Determinants of sugar-induced influx in the mammalian fructose transporter GLUT5
- Department of Biochemistry and Biophysics, Science for Life Laboratory, Stockholm University, Sweden
- Department of Applied Physics, Science for Life Laboratory, KTH Royal Institute of Technology, Sweden
Figures
Figure 1 with 1 supplement
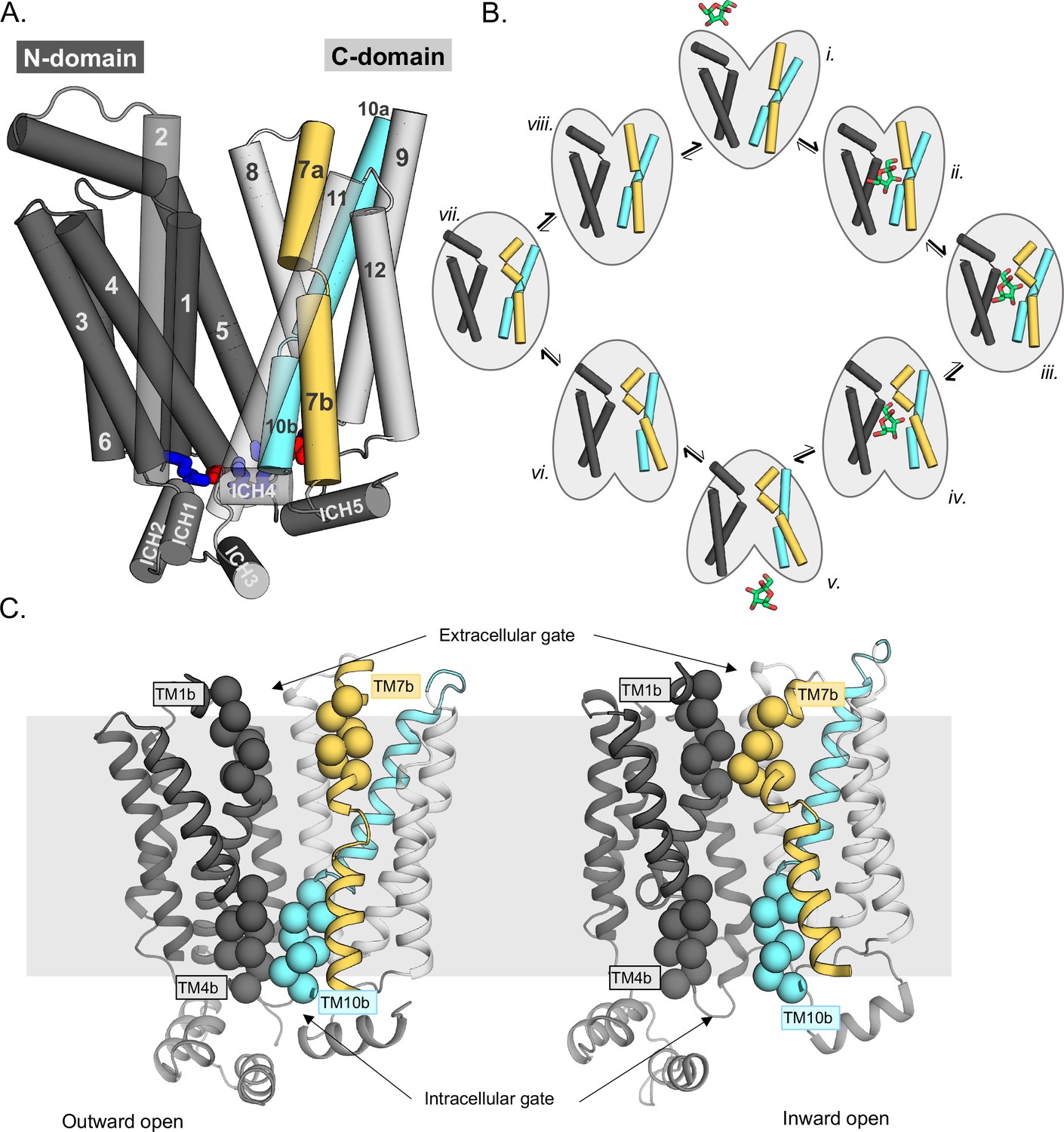
Schematic highlighting the GLUT structural features and their major conformations in the transport cycle.
(A) Structural overview of a sugar porter, GLUT5 (PDB:4ybq). The N-terminal bundle (left, dark gray, transmembrane helices 1–6) and C-terminal bundle (right, light gray, transmembrane helices 7–12) form the separate six-transmembrane bundles, which are connected by the large cytosolic loop comprising intracellular helices (ICH) 1–4. The salt bridge forming residues linking the two bundles are shown as sticks in blue and red, indicating the positive and negative charge of the side chains, respectively. The broken transmembrane helices TM7 (forming TM7a and TM7b) and TM10 (forming TM10a and TM10b) are colored yellow and cyan, respectively. (B) Schematic conformational cycle a sugar porter will undergo, based on currently available protein structures. Briefly, moving clockwise from top middle, the transporter will receive a sugar in the outward-open state (PDB:4ybq) (i). The transporter then undergoes a partial occlusion of the extracellular gate (outward occluded, ii, PDB:4zw9), followed by full occlusion (iii, PDB:6rw3). Once both gates are fully shut, the inner gates begin to open (inward occluded, iv, PDB:4ja3) where the salt-bridge residues begin to lose contact. Finally, the salt bridges are fully broken apart in an inward-open state (v, PDB:4yb9), and the sugar can be released into the cell. The transporter will then go through the same motions in reversed order in the absence of sugar to reset to the outward-open state (vi–viii). (C) The extracellular gate is formed by TM1b and TM7b half-helices, and the intracellular gate is formed by TM4b and TM10b half-helices. Residues defining these gates are shown as spheres. In the outward-open state (left), the extracellular gate is open, and intracellular gate is shut. In the inward-open state (right), the opposite occurs. The gray slab behind the proteins indicates the rough location of the lipid bilayer membrane.
Figure 1—figure supplement 1

Sequence similarity of rGLUT5 to several GLUTs and model organisms.
(A) Sequence alignment of rat GLUT5 to human and bovine GLUT5 (PDB:4yb9), as well as other GLUTs of interest and sugar porters used in homology modeling (GLUT3 – PDB:4zw9, PfHT1 – PDB:6rw3, and XylE – PDB:4ja3). A dark blue color indicates >88% (8/9 matching) sequence identity, and a lighter blue color indicates a >77% (7/9 matching) sequence identity. The transmembrane and intracellular helices (ICH) are indicated by labeled squares above the alignment, colored as in Figure 1. The intracellular (on TM4 and TM10) and extracellular gates (on TM1 and TM7) are colored in pink squares. The boxes below sugar-binding residues are colored in orange, residues involved in coupling of binding to conformational change as well as the GPXPXP motif in turquoise, and salt-bridge residues in green. Residues with multiple features (such as on TM10) have stacked boxes. (B) Sequence identity table for pairwise alignments between sequences from panel A. Sequence identities between 70% and 100% are colored in dark gray, 38% and 70% in light gray, and 0% and 38% in white.
Figure 2 with 11 supplements
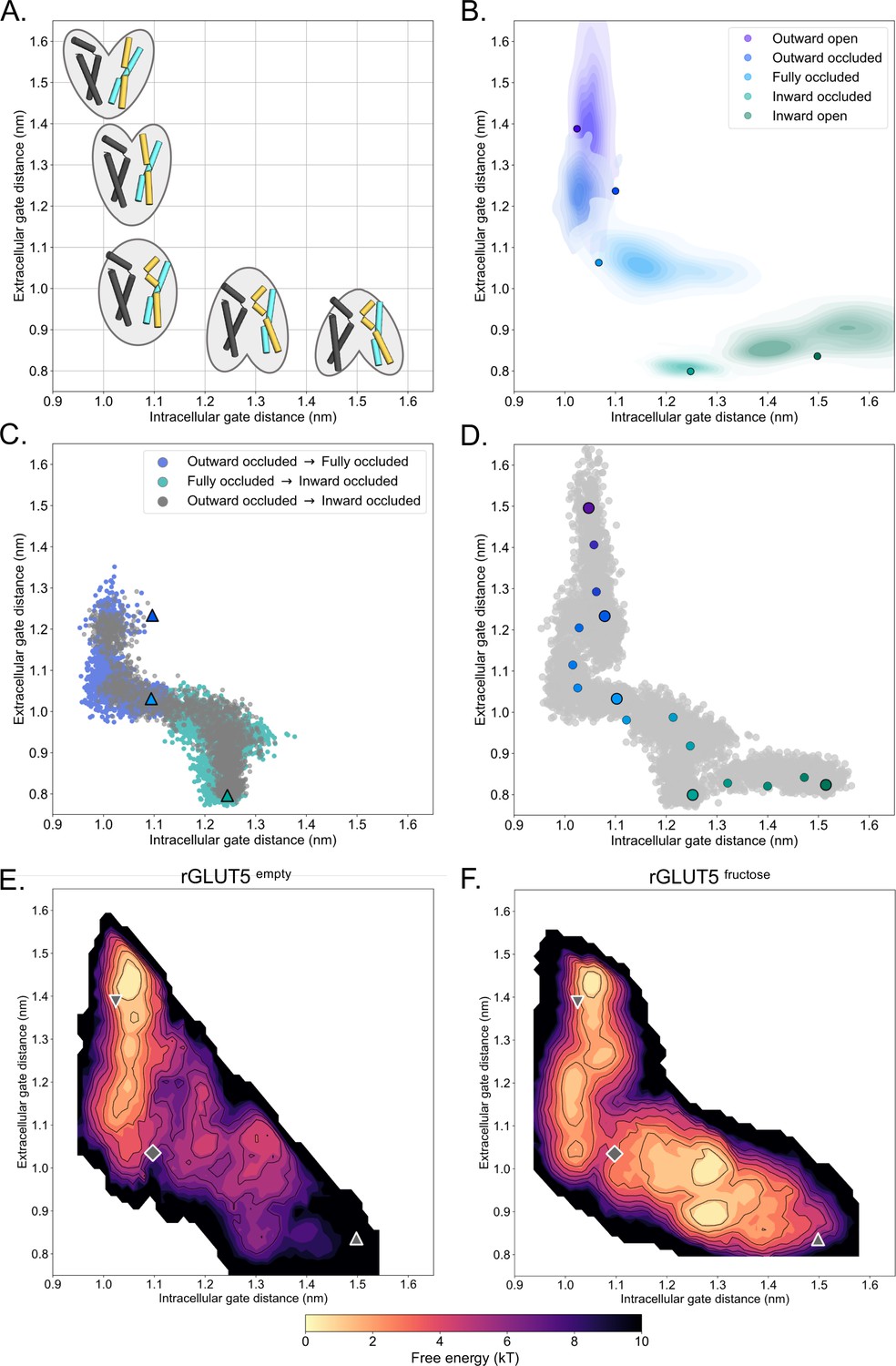
A free energy landscape for D-fructose influx by GLUT5.
