Predicting metabolic modules in incomplete bacterial genomes with MetaPathPredict
- Biology Department, Woods Hole Oceanographic Institution, United States
- Luit Consulting, United States
- Marine Geology and Geophysics Department, Woods Hole Oceanographic Institution, United States
- R. Ken Coit College of Pharmacy, University of Arizona, United States
- Computational Sciences Division, Pacific Northwest National Laboratory, United States
- Department of Molecular Microbiology and Immunology, Oregon Health & Science University, United States
Figures
Figure 1
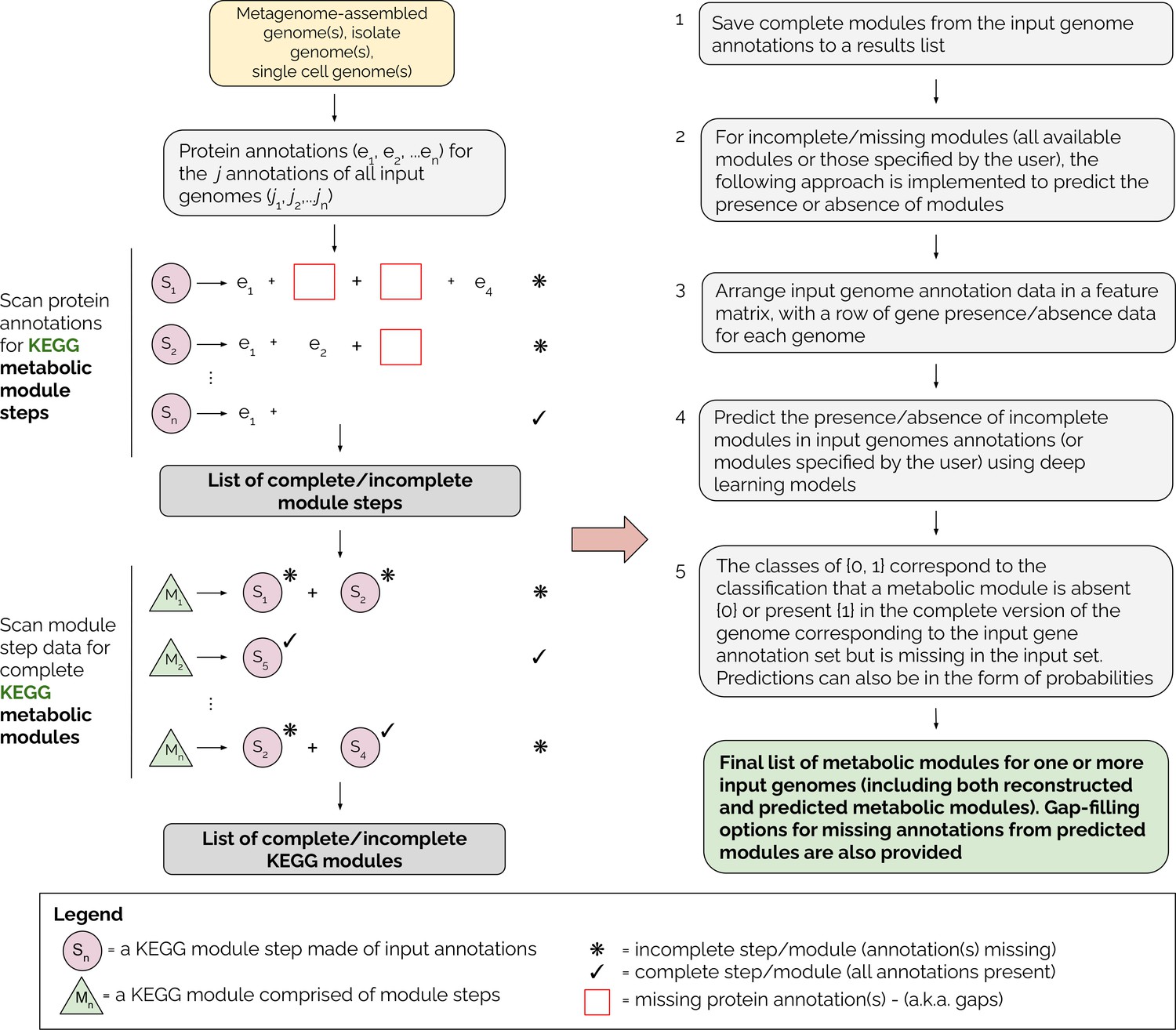
Overview of the MetaPathPredict pipeline.
Input genome annotations are read into MetaPathPredict as a data object. The data are scanned for present KEGG modules and are formatted into a feature matrix. The feature matrix is then used to make predictions for all incomplete modules (or modules specified by the user). A summary and detailed reconstruction and prediction objects, along with gap-filling options are returned in a list as the final output.
