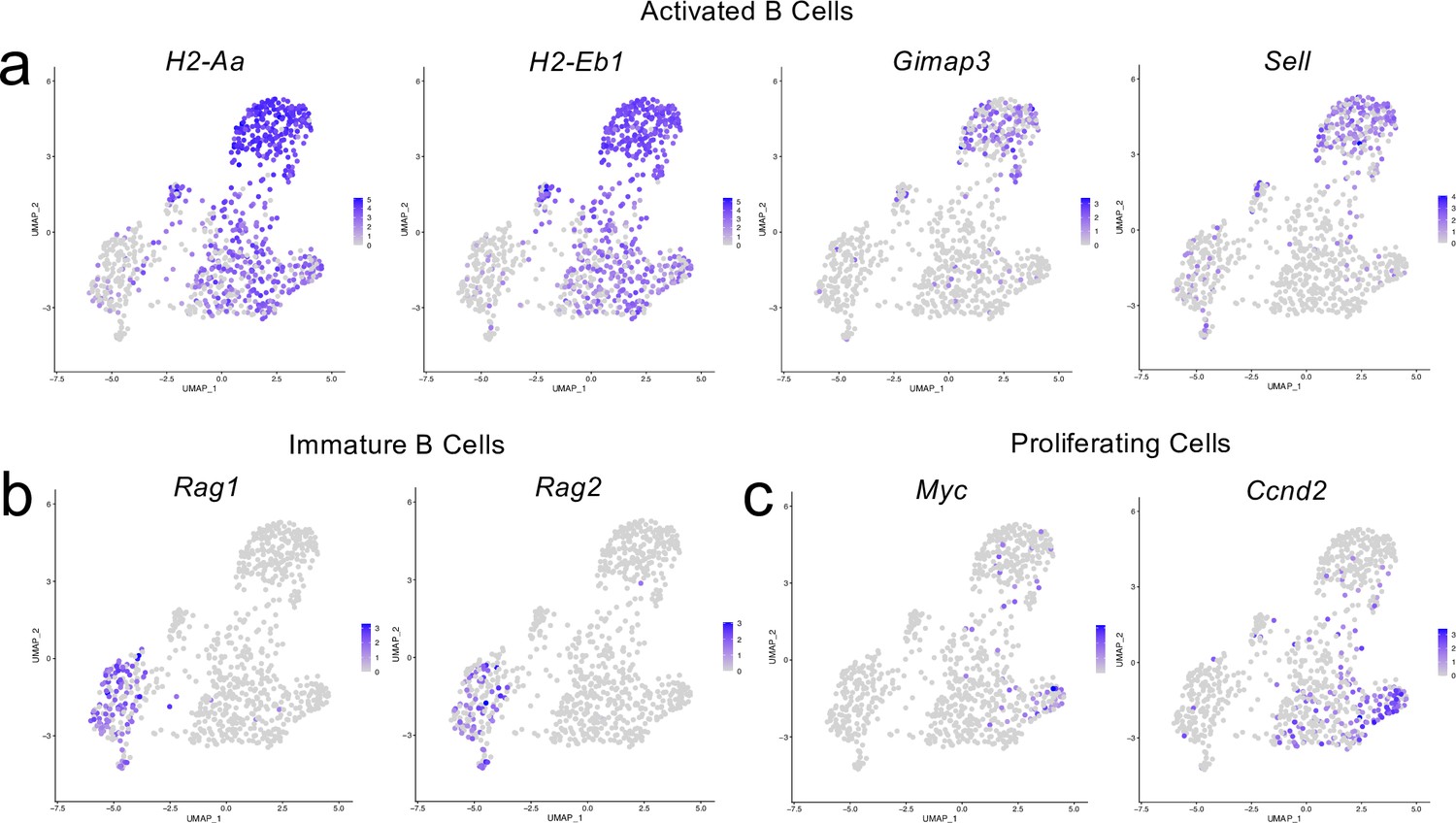
Cluster-defining genes for B cell subpopulations.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. (a–c) Feature plots showing expression patterns of (a) Activated, (b) Immature and (c) Proliferating B cell subset cluster-defining genes. The color of each data point represents the expression level of the indicated gene within that cell. Graphs were calculated using Seurat by normalizing the dataset, finding the variable features of the dataset, scaling the data, and, reducing the dimensionality.