(A) A graphical illustration of the five major states of sugar porters, with the intracellular (IC) and extracellular (EC) gate distances on the x- and y-axis, respectively. Only TM1, TM4, TM7, and TM10 are drawn here, with the intent to highlight only major elements of the rocker switch conformational change. (B) IC and EC gate population densities of atomistic simulations of each rGLUT5 homology model. Filled circles represent the starting configurations from each rGLUT5 homology model. (C) Targeted molecular dynamics (TMD) with bound D-fructose. Individual states are shown in triangles, with the following color schemes: outward occluded: deep blue, occluded: light blue, and inward occluded: green. Gray dots represent all frames corresponding to TMD of outward occluded to inward occluded, skipping the occluded state. This follows a pathway similar to sequential TMD from outward occluded to fully occluded (blue circles), and fully occluded to inward occluded (teal circles). rGLUT5empty TMD results can be found in Figure 2—figure supplement 2A. (D) Beads chosen for the string simulations from TMD projected onto the space defined by the IC and EC gate distances for rGLUT5fructose. The cloud of gray dots represents all gate distance configurations through the TMD simulations, larger colored dots represent each initial homology model, and the smaller colored dots represent the beads between each of these models, which were chosen for the first iteration of the string-of-swarms method. Beads for rGLUT5empty found in Figure 2—figure supplement 2B. (E) Free energy surface for rGLUT5empty. The triangles and diamond illustrate the gate measurements for the following homology models: outward open (top left triangle, structure of rGLUT5, PDB:4ybq), fully occluded (diamond, model of PfHT, PDB:6rw3), and inward open (bottom right, model of bGLUT5, PDB:4yb9). (F) Free energy surface for rGLUT5fructose, with the same homology model projection as in E.
Figure 2—figure supplement 1
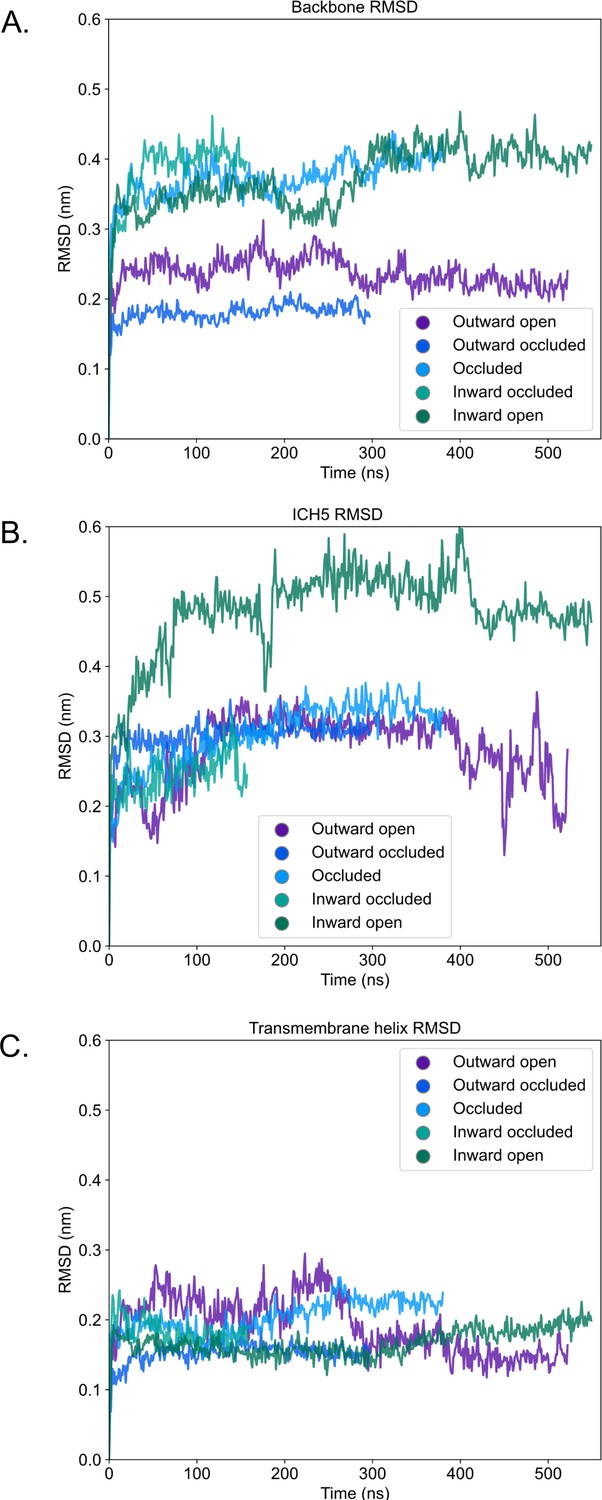
Root mean squared deviation (RMSD) of atomistic simulations of rGLUT5 homology models.
(A) Backbone RMSD calculated by aligning on all C-alpha atoms of the protein upon the original homology model, and reporting their RMSD. (B) RMSD of ICH5 residues after self-aligning. (C) RMSD of transmembrane helices 1–12 (as indicated in Figure 1—figure supplement 1) after self-aligning.
Figure 2—figure supplement 2
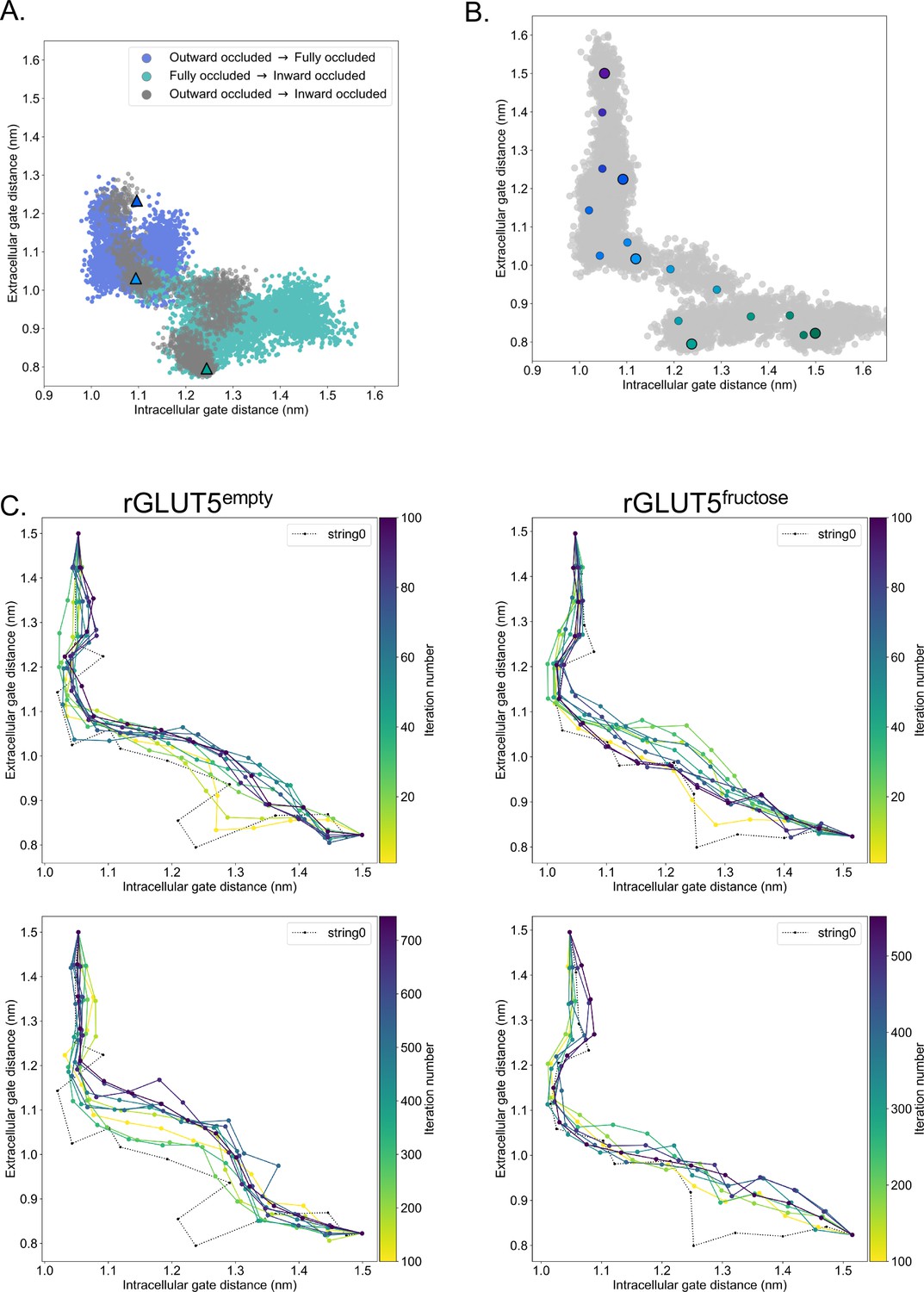
Details of molecular dynamics (TMD), string simulation setup, and convergence.
(A) rGLUT5empty TMD with and without the occluded state as a target. Coloring as in rGLUT5fructose TMD results, Figure 2C. (B) Beads chosen for the string simulations from the TMD along the collective variable (CV) space for rGLUT5empty. The cloud of gray dots represent all CV configurations through the TMD simulations, larger colored dots represent each initial homology model, and the smaller colored dots represent the beads between each of these models, which were chosen for the first iteration of the string-of-swarms method. Beads for rGLUT5fructose found in Figure 2D. (C) String convergence as measured by the change of CV values (gate distances) per iteration. Top: before convergence, iterations 1–100, with every 10th iteration shown. Bottom: after convergence, iterations 100–745 (rGLUT5empty) or 100–552 (rGLUT5fructose), with every 80th iteration shown. The initial string was chosen along the beads highlighted in panel B and Figure 2D, respectively, indicated with a dotted black line labeled ‘string0’.
Figure 2—figure supplement 3
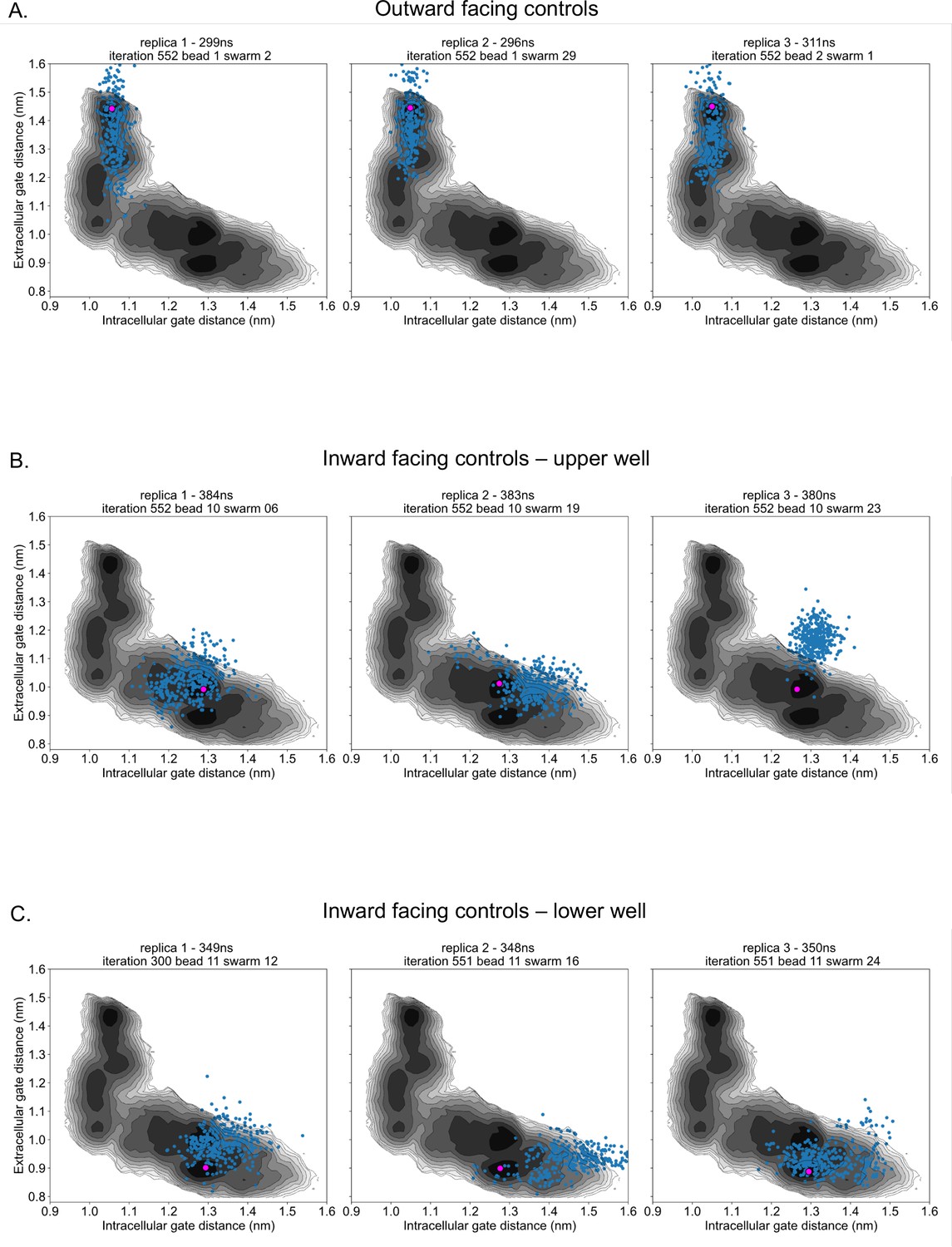
Control simulation distribution along energy surfaces.