Figure 2
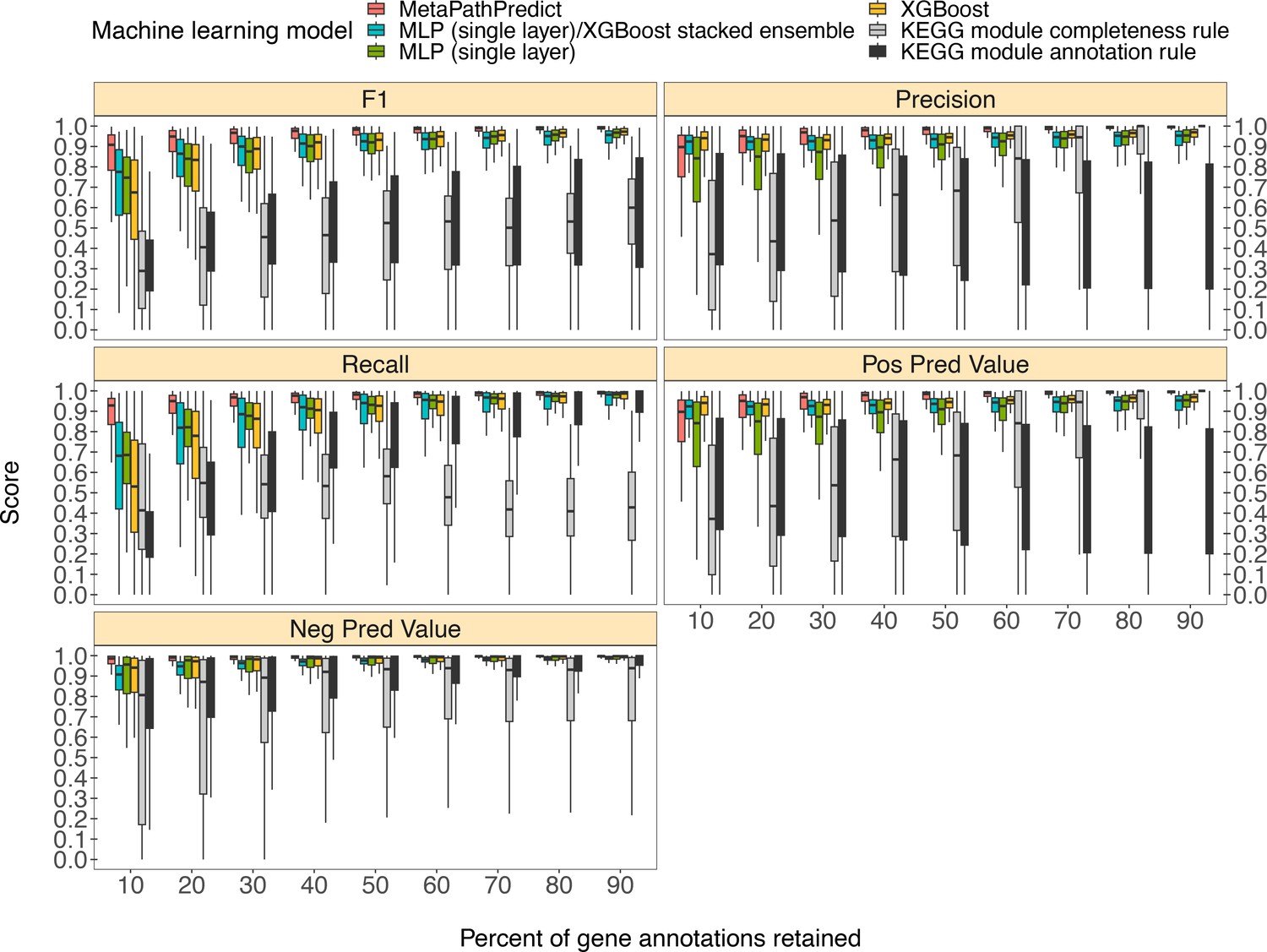
Comparison of performance metrics of MetaPathPredict’s pair of deep learning multi-label classification models to next-best performing XGBoost, single-layer neural network, and XGBoost/single-layer neural network stacked ensemble machine learning models as well as two naïve classification rules.
Down-sampled gene annotations of high-quality genomes used in held-out test sets are from NCBI RefSeq and GTDB. Each boxplot displays the distribution of model performance metrics for predictions on randomly sampled versions of the gene annotation test sets in downsampling increments of 10% (90% down to 10%, from right to left). The binary classifier performances are based on the classification of the presence or absence of KEGG modules in the complete versions of the gene annotations that were down-sampled for model testing.
-
Figure 2—source data 1
Column ‘Module name’ contains the shorthand identifiers from the KEGG database that correspond to KEGG modules; ‘Percent of protein families retained’ contains the percent of protein family presence/absence annotations retained during protein family downsampling; ‘Model type’ corresponds to the machine learning architecture or classification rule; ‘Metric’ lists the performance metric; ‘Score’ contains the value for the performance metric.