Figure 5
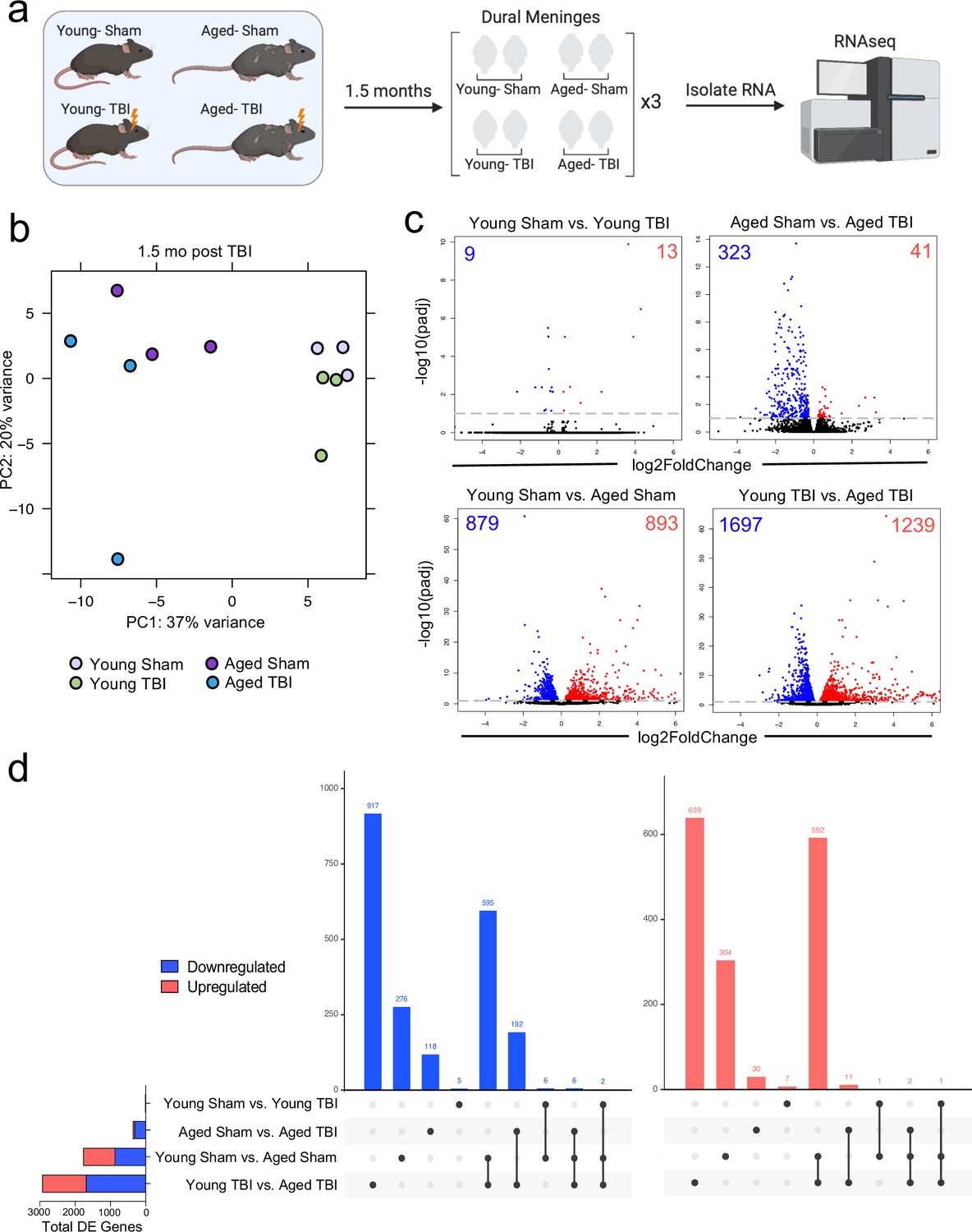
Effects of aging and mild TBI on the meningeal transcriptome.
(a) Schematic depicting experimental layout. Male WT mice at 10 weeks of age or 20 months of age received a TBI or Sham procedure. 1.5 months later, bulk RNA-seq was performed on the four experimental groups with three biological replicates per group (each biological replicate consisted of meningeal RNA samples from 2 to 3 independent mice). (b) Principal component analysis (PCA) showing clustering of samples. (c) Volcano plots illustrate the number of differentially expressed genes with statistically significant differences denoted in blue and red (FDR <0.1). Numbers in each corner depict the number of differentially expressed genes for each comparison. Blue data points represent significantly downregulated genes and red data points represent significantly upregulated genes. (d) Upset plots depicting significantly downregulated (blue) and upregulated (red) genes for each comparison and the number of genes that were shared between comparisons (FDR <0.1). The graph on the left sidebar shows the total number of differentially expressed genes per group. A single black dot indicates the differentially expressed genes are unique to the highlighted comparison. Two or more black dots connected by a line indicate that the differentially expressed genes are shared between the multiple highlighted comparisons. FDR and p-values were calculated with DESeq2 using the Wald test for significance following fitting to a negative binomial linear model and the Benjamini-Hochberg procedure to control for false discoveries. FDR; false discovery rate, DE; differentially expressed.
-
Figure 5—source data 1
Significantly up- and downregulated genes for each bulk RNA-seq comparison.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig5-data1-v1.xlsx
-
Figure 5—source data 2
Raw data of shared and unique differentially expressed genes for each comparison.