(A) Projection of outward-facing control simulations on rGLUT5fructose free energy surfaces of the intra- and extracellular gates (as seen in Figure 2F). The pink dot represents the starting conformation for each control simulation, and blue dots represent the conformation of the protein in collective variable (CV) space every 1 ns. (B) Projection of inward-facing controls from the upper well (well 3 in Figure 2—figure supplement 10), as described in panel A. (C) Projection of inward-facing controls from the lower well (well 4 in Figure 2—figure supplement 10), as described in panel A.
Figure 2—figure supplement 4
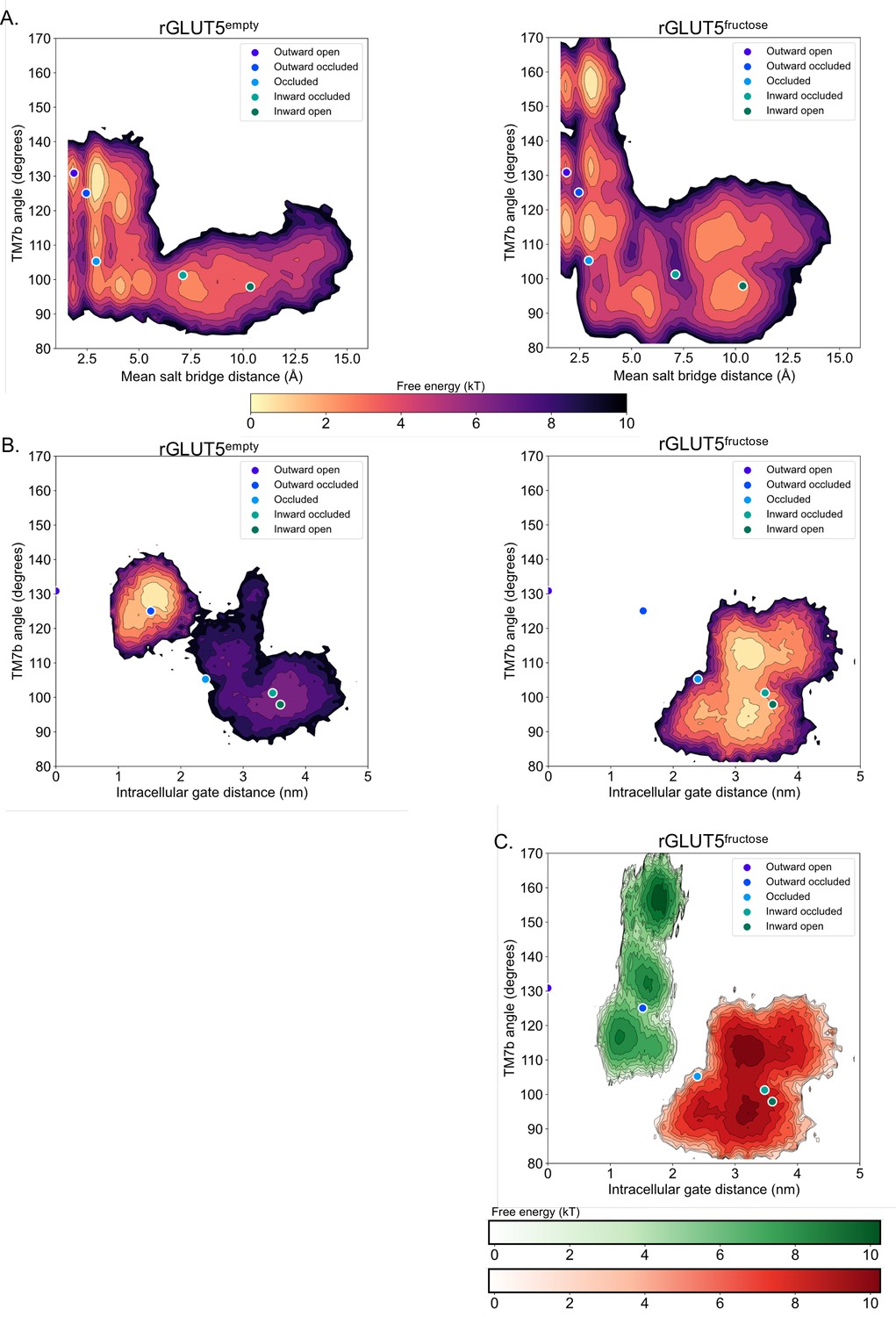
Projection of free energy surfaces onto alternate collective variable (CV) pairs.
(A) Free energy surfaces of rGLUT5empty and rGLUT5fructose using TM7b angle and mean salt-bridge distance as CVs. The projection of each homology model in CV space is also shown. The free energy scale is shown below the image. (B) Free energy surfaces of rGLUT5empty and rGLUT5fructose using TM7b angle and TM10b root mean squared deviation (RMSD) as CVs. The projection of each homology model in CV space is also shown. The free energy scale is shown above the image. (C) Free energy surface of rGLUT5fructose outward-facing beads (green) and inward-facing beads (red) using TM7b angle and TM10b RMSD as CVs.
Figure 2—figure supplement 5
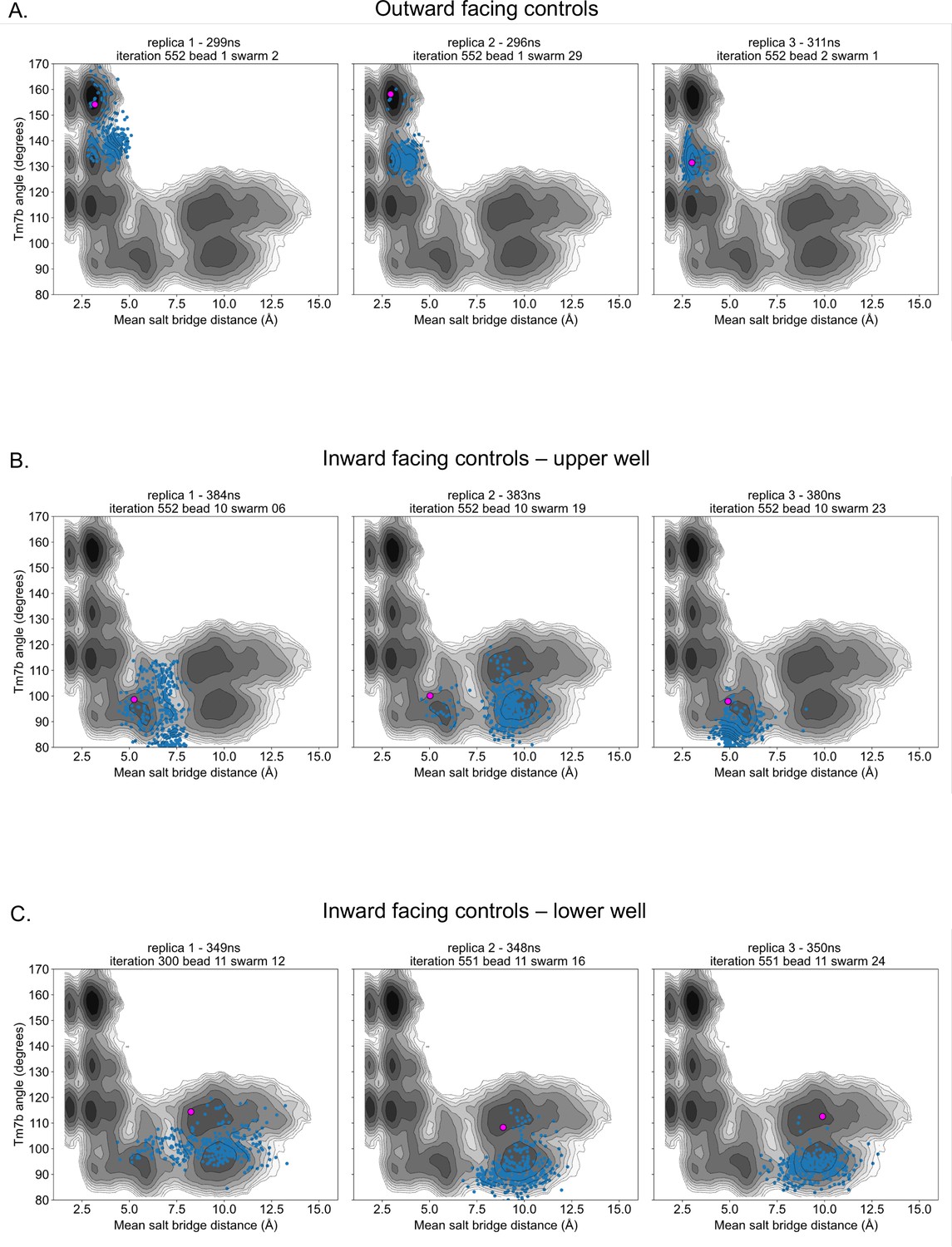
Control simulation distribution along energy surfaces.
(A) Projection of outward-facing control simulations on rGLUT5fructose free energy surfaces of the TM7b angle and mean salt-bridge distances (as seen in Figure 2—figure supplement 4A). The pink dot represents the starting conformation for each control simulation, and blue dots represent the conformation of the protein in collective variable (CV) space every 1 ns. (B) Projection of inward-facing controls from the upper well (well 3 in Figure 2—figure supplement 10), as described in panel A. (C) Projection of inward-facing controls from the lower well (well 4 in Figure 2—figure supplement 10), as described in panel A.