- https://cdn.elifesciences.org/articles/85749/elife-85749-fig2-data1-v1.xlsx
Figure 3
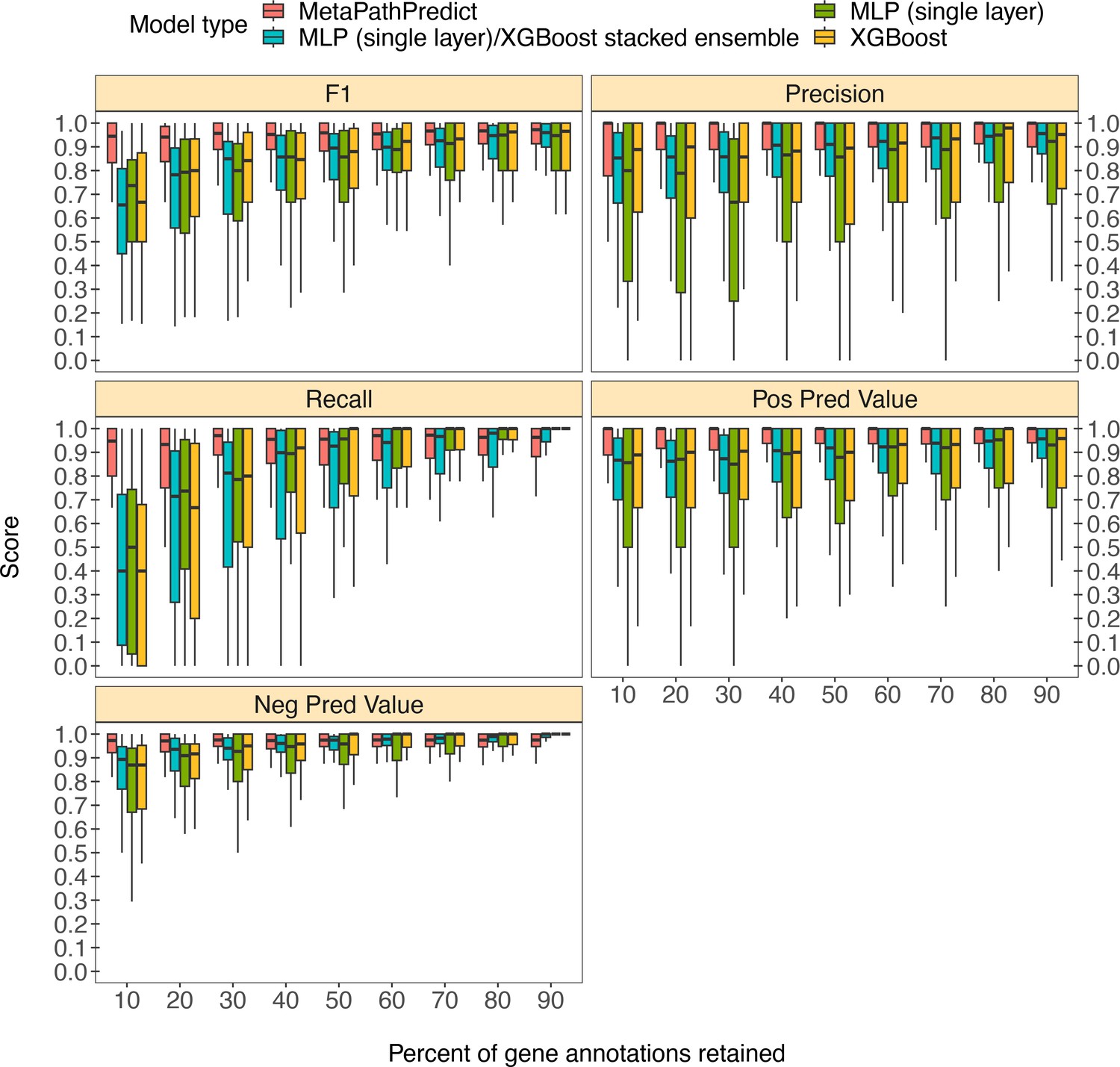
Boxplots of performance metrics of MetaPathPredict models on high-quality bacterial GEM MAGs (n=40).
Model performance metrics are for predictions on down-sampled versions of GEM genome gene annotations in decreasing increments of 10% (retaining 10–90% of the annotations in each test set). MetaPathPredict’s deep learning models were benchmarked against XGBoost and neural network model architectures.
-
Figure 3—source data 1
Column ‘Module name’ contains the shorthand identifiers from the KEGG database that correspond to KEGG modules; ‘Percent of protein families retained’ contains the percent of protein family presence/absence annotations retained during protein family downsampling; ‘Model type’ corresponds to the machine learning architecture; ‘Metric’ lists the performance metric; ‘Score’ contains the value for the performance metric.
- https://cdn.elifesciences.org/articles/85749/elife-85749-fig3-data1-v1.xlsx
Figure 4

Performance metrics boxplots of two deep learning classification models.
Down-sampled sequence reads of high-quality genomes used as a second held-out test set are from NCBI RefSeq and GTDB databases. (Panel a) Boxplots display the distribution of model performance metrics for predictions of KEGG module presence/absence on simulated incomplete genomes down-sampled at the sequence read level by MetaPathPredict models, various next-best performing machine learning architectures, and METABOLIC. Downsampling increments were chosen based on average estimated completeness of the test set genomes at each increment to reflect a range of estimated completeness thresholds. (Panel b) Average estimated genome completeness distributions of test set genomes that were down-sampled at the sequence read level using SeqTK and then assembled with SPAdes.
-
Figure 4—source data 1
Column ‘Module name’ contains the shorthand identifiers from the KEGG database that correspond to KEGG modules; ‘Percent of sequencing reads retained’ contains the percent of sequencing reads retained during sequencing read downsampling; ‘Model type’ corresponds to the machine learning architecture or annotation tool; ‘Metric’ lists the performance metric; ‘core’ contains the value for the performance metric.