Data shows the number of differentially expressed genes in each comparison and whether each gene was differentially expressed in each comparison.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig5-data2-v1.xlsx
Figure 6 with 1 supplement
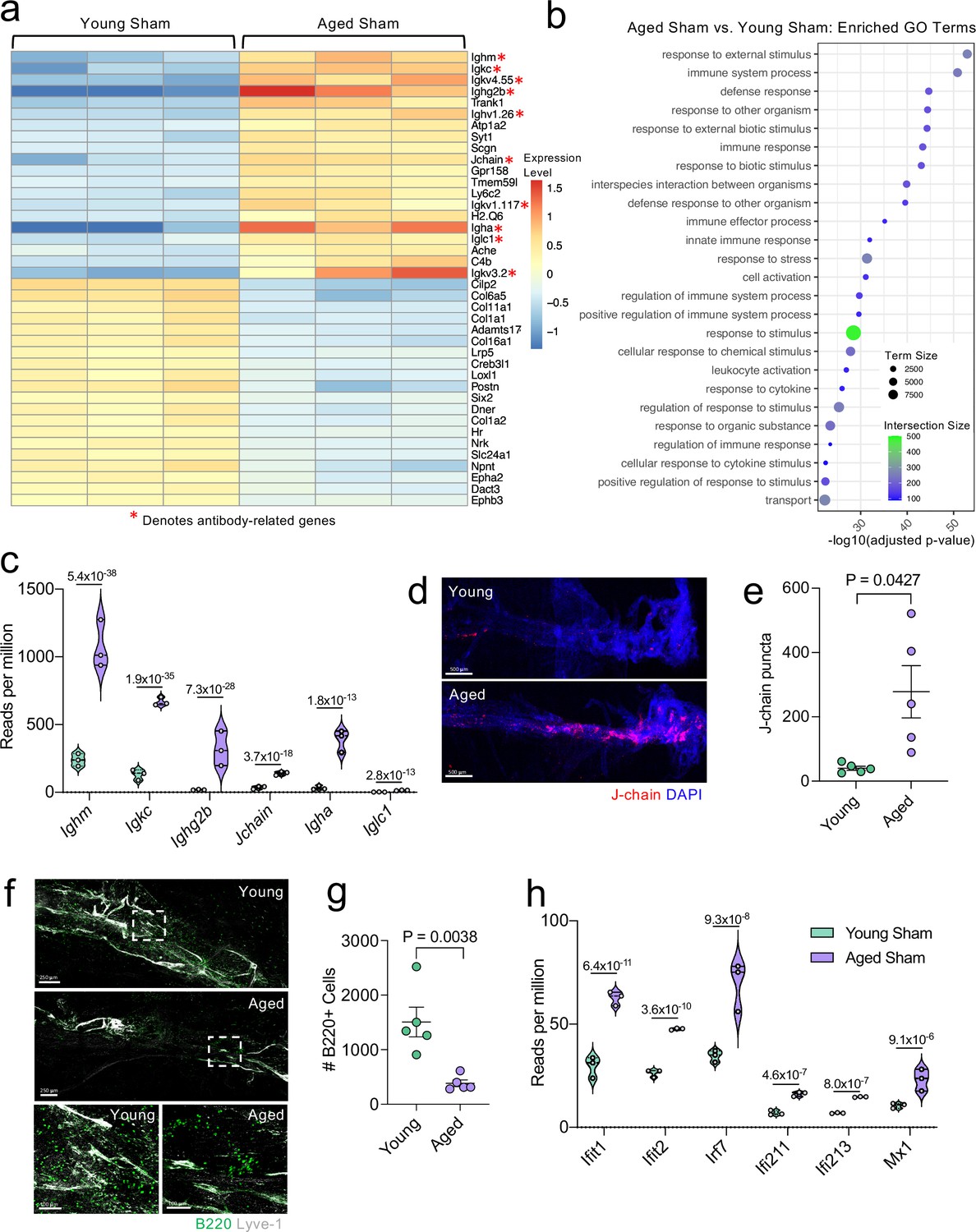
Aging promotes the upregulation of meningeal genes involved in type I IFN and antibody signaling.
Male WT mice at 10 weeks of age or 20 months of age received a TBI or Sham procedure. 1.5 months later, bulk RNA-seq was performed on the four experimental groups with three biological replicates per group (each biological replicate consisted of meningeal RNA samples from 2 to 3 independent mice). (a) Heatmap representation of the top 20 most significantly upregulated and downregulated (FDR <0.1) genes in the Young Sham vs. Aged Sham groups. The red asterisk (*) indicates genes associated with antibody production. (b) Dot plot of GO term biological processes shows enrichment of immune-related pathways with differentially expressed genes between young mice as compared to aged mice. Color and size of each dot represent the size of the GO term and the number of upregulated genes that contribute to each term, respectively. (c) Violin plot depicting counts of significantly activated antibody and B cell related genes in response to age (FDR <0.1). The number above each comparison on the graph represents the adjusted p-value calculated for each gene using DESeq2. The central line within each plot represents the median of the data set. The upper and lower boundaries of the box represent the third (Q3) and first (Q1) quartiles respectively. The violin plot encompasses the three biological replicates. The width of the violin plot represents the frequency of observations at that given y-value. Therefore, the wider the violin plot, the higher the frequency of observations. The meninges of 5 young Sham mice and 5 aged Sham mice were harvested for each immunohistochemical experiment. (d) Representative images from a young Sham mouse and aged Sham mouse showing a region of the SSS stained with J-chain (red) and Lyve-1 (grey) (e) and quantification of J-chain puncta in meningeal whole mounts along the SSS (Sham n=5, TBI n=5, rep = 1). (f) Representative images of the transverse sinus in young and aged mice stained with B220 (green) and Lyve-1 (grey). The dashed box on the top two images corresponds to the higher magnification images depicted below. (g) Quantification of the number of B220 cells along the entire transverse sinus (Sham n=5, TBI n=5, rep = 1). (h) Violin plot depicting counts of significantly activated type-I interferon related genes in response to age (FDR < 0.1). The violin plot parameters are the same as describe for (c). FDR and p-values in (a–c,h) were calculated with DESeq2 using the Wald test for significance following fitting to a negative binomial linear model and the Benjamini-Hochberg procedure to control for false discoveries. Error bars in (e,g) depict mean ± s.e.m. p values in (e,g) were calculated using a two-tailed unpaired two-sample t-test assuming unequal variances. FDR; false discovery rate, SSS; superior sagittal sinus.