Figure 2—figure supplement 6
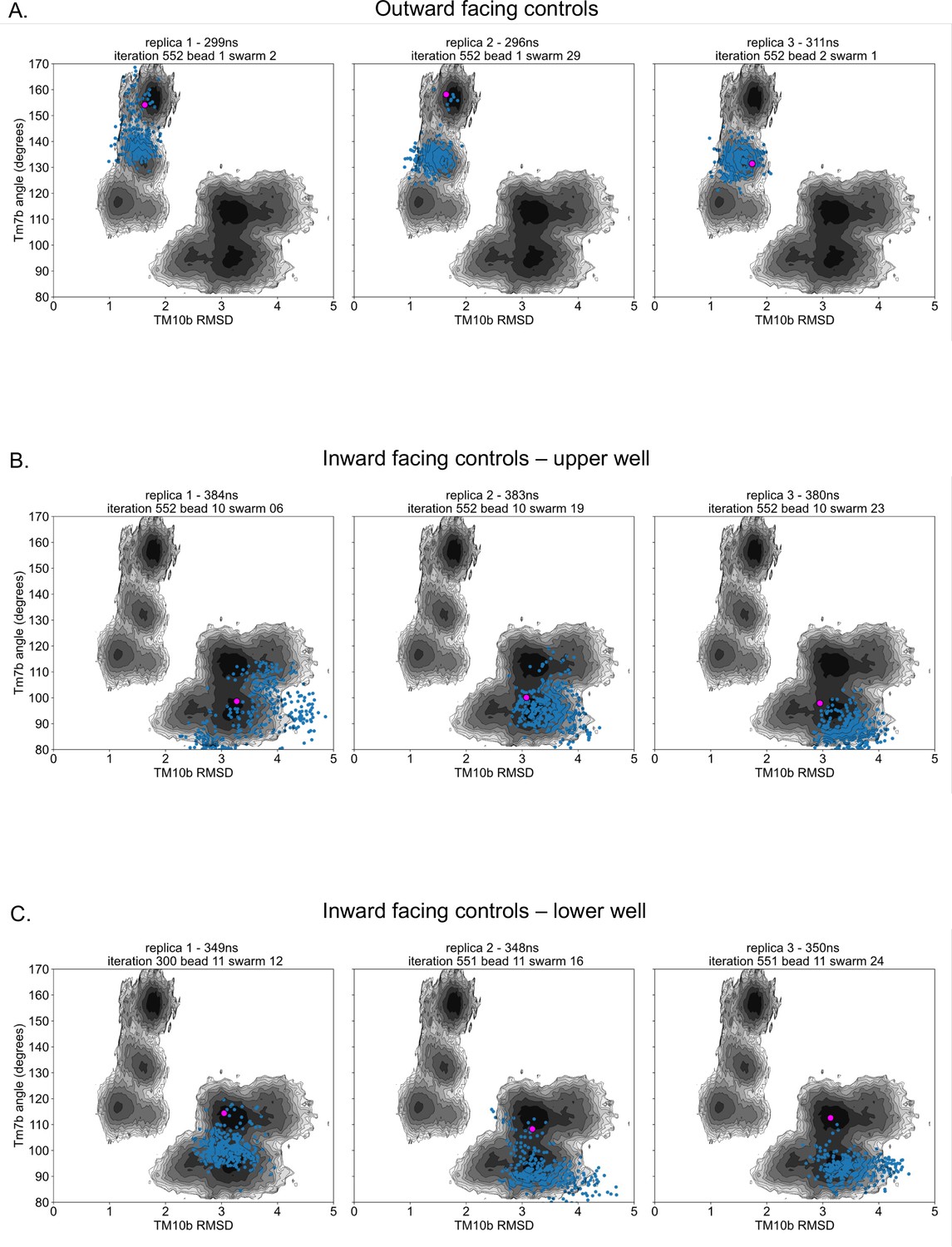
Control simulation distribution along free energy surfaces.
(A) Projection of outward-facing control simulations on rGLUT5fructose free energy surfaces of the combined outward and inward-facing beads (as seen in Figure 2—figure supplement 4C) for TM7b angle and TM10b root mean squared deviation (RMSD). The pink dot represents the starting conformation for each control simulation, and blue dots represent the conformation of the protein in collective variable (CV) space every 1 ns. (B) Projection of inward-facing controls from the upper well (well 3 in Figure 2—figure supplement 10), as described in panel A. (C) Projection of inward-facing controls from the lower well (well 4 in Figure 2—figure supplement 10), as described in panel A.
Figure 2—figure supplement 7
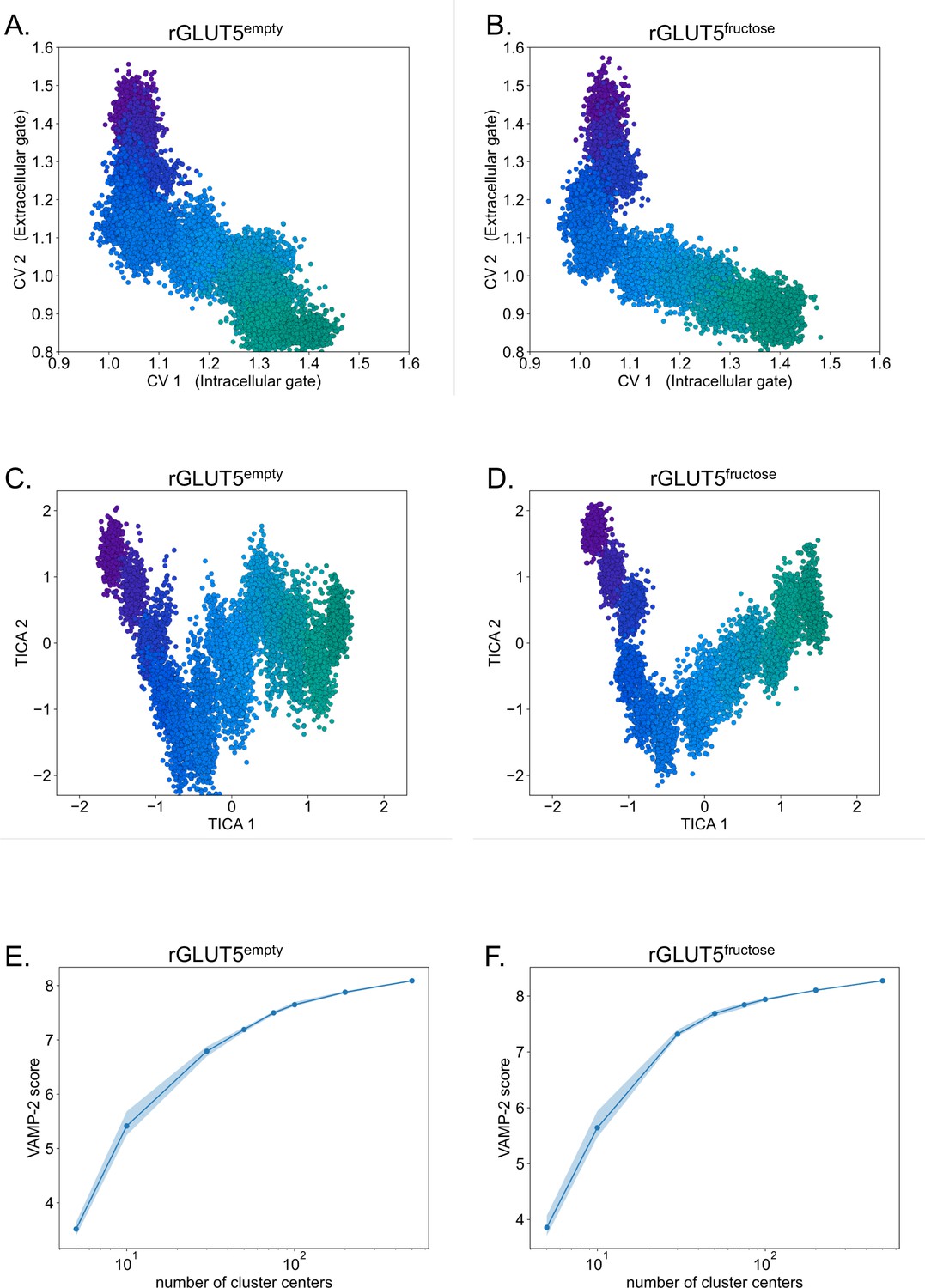
Transformation of CV distribution to MSM calculations.
(A, B) Original collective variable (CV) distribution for rGLUT5empty and rGLUT5fructose simulations, colored by bead as seen in Figure 2D, Figure 2—figure supplement 2B. (C, D) Time-lagged independent component analysis (TICA) space distribution rGLUT5empty and rGLUT5fructose simulations, colored by bead as in panels A and B. (E, F) VAMP-2 scores for rGLUT5empty and rGLUT5fructose simulations, used in determining appropriate number of clusters for the Markov state model (MSM) construction.
Figure 2—figure supplement 8
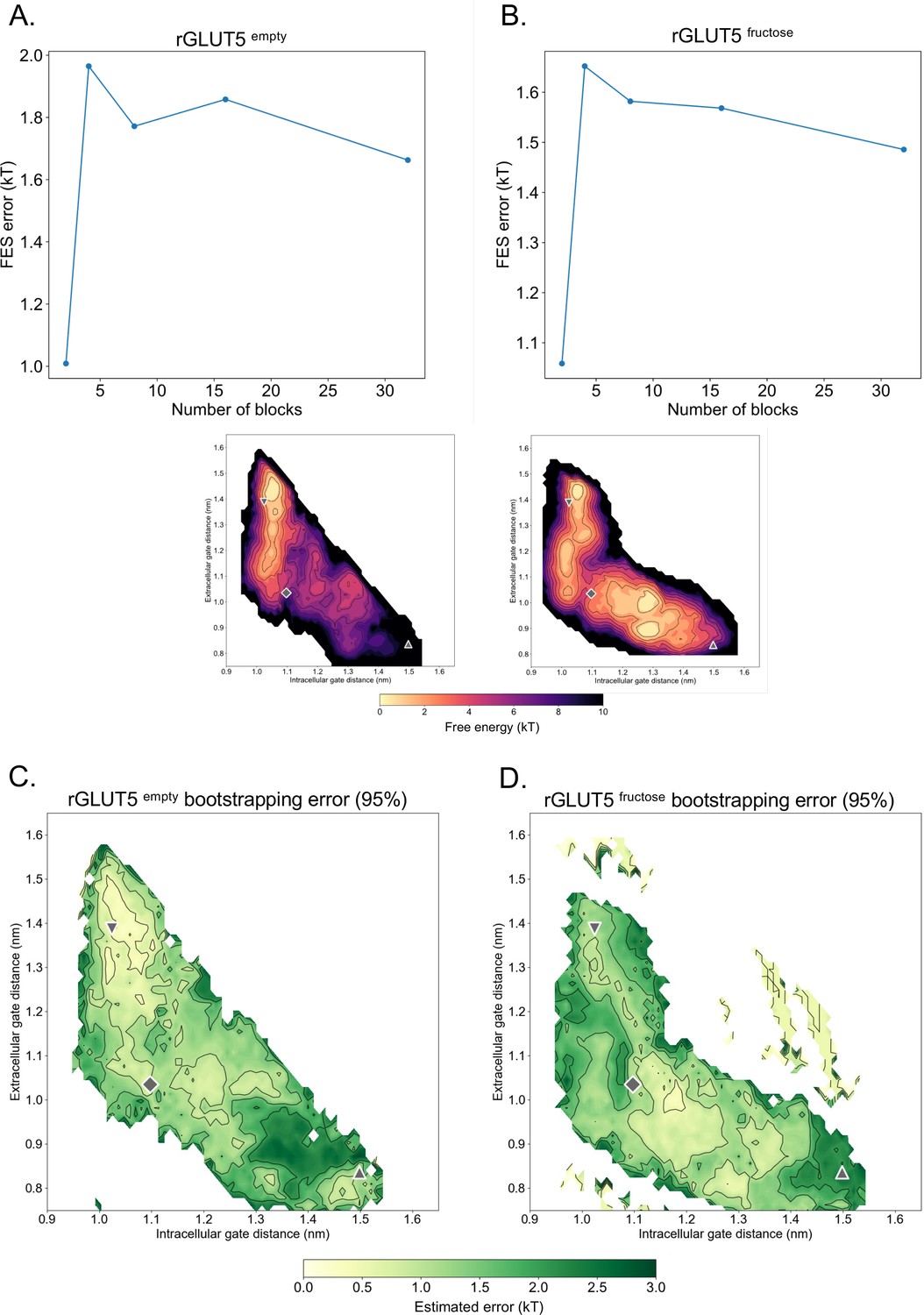
Statistical error of free energy surfaces.