- https://cdn.elifesciences.org/articles/85749/elife-85749-fig4-data1-v1.xlsx
Figure 5
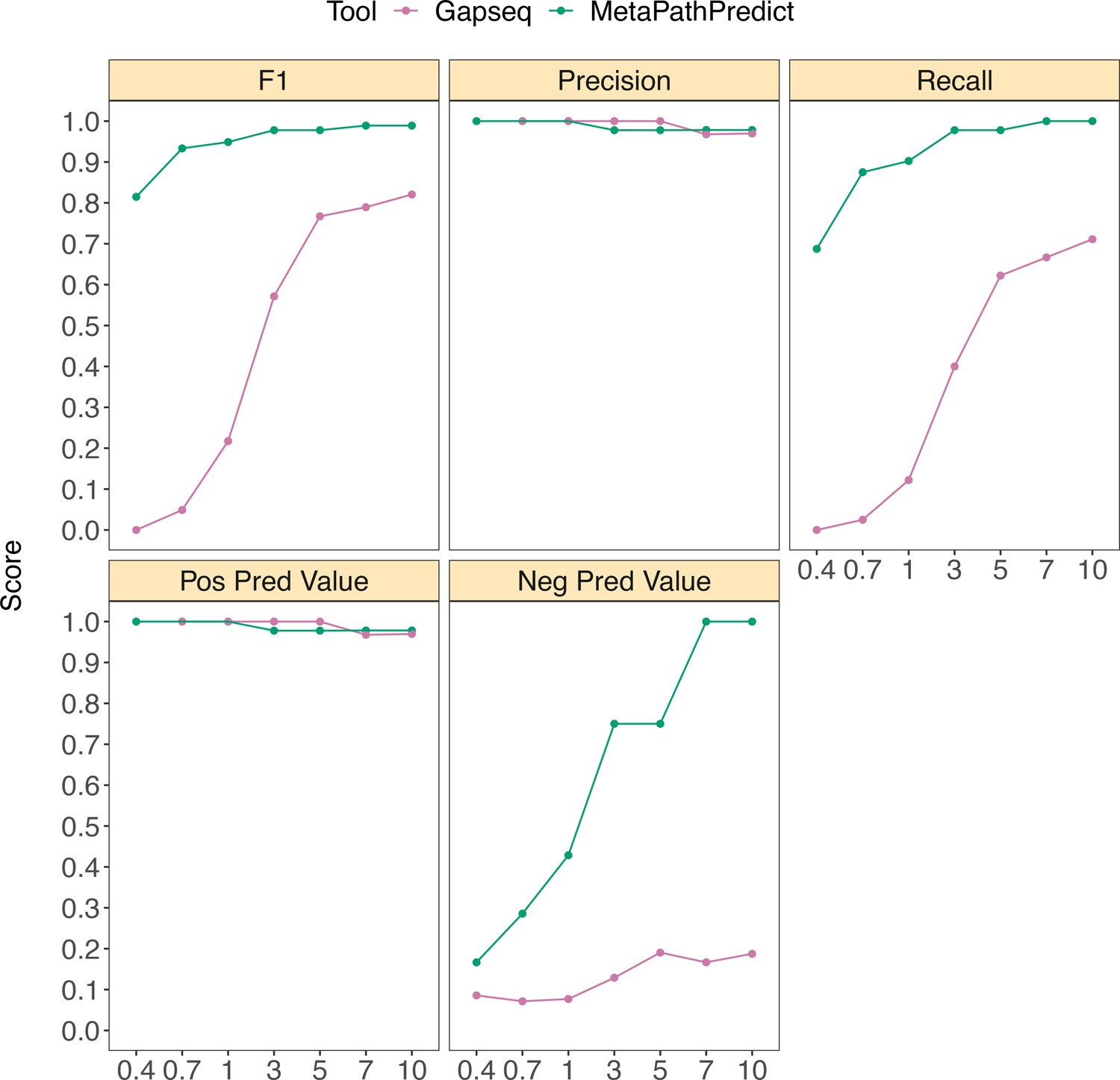
Performance metrics boxplots of MetaPathPredict and Gapseq predictions for KEGG pathway map00290 (Valine, leucine, and isoleucine biosynthesis) which contains KEGG modules M00019, M00432, M00535, and M00570.
For MetaPathPredict predictions, the whole KEGG pathway was considered present if the aforementioned KEGG modules were all present. Down-sampled sequence reads of high-quality genomes used as a second held-out test set are from NCBI RefSeq and GTDB databases. Line segments display model performance metrics for MetaPathPredict and Gapseq predictions of KEGG pathway map00290 presence/absence on simulated incomplete genomes down-sampled at the sequence read level. Downsampling increments were chosen based on average estimated completeness of the test set genomes at each increment to reflect a range of estimated completeness thresholds.
-
Figure 5—source data 1
‘Percent of sequencing reads retained’ contains the percent of sequencing reads retained during sequencing read downsampling; ‘Model type’ corresponds to the machine learning architecture or annotation tool; ‘Metric’ lists the performance metric; ‘Score’ contains the value for the performance metric.