-
Figure 6—source data 1
Table depicting all differentially regulated genes in the Young Sham vs. Aged Sham comparison.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data1-v1.xlsx
-
Figure 6—source data 2
Raw data depicting enriched GO-terms and contributory genes that were upregulated in aging.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data2-v1.xlsx
-
Figure 6—source data 3
Number of reads per million of antibody-related genes upregulated in aging in the bulk RNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data3-v1.xlsx
-
Figure 6—source data 4
Table depicting the number of J-chain puncta quantified in young and aged mice.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data4-v1.xlsx
-
Figure 6—source data 5
Table depicting the number of B220 + cells along the transverse sinus in young and aged mice.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data5-v1.xlsx
-
Figure 6—source data 6
Number of reads per million of interferon-related genes upregulated in aging in the bulk RNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-data6-v1.xlsx
Figure 6—figure supplement 1
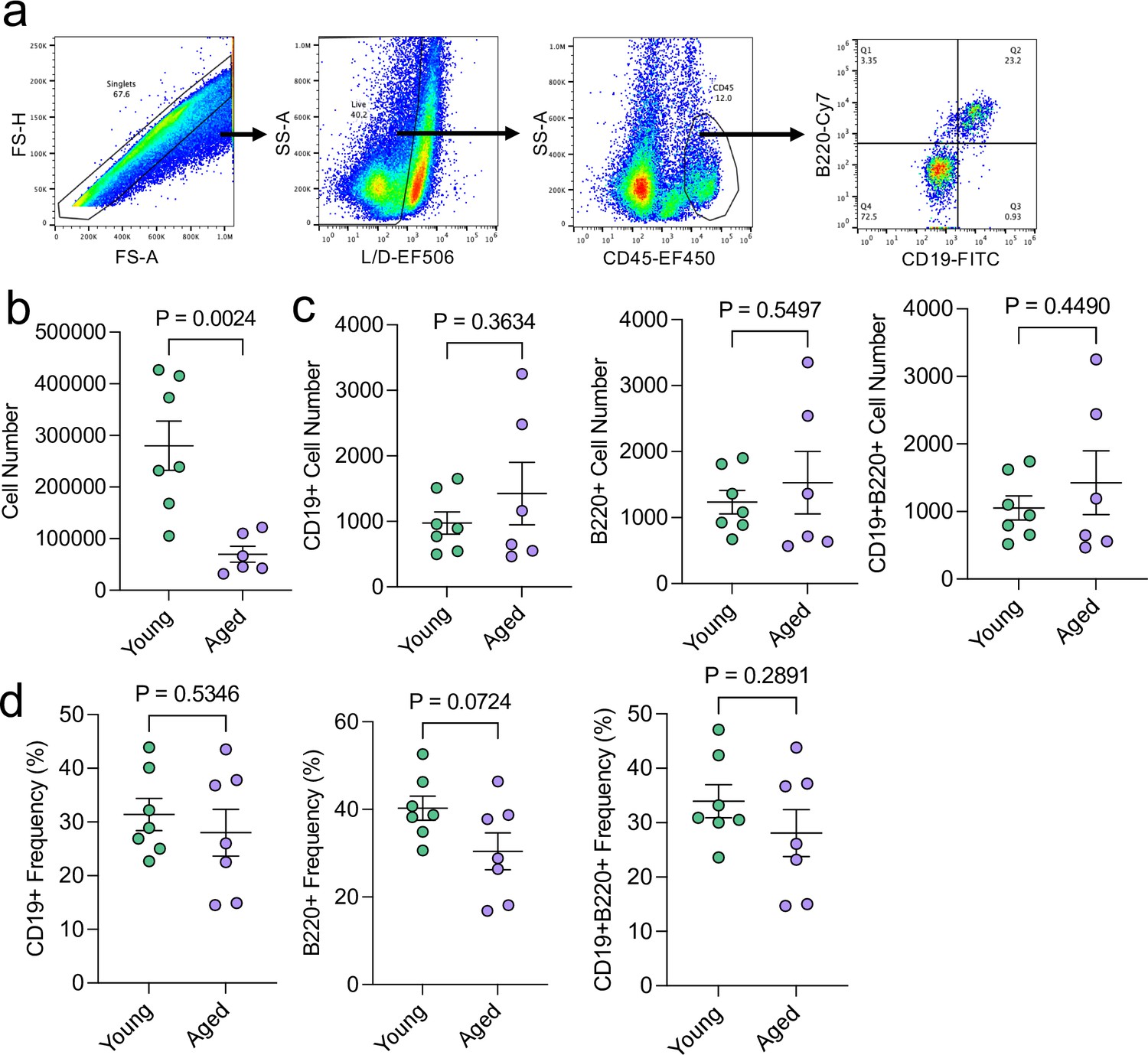
Aging does not appreciably influence the total numbers of meningeal CD19 + and B220 + cells.