(A, B) Error per block size tested for rGLUT5empty and rGLUT5fructose. (C, D) Estimated error of free energy surface (FES) calculations after bootstrapping for rGLUT5empty and rGLUT5fructose, alongside the FESs as shown in Figure 2E, F. A maximum error of 3 kT is shown here. The outward-open, occluded, and inward-open states are shown here as seen in Figure 2, to facilitate comparison with the location of the minima in the FES.
Figure 2—figure supplement 9
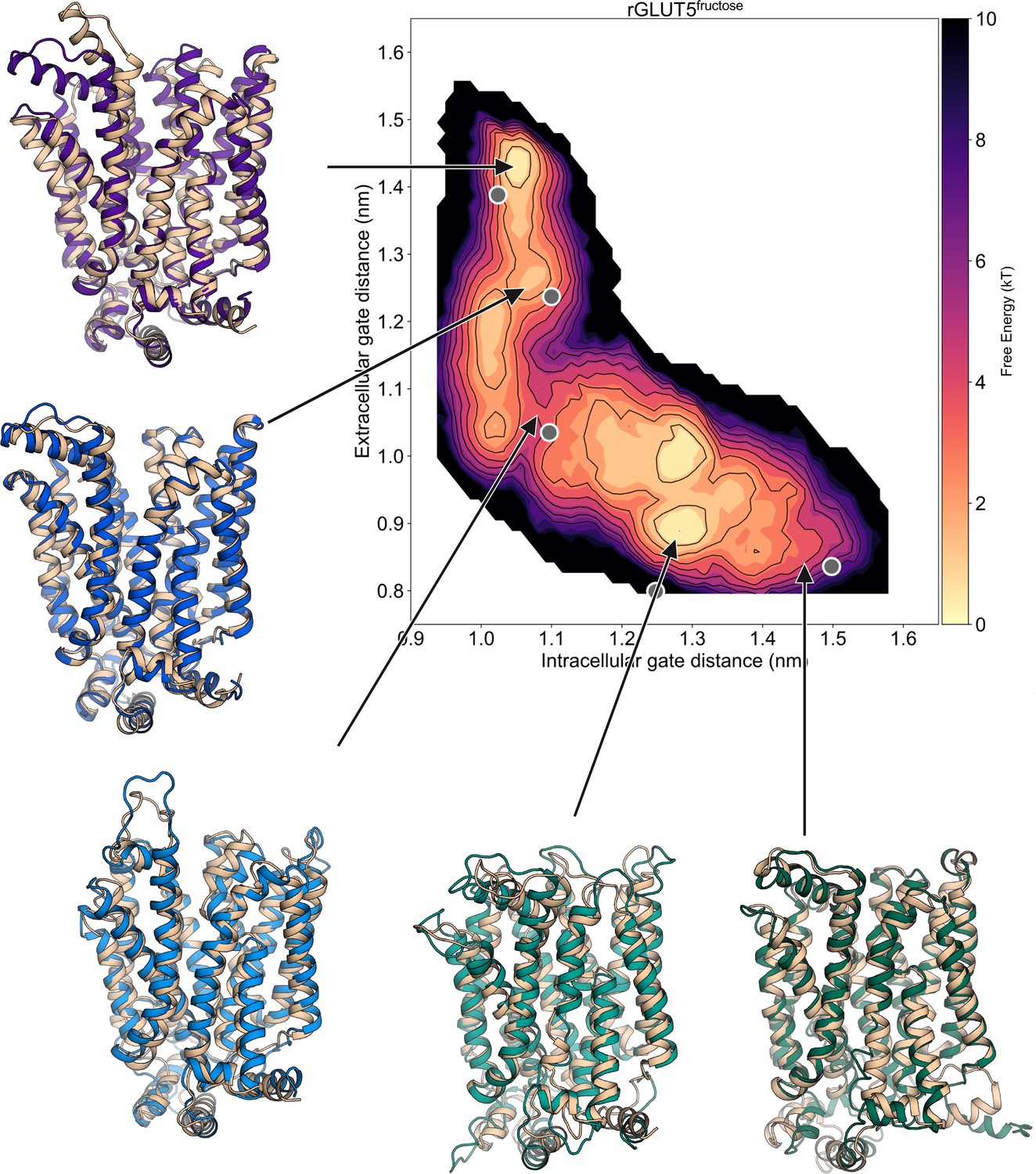
Average local energy minima conformation superimposed on homology models.
The 5 states’ homology models (outward open – PDB:4ybq, outward occluded – PDB:4zw9, fully occluded – PDB:6rw3, inward occluded – PDB:4ja3, inward open – PDB:4yb9) aligned to the average structure of each state’s local environment indicated by the arrow on the free energy surface of rGLUT5fructose (as seen in Figure 2F). Each state’s original model is colored in pale brown, and the conformation representative of each free energy minimum is colored as in Figure 2D.
Figure 2—figure supplement 10
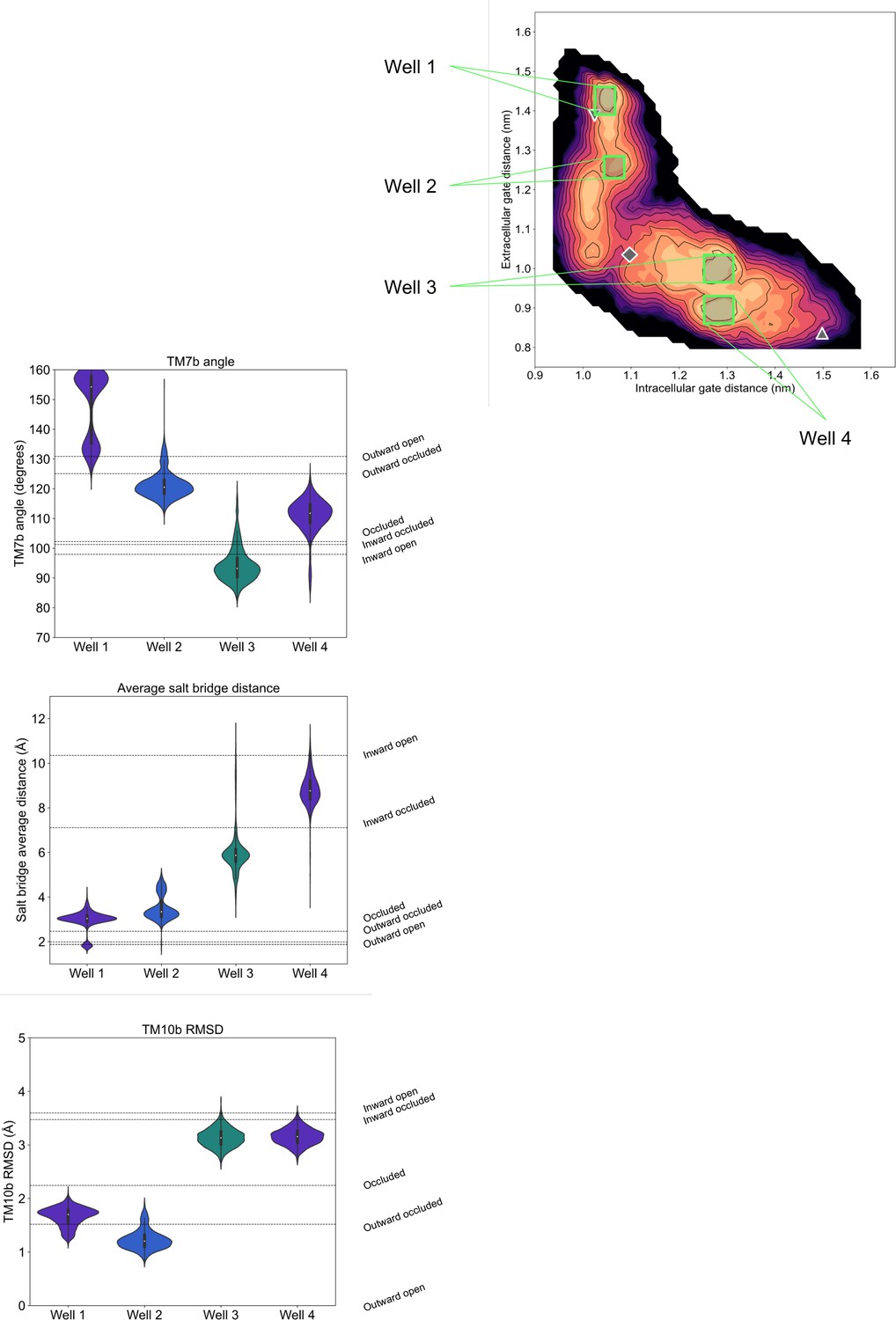
Violin plots of alternative collective variables (CVs) calculated for conformations extracted from the D-fructose-bound free energy landscape local minima.
Configurations from four wells along the energy surface were extracted and the distribution of other state-defining features were measured. The wells have a sample size of 6000, 4000, 5979, and 6000 frames, respectively. The dotted lines along the plot horizon indicate the measurement of this feature for each state’s initial homology model.
Figure 2—figure supplement 11
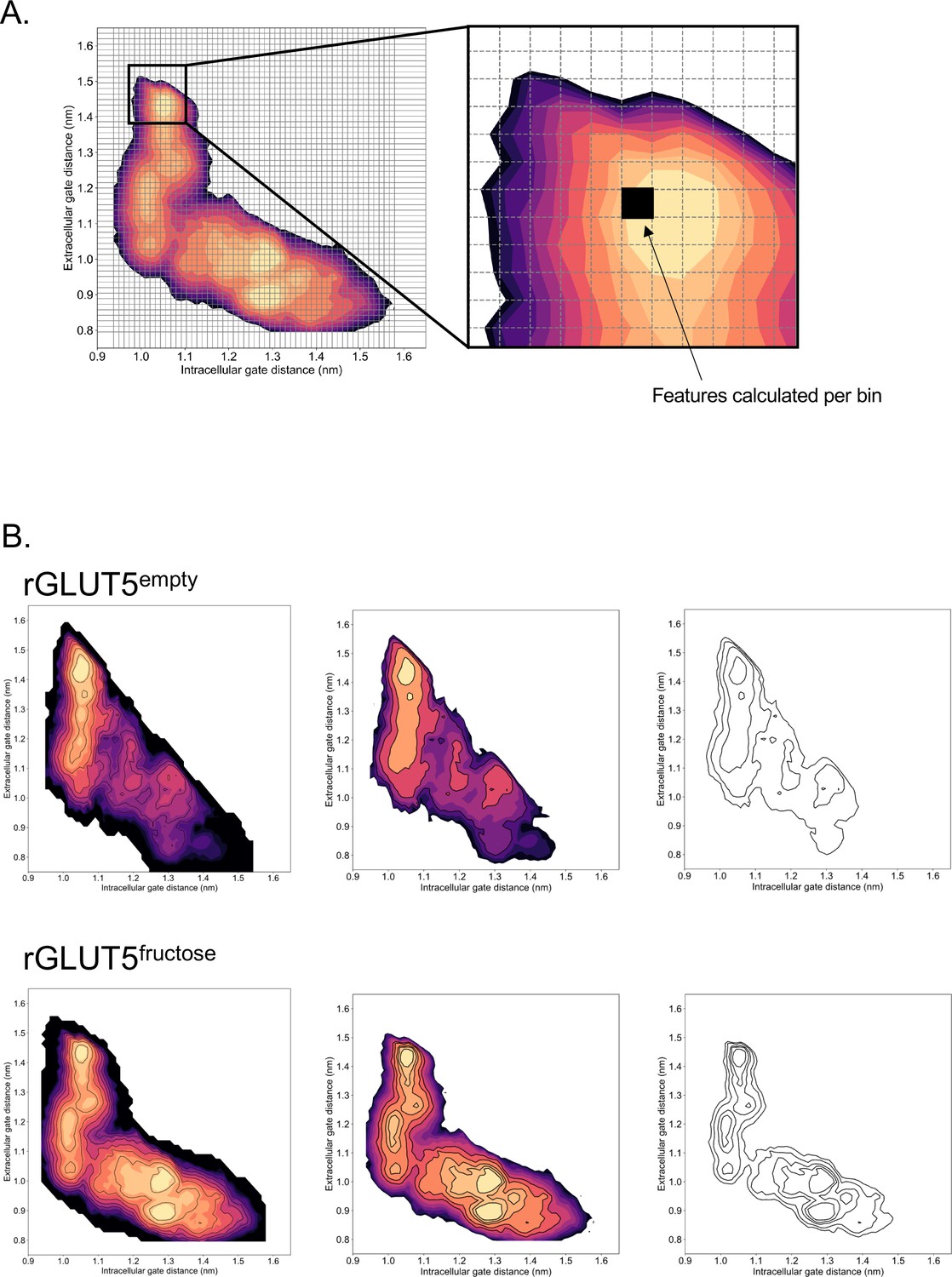
Calculation of features along energy surfaces.