- https://cdn.elifesciences.org/articles/85749/elife-85749-fig5-data1-v1.xlsx
Appendix 1—figure 1
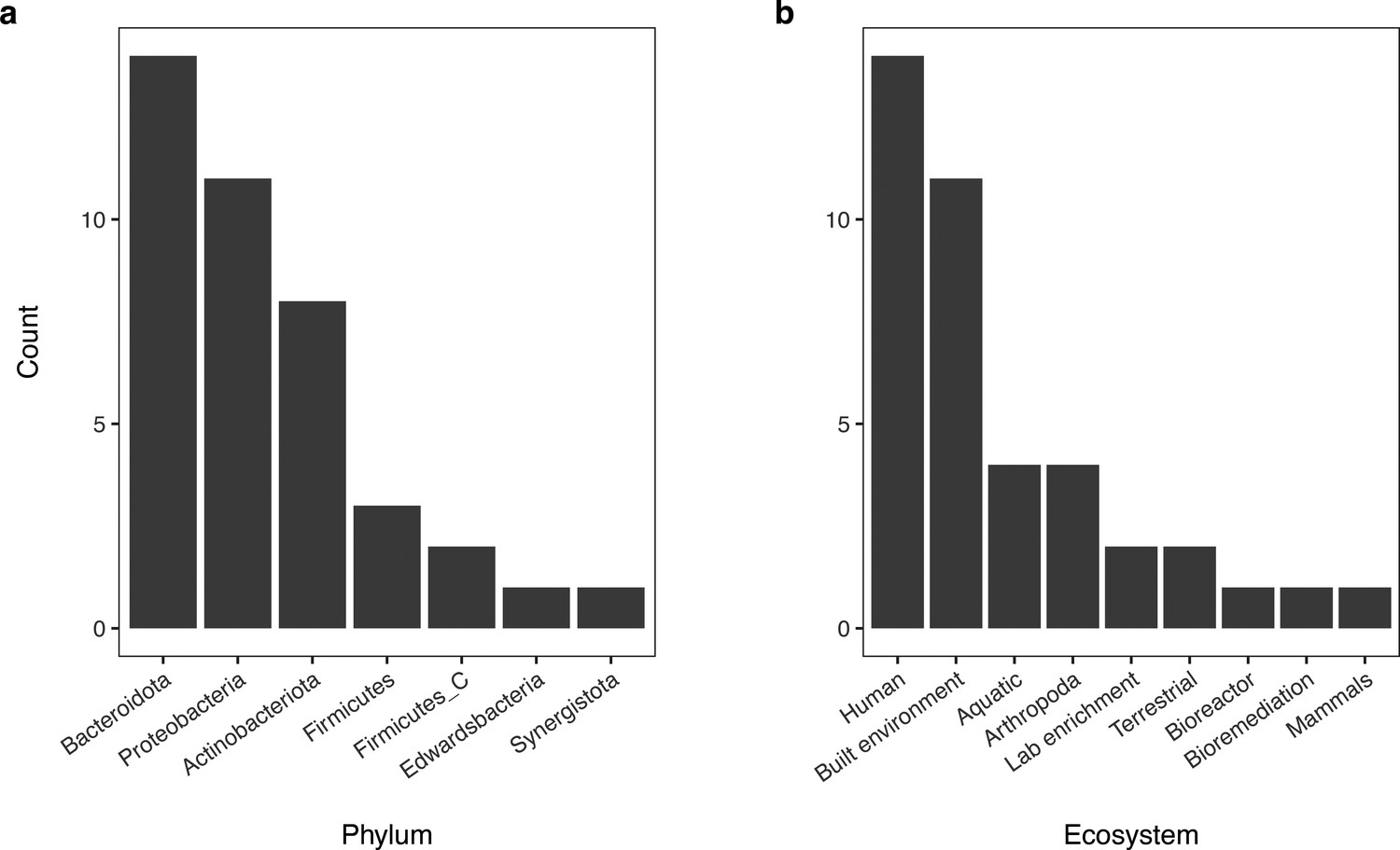
Distribution of phyla of bacterial genomes from the GEM repository used during model validation and associated environments they were recovered from.
(Panel a) Bar chart of the taxonomic distribution of genomes (n = 40) from the GEM repository used during model validation. (Panel b) Bar chart of the environmental sources of metagenomes the MAGs from this test set were recovered from.
-
Appendix 1—figure 1—source data 1
The column “Genome ID” lists the names of genomes downloaded from the GEM database; columns “Phylum”, “Class”, “Order”, “Family”, “Genus”, and “Species” contain the associated taxonomic information for each genome; “Ecosystem category” lists the environment the genome was recovered from.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig1-data1-v1.xlsx