Male WT mice at 10 weeks of age or 20 months of age were sacrificed and the dural meninges were processed for flow cytometry. (a) Flow cytometry gating strategy. Warmer colors indicate higher concentration of cells. (b) Quantification of total cell numbers in the dural meninges in young and aged mice (Young n=7, Aged n=6, rep = 1). (c) Quantification of the numbers of CD19+, B220+, and CD19 + B220 + cells in the dural meninges of young and aged mice (Young n=7, Aged n=6, rep = 1). (d) Quantification of the frequency of CD19+, B220+, and CD19 + B220 + cells of total CD45 + cells in young and aged dural meninges (Young n=7, Aged n=7, rep = 1). Two aged mice were excluded from this experiment: one mouse was excluded entirely due to gross hepatosplenomegaly and another mouse was excluded from the absolute cell count quantifications due to a suboptimal dissection. p values for were calculated using unpaired two-sample students t-tests.
-
Figure 6—figure supplement 1—source data 1
Tables depicting flow cytometry cell counts and frequencies of B cells in the dural meninges in young and aged mice.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig6-figsupp1-data1-v1.xlsx
Figure 7
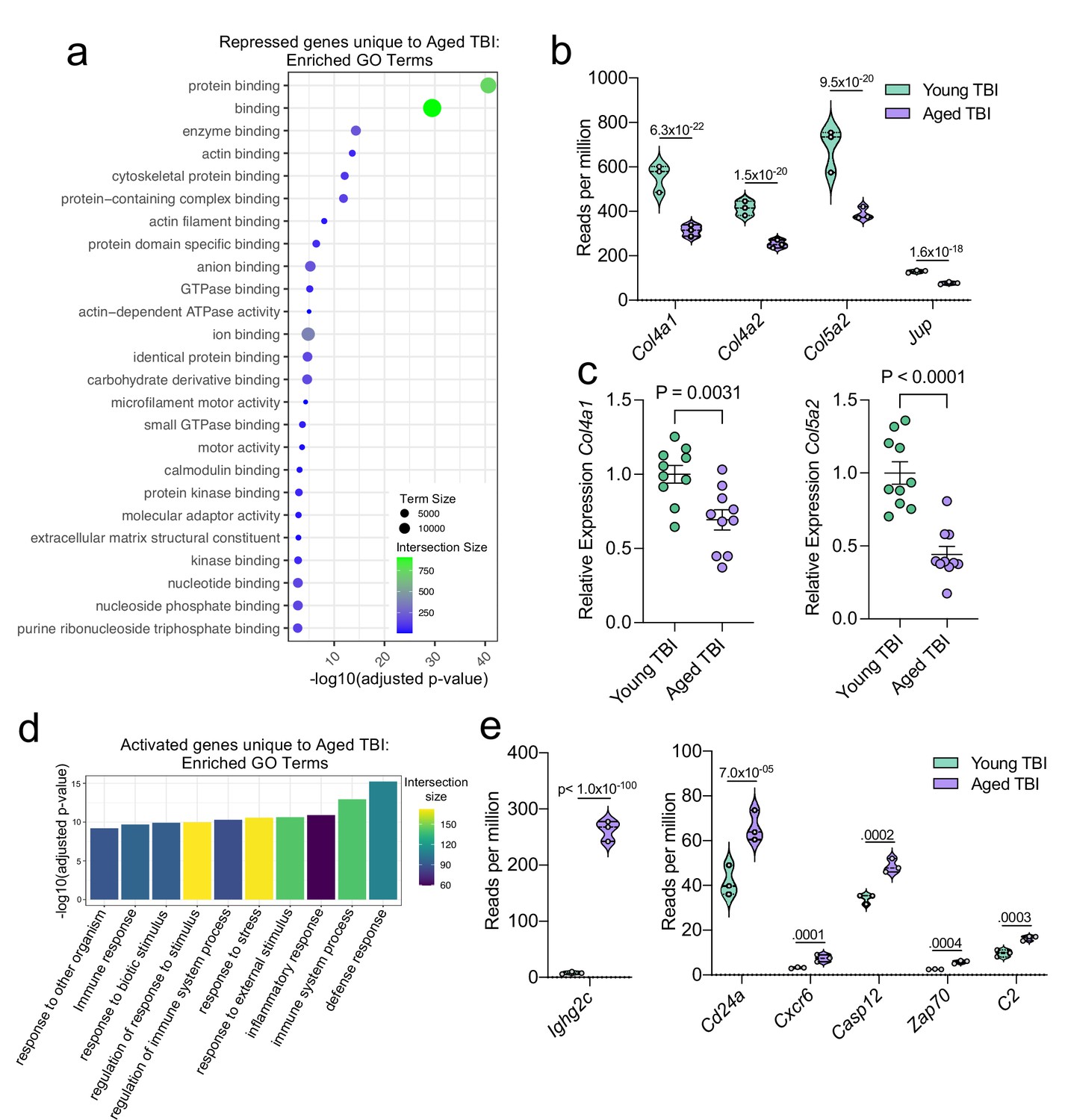
Aging and mild TBI together promote a unique meningeal transcriptional signature.