(A) A simplified graphic of how each bin is extracted from the free energy surface. (B) A simplified graphic of how the black line abstractions used for analysis originate from the free energy surfaces for Figures 3A and 4C.
Figure 3 with 1 supplement
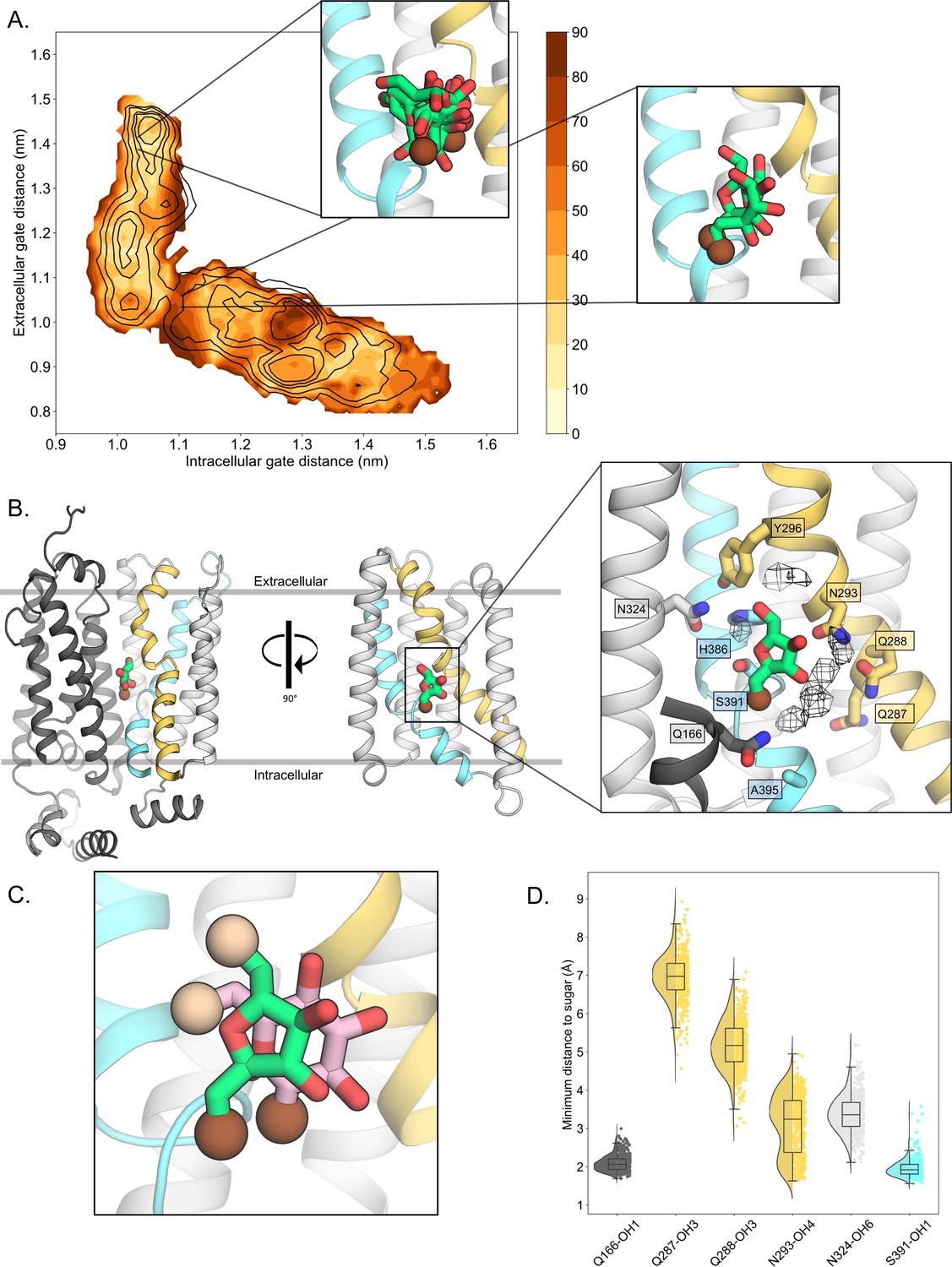
D-fructose becomes highly coordinated in the occluded conformation.
(A) Sugar coordination of the D-fructose-bound simulations superimposed onto the free energy landscape for rGLUT5fructose, colored according to the frequency of the most populated cluster in each bin, therefore darker colors indicate a more consistent pose (see Methods for bin description). Snapshots extracted from two bins, corresponding to the outward-open or occluded states, respectively, depicting ~70% of total pose variability (see Table 3 in Methods) are shown as inserts. GLUT5 helices, D-fructose, and C1 hydroxyl group shown as in panel B. (B) An overview of a representative binding pose of D-fructose in occluded GLUT5, colored as in Figure 1. The D-fructose C1 hydroxyl group is colored as a brown sphere for orientation purposes. Selected interacting residues shown as sticks, and the density of waters near the D-fructose in the occluded state is shown as a black mesh. (C) The coordination of D-fructose is oriented the same as previously determined glucose positions, such as in PfHT1 (PDB:6rw3, pink). The C6 hydroxyl group is shown as an enlarged sphere colored tan, and the C1 hydroxyl group in brown as in panel B. The D-fructose pose chosen here is the most populated cluster, as seen in Methods, Table 3. (D) The distribution of distances of indicated D-fructose hydroxyl groups to certain side chains. Distances shown are calculated from the most populated cluster in the occluded state bin (see Table 3).
Figure 3—figure supplement 1
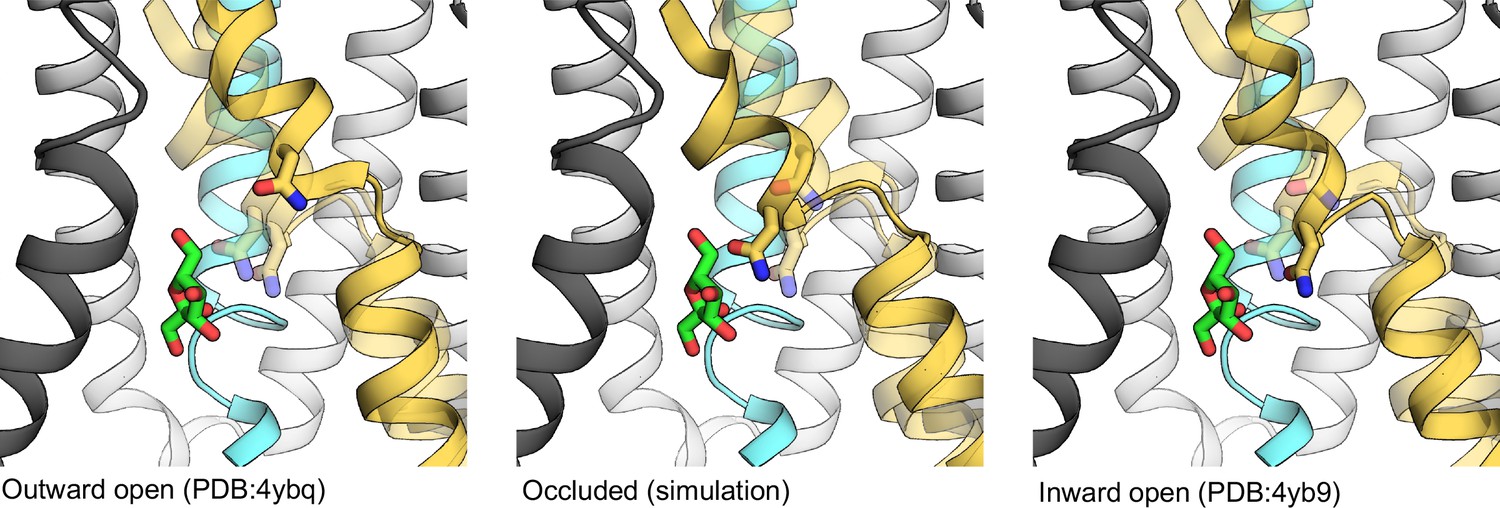
The coordination of N293 (licorice) through the transport cycle.
The TM7 is shown for all states, altering transparency for each described state to easier show the motion through the transport cycle. The other transmembrane (TM) helices are shown only for the occluded state. Coloring for each TM helix is as shown in Figure 1B.
Figure 4 with 3 supplements
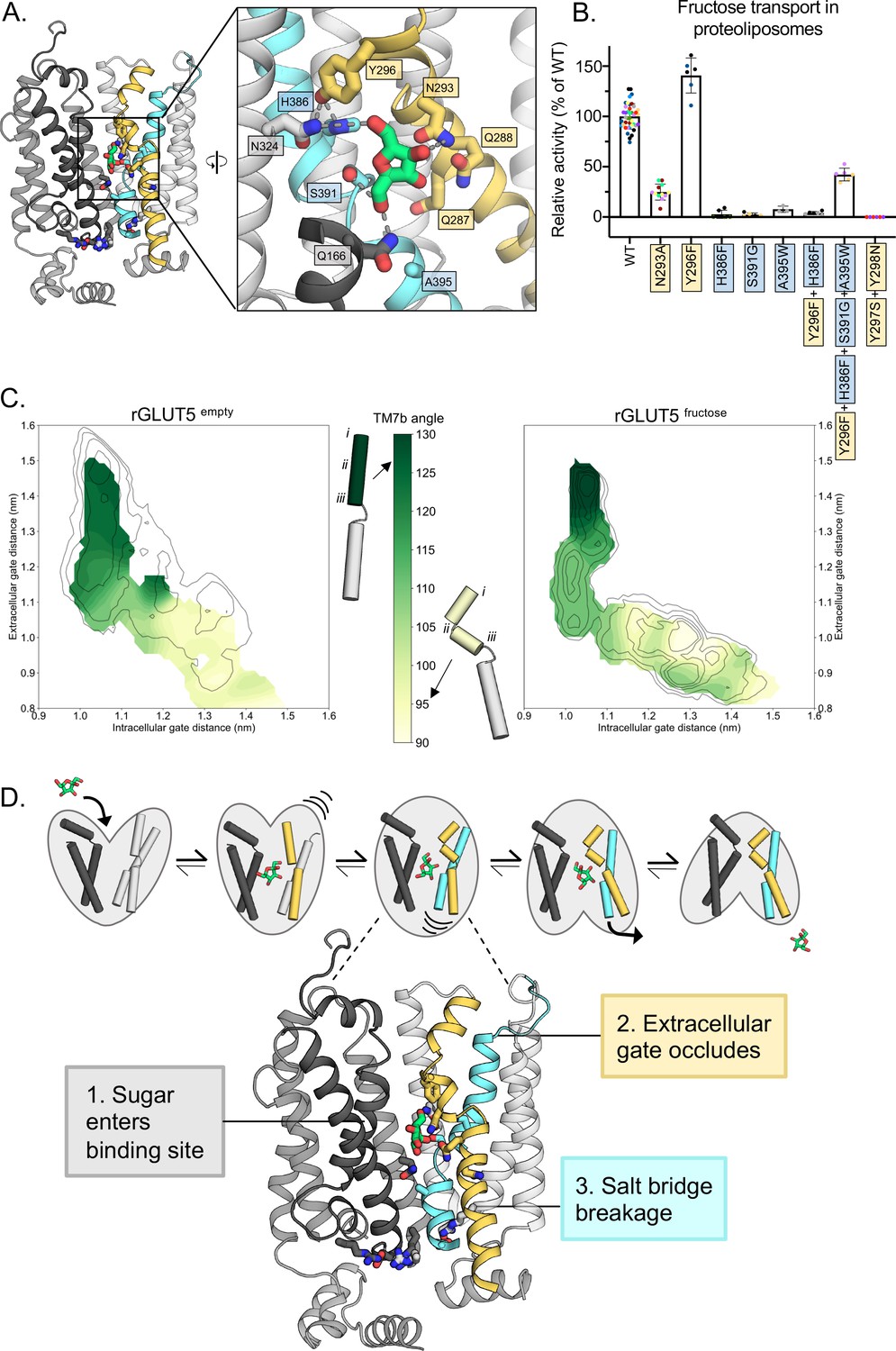
Confirming the residues required for coupling sugar binding to the extracellular gate TM7b and the intracellular gate TM10b.