Appendix 1—figure 2
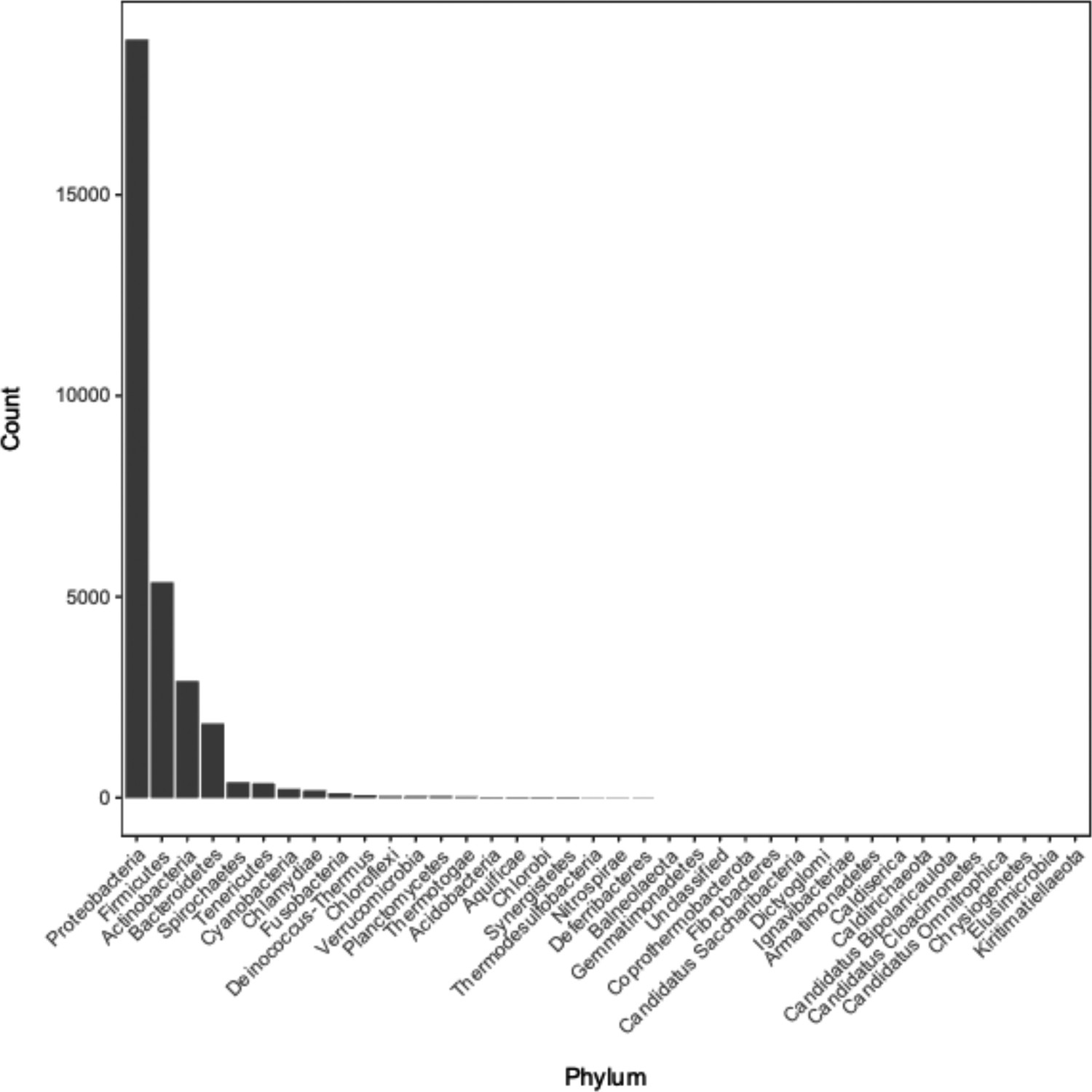
Distribution of phyla of bacterial genomes from which annotation data was used during model training and testing.
See Supplementary file 1C for the full metadata table.
-
Appendix 1—figure 2—source data 1
The column “Genome ID” lists the names of genomes downloaded from the NCBI and GTDB databases; “Database” lists which database each genomewas downloaded from (NCBI or GTDB); columns “Phylum”, “Class”, “Order”, “Family”, “Genus”, and “Species” contain the associated taxonomic information for each genome.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig2-data1-v1.xlsx
Appendix 1—figure 3
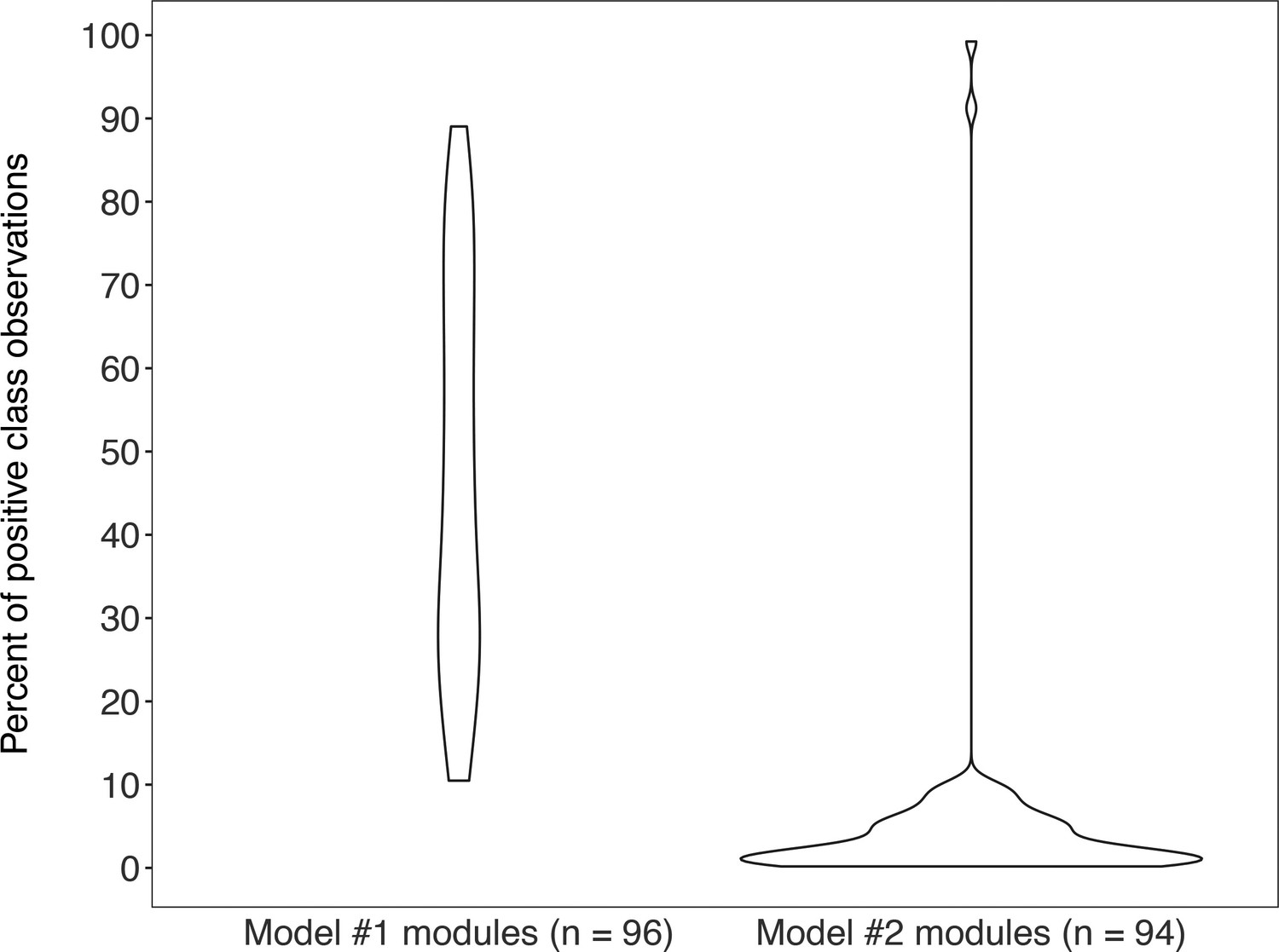