Male WT mice at 10 weeks of age or 20 months of age received a TBI or Sham procedure. 1.5 months later, bulk RNA-seq was performed on the 4 experimental groups with 3 biological replicates per group (each biological replicate consisted of meningeal RNA samples from 2 to 3 independent mice). (a) Dot plot showing GO term molecular functions enriched by the repressed genes unique to the Young TBI vs Aged TBI comparison. The color and size of each dot represents the size of the GO term and the number of upregulated genes that contribute to each term, respectively. (b) Violin plot depicting counts of significantly repressed extracellular matrix related genes (FDR <0.1). (c) Quantitative PCR relative expression of Col4a1 and Col5a2 within the dural meninges 1.5 months after TBI (Sham n=10, TBI n=10, rep = 1). (d) Bar plot shows enrichment of GO term biological processes related to the immune system with the genes unique to the Young TBI vs Aged TBI comparison. The color of each bar represents the number of upregulated genes that contribute to each GO term. (e) Violin plots depicting counts of significantly activated immune-related genes (FDR <0.1). (b,e) Each statistic represents the adjusted p-value calculated for each gene using DESeq2. The central line within each plot represents the median of the data set. The upper and lower boundaries of the box represent the third (Q3) and first (Q1) quartiles respectively. The violin plot encompasses the three biological repeats. The width of the violin plot represents the frequency of observations at that given y-value. Therefore, the wider the violin plot, the higher the frequency of observations. FDR and p-values for (a,b,d,e) were calculated with DESeq2 using the Wald test for significance following fitting to a negative binomial linear model and the Benjamini-Hochberg procedure to control for false discoveries. Error bars in (c) depict mean ± s.e.m. p values for (c) were calculated using unpaired two-sample students t-tests.
-
Figure 7—source data 1
Raw data showing enriched GO-terms and contributory genes that were uniquely downregulated in aging after TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig7-data1-v1.xlsx
-
Figure 7—source data 2
Number of reads per million of collagen-related genes downregulated in aging after injury in the bulk RNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig7-data2-v1.xlsx
-
Figure 7—source data 3
Relative expression of collagen-related genes by qPCR in whole meninges one week post-TBI.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig7-data3-v1.xlsx
-
Figure 7—source data 4