(A) An overview of the residues connecting the coupling of TM7b breakage during sugar binding to the movement of TM10b. The occluded state is represented here. When N293 is pulled toward the D-fructose hydroxyl group on C4, TM7b is kinked, and Y296 is rotated toward TM10. Y296 is then able to interact with H386 on TM10a. Coloring and objects shown as described in Figure 1. TM2, TM11, and ICH4 are omitted for clarity, though the salt-bridge residue R407 on TM11 remains. The inlay highlights key residue interactions with the sugar, the Y296–H386 interactions, and the Y296–N324 interactions. Dashes represent possible hydrogen-bond interactions (as shown in Figure 3B, D, not including Y296–H386 or Y296–N324). (B) 4C-D-fructose transport activity of wild-type (WT) rGLUT5 and mutations related to the coupling of D-fructose binding with TM7 breakage and TM10 movement, residue names are colored as in panel A. Uptake of 14C-D-fructose (black bars) in proteoliposomes of rat GLUT5 variants relative to WT. Errors bars represent standard error of the mean (SEM) of n ≥ six independent measurements taken from two to three independent liposome reconstitutions. Each independent reconstitution is colored differently. Colors shared between different bars indicate shared liposome preparation prior to reconstitution. (C) TM7b angle for rGLUT5empty (left) and rGLUT5fructose (right) superimposed onto the respective free energy landscapes. This angle is calculated by measuring the vector formed between residue groups i, ii, and iii as shown on the protein cartoons (see Methods). The angle in the outward-open state is approximately 130 degrees, and will bend to nearly a 90 degree angle toward the inward-facing states. (D) Our updated view on the molecular determinants of sugar-induced conformational change in GLUT5. Residues moving to interact with the sugar in the binding site induce a conformational change in TM7b (yellow), occluding the extracellular gate. This breakage of TM7b in a fully occluded state is communicated to an inward opening via TM10b (cyan). Interactions between TM7b (Y296) to TM10a (H386) and TM8 (N324), as well as between the D-fructose and S391, stabilize TM10a. The TM10b, which is separated from TM10a by the GPXPXP motif, is uncoupled from TM10a, and will move away from TM4b. This movement will then induce breakage of the salt-bridge residues to an inward-open state. Coloring as in Figure 4—source data 1 for 4B is supplied.
-
Figure 4—source data 1
Raw data from sugar transport assays of GLUT5.
- https://cdn.elifesciences.org/articles/84808/elife-84808-fig4-data1-v1.xlsx
Figure 4—figure supplement 1
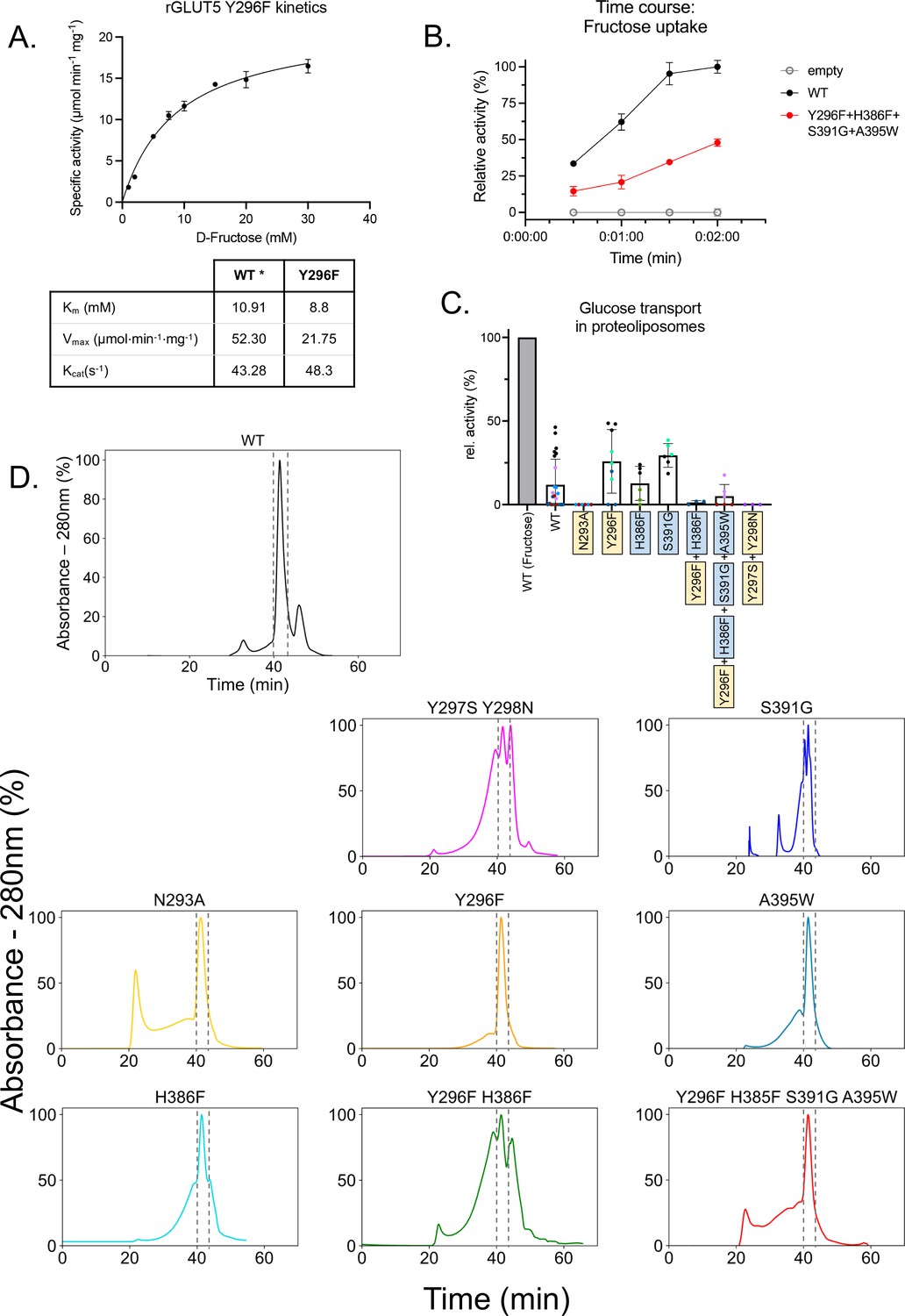
GLUT5 kinetics and size exclusion chromatography (SEC) traces for purified rGLUT5 proteins.
(A) Zero trans kinetics (influx) of the rat GLUT5 mutation Y296F. Error bars represent the standard error of the mean (SEM) of n = 3 individual experiments. The KM is an average calculated from two independent reconstitutions and titrations. The kinetic parameters for rat GLUT5 wild-type (WT; indicated by *) originate from our previously measured kinetics Qureshi et al., 2020. (B) Time course of the low-activity rGLUT5 mutation Y296F–H396F–S391G–A395W (Figure 4B), normalized between 100% (signal from WT transport after 2 min) and 0% (noise from empty liposomes as background). Error bars represent the SEM of n = 3 individual experiments. (C) 14C-D-glucose transport activity of WT rGLUT5 and mutations as seen in Figure 4B. normalized between 100% (signal from WT D-fructose transport after 2 min) and 0% (noise from empty liposomes as background). Errors bars represent SEM of at least three independent measurements taken from at least one independent liposome reconstitution. Each independent reconstitution is colored differently. Colors shared between different bars indicate shared liposome preparation prior to reconstitution. (D) Relative UV absorbance of n-dodecyl-β-D-maltopyranoside (DDM)-purified rGLUT5 proteins during size exclusion chromatography of the final purified sampled. The rGLUT5 peak between gray dashes was collected for proteoliposome transport assays. Each curve is normalized to the maximum intensity of each trace as 100%. Source data for this figure are supplied.
Figure 4—figure supplement 2

Sequence logos for transmembrane helices of putative GLUT5 proteins (see Methods) in other organisms.
Column numbering corresponds to rat GLUT5 residue numbers. TM7 and TM10 helices are colored as in Figure 1. Following coloring as in Figure 1—figure supplement 1A, residues involved in D-fructose binding are indicated with an orange circle, the GPXPXP motif as well as residues involved in coupling D-fructose binding to conformational change are indicated with a cyan circle. The TM7b tyrosine or phenylalanine can be found at column 296.