Violin plots of the percent of positive “KEGG module present” classes for genomes from MetaPathPredict’s deep learning training and test datasets for both of its models (model #1 on the left-hand side; model #2 on the right-hand side).
Each train/test split contains the same distribution of positive and negative classes.
-
Appendix 1—figure 3—source data 1
The column “Module name” contains the name of all KEGG Modules that MetaPathPredict is trained to predict; “Prevalence” lists the percent of training genomes the complete module was detected in; “Model” lists the model the prevalence data corresponds to (Model 1/Model 2).
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig3-data1-v1.xlsx
Appendix 1—figure 4
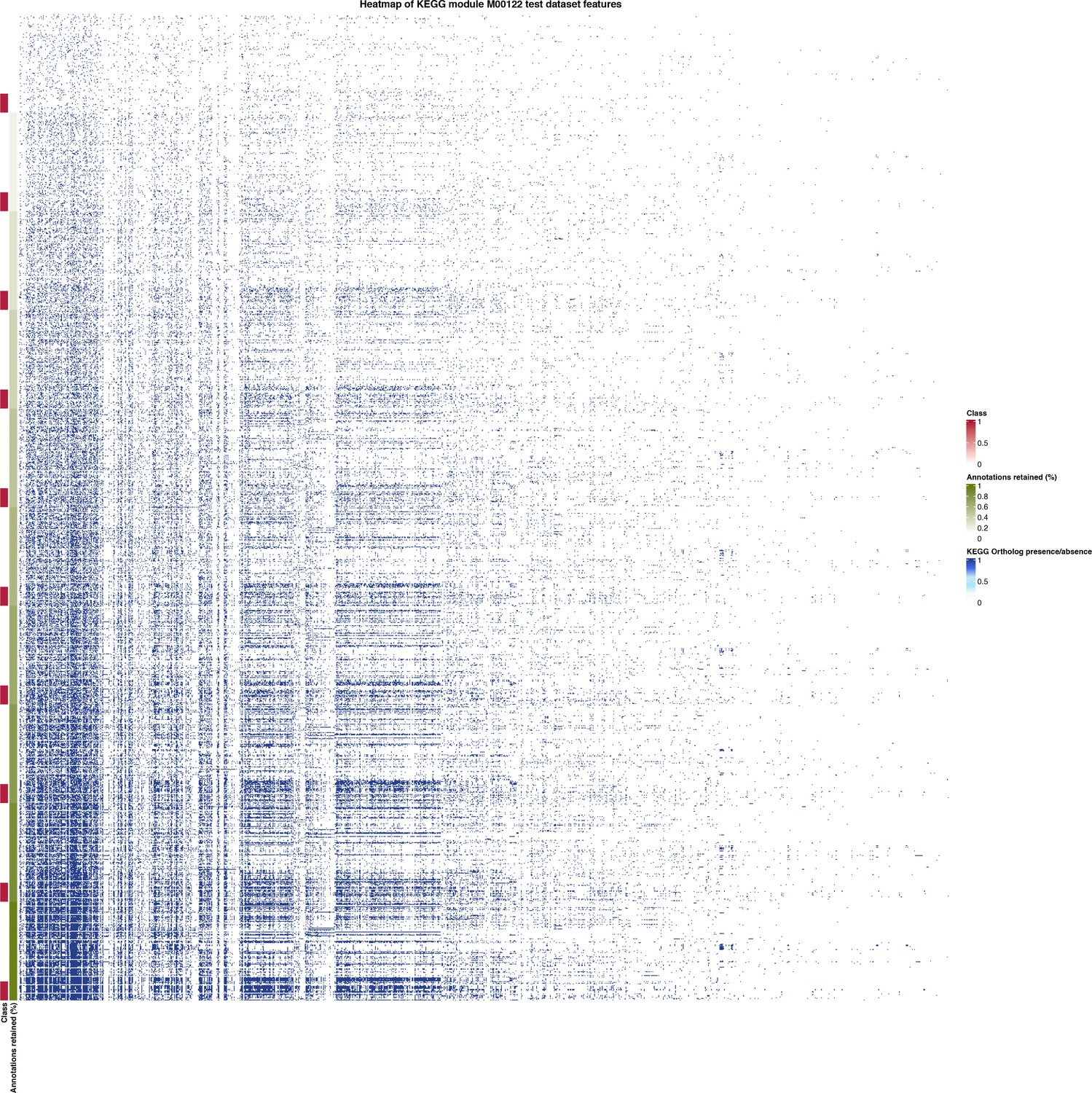
Heatmap of held-out test data for the set of features (KEGG Ortholog presence/absence) used by MetaPathPredict’s deep learning models.