Table depicting all differentially regulated genes unique to the Young TBI vs. Aged TBI comparison.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig7-data4-v1.xlsx
-
Figure 7—source data 5
Number of reads per million of immune-related genes downregulated in aging after injury in bulk RNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig7-data5-v1.xlsx
Figure 8
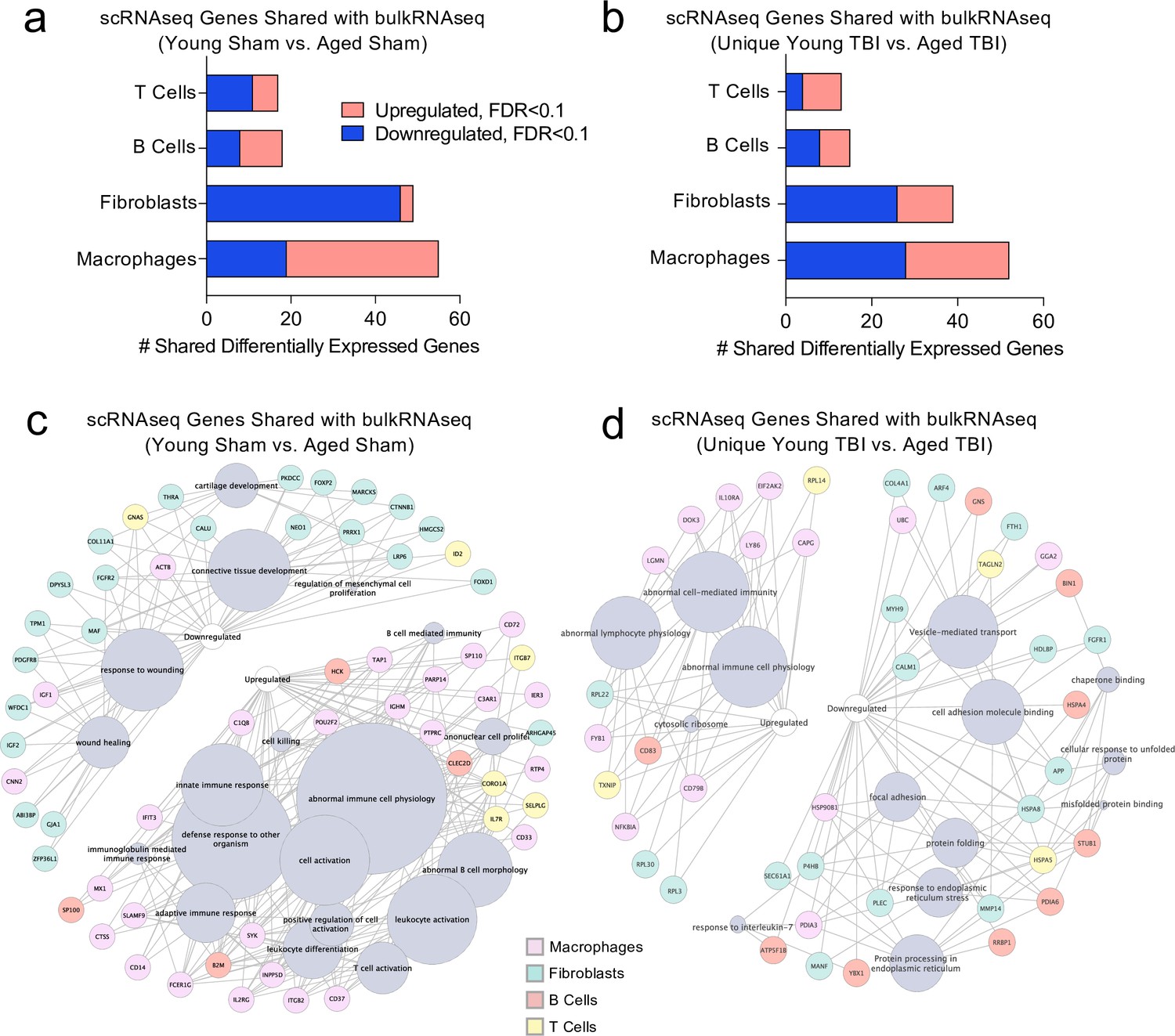
Shared gene signatures show dysregulated immune activation.
Differential gene expression was compared between the scRNA-seq dataset in young mice (1 week post-TBI) and the bulk RNA-seq dataset in aged mice (1.5 months post-TBI). Quantification of the number of differentially regulated genes shared between T cells, B cells, Fibroblasts, and Macrophages in the scRNA-seq dataset with the differentially expressed genes seen in (a) aging alone and (b) 1.5 months after TBI in aged mice (FDR < 0.1). Modified circos plots depicting the shared up- and down-regulated genes and the cellular processes to which they contribute, between the T Cells, B Cells, Fibroblasts, and Macrophages from the scRNA-seq dataset and the bulk RNA-seq dataset in (c) aging alone and (d) 1.5 months after TBI. The color of the circle with each gene represents the cell population to which the gene belongs. The size of the grey circles corresponds to the number of genes contributing to the term, which is shown by the intersecting line from each gene. FDR values in the bulk RNA-seq dataset were calculated with DESeq2 using the Wald test for significance following fitting to a negative binomial linear model and the Benjamini-Hochberg procedure to control for false discoveries. Differential gene expression in the scRNA-seq dataset was calculated using the ZINB-WaVE function for zero-enriched datasets and DESeq2. Graphs in (c–d) were constructed using ToppCluster and Cytoscape. FDR; false discovery rate.
-
Figure 8—source data 1
Raw data showing differentially expressed genes in scRNA-seq dataset and bulk RNA-seq dataset used for comparisons.
The up- and downregulated genes for macrophages, fibroblasts, B Cells and T cells were assessed for the scRNA-seq dataset. The up- and downregulated genes for the Young Sham vs. Aged Sham and those unique to the Young TBI vs. Aged TBI were assessed for the bulk RNA-seq dataset.
- https://cdn.elifesciences.org/articles/81154/elife-81154-fig8-data1-v1.xlsx
Tables
Table 1
Counts of each cell population separated by Sham and TBI.
Male WT mice at 10 weeks of age received a TBI or Sham procedure. One week later, the meninges from 5 mice per group were harvested, pooled, and processed for scRNA-seq. The cell counts for each cell population are shown after data processing.