Figure 4—figure supplement 3
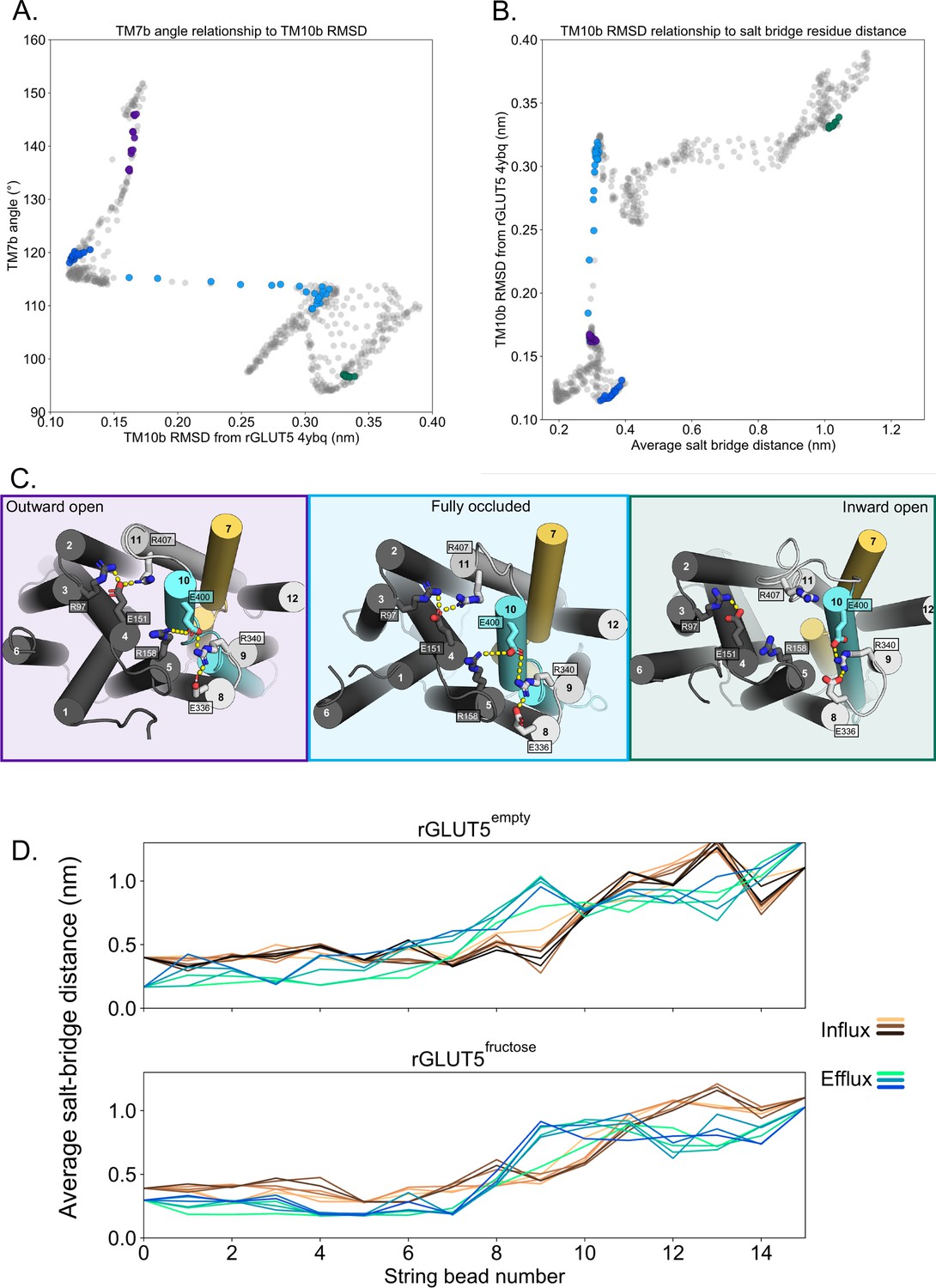
Salt-bridge formation during the conformational cycle.
(A) Relationship between TM7b angle and TM10b root mean squared deviation (RMSD). Circles colored correspond to bins with extracellular (EC) and intracellular (IC) values near the outward-open (purple), outward-occluded (deep blue), occluded (light blue), and inward-open (green) conformations, respectively. The trajectory does not sample enough around the original inward-occluded model to enable its inclusion in the analysis. (B) Relationship between TM10b RMSD and the average of the two state-dependent salt-bridge network distances. Coloring as in panel A. (C) The salt-bridge residues of GLUT5 in three of the major conformations, forming two main salt-bridge groups, where interactions are indicated by yellow dashes. State-dependent interactions are observed for the salt bridge between E151–R401 (TM4–TM11) and E400–R158 (TM10–TM5), which are not observed in the inward-open state (right, green). Protein coloring as in Figure 1, and background coloring indicates states representing the same colors in panels A and B. (D) Measuring the average of the two state-dependent salt-bridge distances during the transport cycle (panel C) in several beads, through iterations. Earlier iterations are shown in a light shade, and the final iteration is darkest (rGLUT5empty/influx: 745, rGLUT5empty/efflux: 450, rGLUT5fructose/influx: 552, rGLUT5fructose/efflux: 553), with every 50th iteration in between displayed.
Author response image 1

Crystal structure of PfHT bound to 2,5 anhydro-D-mannitol.
Author response image 2

Overlay of initial simulations with the rGLUT5empty energy surface.
The last 100ns of each simulation is shown as a small colored dot, 1 dot per nanosecond, the initial conformation of the homology model in the larger colored dot. The energy surface is projected with the same levels as shown in Figure 2E, with the colors changed to inverted grayscale for simplicity.
Author response image 3

The effect of hysteresis on two string simulations with starting paths generated by steered MD.
Both have a strong memory of their initial starting states which are predicted to be the most energetically favorable (red), moving only uphill in free energy.
Author response image 4

(A) Free energy surfaces from the MSM for rGLUT5empty with a pathway generated from TMD in the direction opposite to the one reported in the manuscript (in open → out open).
Iterations 100 – 420 shown here. B. Free energy surfaces from the MSM for rGLUT5fructose with a pathway generated from TMD in the opposite direction as reported in the manuscript (in open → out open). Iterations 100 – 553 shown here.
Author response image 5

(A) Movement of three replicas of the inward occluded state homology model on the gate CV space on top of the free energy surface of rGLUT5fructose.
A pink dot indicates the conformation for the starting model, and blue dots show the conformation for every 1ns. B. As in panel A, but using TM7b angle vs mean salt bridge distance as the CVs. C. As in panel A, but using TM7b angle vs TM10b RMSD as the CVs.
Author response image 6

.
Movement of inward open simulations superimposed on the rGLUT5fructose free energy surface. The model of rGLUT5 in the inward open state was simulated several times to assess the stability of this state in the intracellular/extracellular gate projection. The magenta dot represents the conformation of the model prior to simulation (which does not vary between conditions/replicas), blue dots represent the simulation frames (one per nanosecond), and the yellow dot represents the end frame of the simulation. Simulation time can be found in Table 1.
Author response image 7

The distribution of the distances of the two groups of the asparagine side chains in the clustered sugar position (see Methods).
Tables
Table 1
GLUT5 model and simulation details.
| State modeled | Type of model | ICH5 resolved in template? | Template structure (if applicable) | PDB code | Percentage identity to rat GLUT5 | Time run (ns) | Bilayer used |
|---|---|---|---|---|---|---|---|
| Outward open | Structure | Yes | 4ybq | 100% | 523 | POPC | |
| Outward occluded | Model | Yes | Human GLUT3 | 4zw9 | 40.8% | 298 | POPC |
| Fully occluded | Model | No | Plasmodium falciparum hexose transporter (PfHT1) | 6rw3 | 26.0% | 381 | POPC |
| Inward occluded | Model | No | E. coli xylose transporter (XylE) | 4ja3 | 23.3% | 158 + (200) | POPC |
| 231 | |||||||
| 188 | |||||||
| Inward open | Model | No | Bovine GLUT5 | 4yb9 | 76.7% | 550 | POPC |
| 144 | |||||||
| 83 | |||||||
| 244 | POPE | ||||||
| 232 | 70% POPE, 20% POPG, 10% DOPA |
Table 2
Summary of targeted MD simulations.
| rGLUT5empty Outward open – Inward open (condition 1) | ||||
|---|---|---|---|---|
| TMD number | Starting configuration | Target state | Time (ps) | Final heavy atom RMSD (nm) |
| 1.1 | Out open structure | Outward occluded | 11,220 | 0.048415 |
| 1.2 | TMD 1.1 final frame | Fully occluded | 10,890 | 0.050022 |
| 1.3 | TMD 1.2 final frame | Inward occluded | 8120 | 0.062960 |
| 1.4 | TMD 1.3 final frame | Inward open | 10,780 | 0.052656 |
| 1.5 (skipping occluded state validation) | TMD 1.1 final frame | Inward occluded | 10,440 | 0.051464 |
| rGLUT5empty Inward open – Outward open (condition 2) | ||||
| TMD number | Starting configuration | Target state | Time (ps) | Final heavy atom RMSD (nm) |
| 2.1 | In open homology model | Inward occluded | 9620 | 0.066614 |
| 2.2 | TMD 2.1 final frame | Fully occluded | 8620 | 0.053146 |
| 2.3 | TMD 2.2 final frame | Outward occluded | 7760 | 0.058147 |
| 2.4 | TMD 2.3 final frame | Outward open | 9120 | 0.054561 |
| 2.5 (skipping occluded state validation) | TMD 2.1 final frame | Outward occluded | 11,840 | 0.048901 |
| rGLUT5fructose Outward open – Inward open (condition 3) | ||||
| TMD number | Starting configuration | Target state | Time (ps) | Final heavy atom RMSD (nm) |
| 3.1 | Out open structure with D-fructose bound | Outward occluded | 8500 | 0.065881 |
| 3.2 | TMD 3.1 final frame | Fully occluded | 10,780 | 0.052316 |
| 3.3 | TMD 3.2 final frame | Inward occluded | 12,630 | 0.059851 |
| 3.4 | TMD 3.3 final frame | Inward open | 10,260 | 0.045643 |
| 3.5 (skipping occluded state validation) | TMD 3.1 final frame | Inward occluded | 12,480 | 0.055612 |
| rGLUT5fructose Inward open – Outward open (condition 4) | ||||
| TMD number | Starting configuration | Target state | Time (ps) | Final heavy atom RMSD (nm) |
| 4.1 | In open homology model with D-fructose bound | Inward occluded | 10,000 | 0.075750 |
| 4.2 | TMD 4.1 final frame | Fully occluded | 12,980 | 0.048203 |
| 4.3 | TMD 4.2 final frame | Outward occluded | 11,780 | 0.049837 |
| 4.4 | TMD 4.3 final frame | Outward open | 12,420 | 0.049001 |
| 4.5 (skipping occluded state validation) | TMD 4.1 final frame | Outward occluded | 12,480 | 0.049699 |
Table 3
Analysis of sugar-binding pose in outward-open and occluded states.
| Bin closest to: outward-open stateTotal frames in bin: 1000Total possible clusters for sugar pose: 217 | Bin closest to: occluded stateTotal frames in bin: 690Total possible clusters for sugar pose: 116 | ||||
|---|---|---|---|---|---|
| Cluster number | Number of frames in cluster | Percentage of total frames in bin | Cluster number | Number of frames in cluster | Percentage of total frames in bin |
| 1 | 234 | 23.40% | 1 | 447 | 64.78% |
| 2 | 123 | 12.30% | 2 | 52 | 7.54% |
| 3 | 106 | 10.60% | 3 | 24 | 3.48% |
| 4 | 86 | 8.60% | 4 | 23 | 3.33% |
| 5 | 59 | 5.90% | 5,6,7 | 3 | 0.43% |
| 6 | 38 | 3.80% | 8–12 | 2 | 0.29% |
| 7 | 36 | 3.60% | 13–116 | 1 | 0.14% |
| 8–12 | 32–10 | 3.20–1.00% | |||
| 13–16 | 5–2 | 0.50–0.20% | |||
| 17–217 | 1 | 0.10% | |||
Table 4
Details of conventional simulations from points along the free energy surface.
| Outward-facing controls | ||||
|---|---|---|---|---|
| Replica | Iteration (out of 552) | Bead (out of 16) | Swarm (out of 32) | Simulation length (ns) |
| 1 | 552 | 1 | 2 | 299 |
| 2 | 552 | 1 | 29 | 296 |
| 3 | 552 | 2 | 1 | 311 |
| Inward-facing controls – upper well | ||||
| Replica | Iteration (out of 552) | Bead (out of 16) | Swarm (out of 32) | Simulation length (ns) |
| 1 | 552 | 10 | 6 | 384 |
| 2 | 552 | 10 | 19 | 383 |
| 3 | 552 | 10 | 23 | 380 |
| Inward-facing controls – lower well | ||||
| Replica | Iteration (out of 552) | Bead (out of 16) | Swarm (out of 32) | Simulation length (ns) |
| 1 | 300 | 11 | 12 | 349 |
| 2 | 551 | 11 | 16 | 348 |
| 3 | 551 | 11 | 24 | 350 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Determinants of sugar-induced influx in the mammalian fructose transporter GLUT5
eLife 12:e84808.
https://doi.org/10.7554/eLife.84808




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}