The annotation row on the left-hand side of the plot is annotated with classes and predictions for KEGG module M00122 (cobalamin biosynthesis), and is sorted by the percentage of protein annotations retained in each observation (increasing in protein annotations retained from top to bottom).
-
Appendix 1—figure 4—source data 1
The column “Module M00122” contains the class labels for the presence or absence of KEGG Module M00122 in the training dataset; “Proportion of protein families retained” contains the proportion of protein family presence/absence annotations retained during protein family downsampling; the remain columns correspond to the presence or absence of protein families in the training genomes.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig4-data1-v1.zip
Tables
Table 1
Definitions of machine learning model performance metrics used to assess MetaPathPredict models.
| Metric | Definition |
|---|---|
| Precision | true positive/(true positive +false positive) |
| Recall | true positive/(true positive +false negative) |
| Specificity* | true negative/(true negative +false positive) |
| F1 score | 2 × ((precision ×recall)/(precision +recall)) |
| Positive predictive value | recall ×prevalence / (recall ×prevalence) + (1 – specificity) × (1 – prevalence) |
| Negative predictive value | specificity × (1 – prevalence) / ((1 – recall)×prevalence) + (specificity × (1 – prevalence)) |
| Prevalence* | (true positive +false negative) / (true positive +false positive +true negative +false negative) |
-
*
Specificity and prevalence are defined due to their use in the definitions of negative and positive predictive value.
Additional files
-
Supplementary file 1
Metadata for KEGG modules available within MetaPathPredict, mean SHAP values for both of MetaPathPredict’s models, and training genome metadata.
(a) MetaPathPredict KEGG Module information. The column “Module name” contains the name of all KEGG Modules that MetaPathPredict is trained to predict; “Module number” contains the module identifier; “Module class” contains module metadata including which group of KEGG metabolism the module belongs to. (b) SHAP results for the features in MetaPathPredict’s models. The column “K number (Model 1)” contains the KEGG Ortholog gene identifier for each feature in Model 1. “Mean SHAP value (Model 1)” corresponds to the mean SHAP value for features in Model 1; highest mean SHAP values are listed at the top of the column in descending order. “KEGG Module (Model 1)” shows which KEGG module(s), if any, the features of Model 1 are present in; “Gene definition (Model 1)” contains gene annotation information for each feature in Model 1. The same column definitions are repeated for Model 2 and correspond to the same information as for Model 1. (c) Training genome metadata. The column “Genome ID” lists the names of genomes downloaded from the NCBI and GTDB databases; “Database” lists which database each genome was downloaded from (NCBI or GTDB); columns “Phylum”, “Class”, “Order”, “Family”, “Genus”, and “Species” contain the associated taxonomic information for each genome.
- https://cdn.elifesciences.org/articles/85749/elife-85749-supp1-v1.xls
-
MDAR checklist
- https://cdn.elifesciences.org/articles/85749/elife-85749-mdarchecklist1-v1.pdf
-
Appendix 1—figure 1—source data 1
The column “Genome ID” lists the names of genomes downloaded from the GEM database; columns “Phylum”, “Class”, “Order”, “Family”, “Genus”, and “Species” contain the associated taxonomic information for each genome; “Ecosystem category” lists the environment the genome was recovered from.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig1-data1-v1.xlsx
-
Appendix 1—figure 2—source data 1
The column “Genome ID” lists the names of genomes downloaded from the NCBI and GTDB databases; “Database” lists which database each genomewas downloaded from (NCBI or GTDB); columns “Phylum”, “Class”, “Order”, “Family”, “Genus”, and “Species” contain the associated taxonomic information for each genome.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig2-data1-v1.xlsx
-
Appendix 1—figure 3—source data 1
The column “Module name” contains the name of all KEGG Modules that MetaPathPredict is trained to predict; “Prevalence” lists the percent of training genomes the complete module was detected in; “Model” lists the model the prevalence data corresponds to (Model 1/Model 2).
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig3-data1-v1.xlsx
-
Appendix 1—figure 4—source data 1
The column “Module M00122” contains the class labels for the presence or absence of KEGG Module M00122 in the training dataset; “Proportion of protein families retained” contains the proportion of protein family presence/absence annotations retained during protein family downsampling; the remain columns correspond to the presence or absence of protein families in the training genomes.
- https://cdn.elifesciences.org/articles/85749/elife-85749-app1-fig4-data1-v1.zip
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Predicting metabolic modules in incomplete bacterial genomes with MetaPathPredict
eLife 13:e85749.
https://doi.org/10.7554/eLife.85749




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}