| Cell Type | Cell Counts Sham | Cell Counts TBI |
|---|---|---|
| Activated Macs 1 | 185 | 499 |
| Activated Macs 2 | 199 | 319 |
| Macs 3 | 24 | 31 |
| B Cells 1 | 217 | 172 |
| B Cells 2 | 172 | 137 |
| Immature/Diff B Cells | 63 | 92 |
| CD3+T Cells | 150 | 240 |
| Activated T Cells | 148 | 169 |
| NK Cells | 68 | 55 |
| Dendritic Cells | 131 | 200 |
| Plasmacytoid Dendritic Cells | 18 | 42 |
| Neutrophils | 16 | 26 |
| Proliferating Cells | 36 | 58 |
| Fibroblasts | 124 | 781 |
| Endothelial Cells 1 | 235 | 380 |
| Endothelial Cells 2 | 78 | 199 |
| Pericytes | 20 | 69 |
| Choroid Plexus | 58 | 135 |
| Pineal Gland Cells | 31 | 75 |
| Schwann Cells | 35 | 47 |
| Clotting Related | 34 | 43 |
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | Anti-CD45.2 EF450 (rat monoclonal) | Thermo Scientific | Catalog # 11-0451-82, Clone 30-F11 | Flow(1:200) |
| Antibody | Anti-B220 PE-Cy7 (rat monoclonal) | BioLegend | Catalog # 103222, Clone RA3-6B2 | Flow(1:200) |
| Antibody | Anti-CD19 FITC (rat monoclonal) | eBioscience | Catalog # 11-0193-81, Clone eBio1D3 | Flow(1:200) |
| Antibody | Anti-J chain (rabbit monoclonal) | Invitrogen | Catalog # MA5-16419, Clone: SP105 | IF(1:200) |
| Antibody | Anti-Collagen I (rabbit polyclonal) | Abcam | Catalog # ab21286 | IF(1:200) |
| Antibody | Anti-Lyve-1-Alexa Fluor 488 (rat monoclonal) | eBioscience | Catalog # 53-0443-82, Clone ALY7 | IF(1:200) |
| Antibody | Anti-Iba1 (goat polyclonal) | Abcam | Catalog # ab5076 | IF(1:300) |
| Antibody | Anti-GFAP (rat monoclonal) | Thermo Fisher Scientific | Catalog # 13–0300, Clone 2.2B10 | IF(1:1000) |
| Antibody | Anti-MHC Class II 660 (rat monoclonal) | eBioscience | Catalog # 14-5321-82, Clone M5/114.12.2 | IF(1:100) |
| Antibody | Anti-CD31 (armenian hamster monoclonal) | Millipore Sigma | Catalog # MAB1398Z, Clone 2H8 | IF(1:200) |
| Antibody | Anti-B220 (rat monoclonal) | Thermo Fisher Scientific | Catalog # 14-0452-82, Clone RA3-6B2 | IF(1:200) |
| Antibody | Anti-NeuN (mouse monoclonal) | EMD Millipore | Catalog # Mab277, Clone A60 | IF(1:500) |
| Antibody | Donkey anti- rat Alexa Fluor 488 (donkey polyclonal) | Thermo Fisher Scientific | Catalog # A-21208 | IF(1:1000) |
| Antibody | Donkey anti- goat Alexa Fluor 647 (donkey polyclonal) | Thermo Fisher Scientific | Catalog # A-21447 | IF(1:1000) |
| Antibody | Donkey anti-rat Alexa Fluor 594 (donkey polyclonal) | Thermo Fisher Scientific | Catalog # A-21209 | IF(1:1000) |
| Antibody | Donkey anti-rabbit Alexa Fluor 647 (donkey polyclonal) | Thermo Fisher Scientific | Catalog # A-31573 | IF(1:1000) |
| Antibody | Alexa Fluor 488 anti-Armenian Hamster (goat polyclonal) | Jackson ImmunoResearch | Catalog # 127-545-160, RRID: AB_2338997 | IF(1:1000) |
| Other | Fixable Viability Dye eFlour 506 | eBioscience | Catalog # 65-0866-18 | Flow(1:800) |
| Other | Absolute counting beads | Life Technologies | Catalog # C36950 | Used for counting cells in flow cytometry. See ‘Flow cytometry’. |
| Other | Prolong Gold antifade reagent | Invitrogen | Catalog # P36930 | Used for mounting tissues for confocal imaging. See ‘Immunohistochemistry, imaging, and quantification’. |
| Commercial assay, kit | RNeasy Micro Kit | Qiagen | Catalog # 74004 | |
| Commercial assay, kit | SensiFAST cDNA synthesis kit | Bioline | Catalog # BIO-65054 | |
| Commercial assay, kit | SensiFAST Probe No-ROX Kit | Bioline | Catalog # BIO-86005 | |
| Sequence-based reagent | Gapdh | Life Technologies | Catalog # 4331182 | Assay ID Mm99999915_g1 |
| Sequence-based reagent | Ifnar1 | Life Technologies | Catalog # 4331182 | Assay ID Mm00439544_m1 |
| Sequence-based reagent | Irf5 | Life Technologies | Catalog # 4331182 | Assay ID Mm00496477_m1 |
| Sequence-based reagent | Ifnb1 | Life Technologies | Catalog # 4331182 | Assay ID Mm00439552_s1 |
| Sequence-based reagent | Ifi203 | Life Technologies | Catalog # 4331182 | Assay ID Mm00492601_m1 |
| Sequence-based reagent | Col4a1 | Life Technologies | Catalog # 4331182 | Assay ID Mm01210125_m1 |
| Sequence-based reagent | Col5a2 | Life Technologies | Catalog # 4331182 | Assay ID Mm01254423_m1 |
| Other | Collagenase VIII | Sigma Aldrich | Catalog # 9001-12-1 | Used for meningeal digestion for flow cytometry. See ‘Meningeal tissue collection’. |
| Other | Collagenase D | Sigma Aldrich | Catalog # 11088866001 | Used for meningeal digestion for flow cytometry. See ‘Meningeal tissue collection’. |
| Other | DAPI | Sigma Aldrich | Catalog # D9542 | IF(1:1000), See ‘Immunohistochemistry, imaging, and quantification’. |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The meningeal transcriptional response to traumatic brain injury and aging
eLife 12:e81154.
https://doi.org/10.7554/eLife.81154




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}