A generic binding pocket for small molecule IKs activators at the extracellular inter-subunit interface of KCNQ1 and KCNE1 channel complexes
- Department of Anesthesiology, Pharmacology and Therapeutics, University of British Columbia, Canada
- Laboratory of Computational Modeling of Biological Processes, Institute of Molecular Biology, Armenia
- Molecular Neuroscience Group, Institute of Molecular Biology, Armenia
Figures
Figure 1
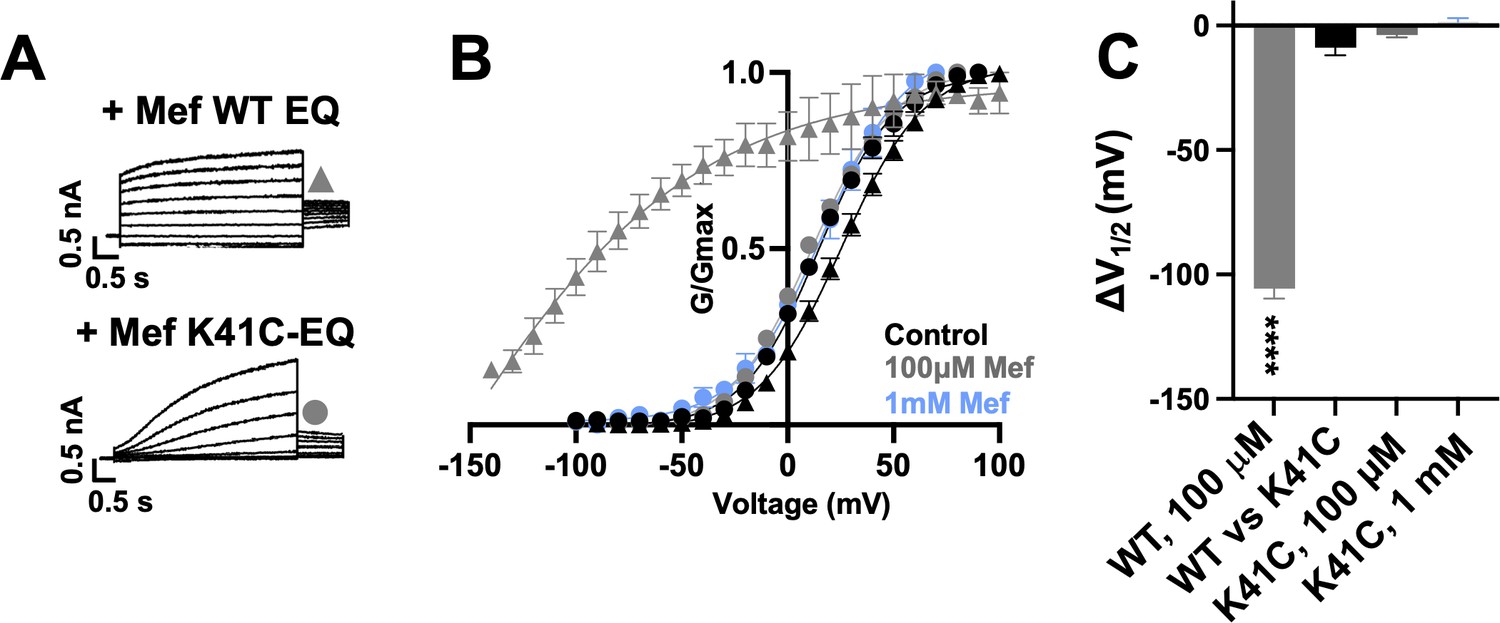
K41C-KCNE1 mutants prevent the agonist effect of mefenamic acid.
(A) Current traces of WT EQ (top) and K41C-EQ (bottom) in the presence of 100 µM mefenamic acid (Mef). A 4 s protocol was used with pulses from –150 mV or higher to +100 mV, in 10 mV steps, followed by a repolarization step to –40 mV for 1 s. Holding potential and interpulse interval were –80 mV and 15 s, respectively. (B) G-V plots obtained from WT EQ tail currents (triangles) and K41C-EQ (circles) in the absence (control: black) and presence of Mef (100 µM: grey; 1 mM: light blue). Boltzmann fits were: WT EQ control (n=6): V1/2 = 25.4 mV, k=19.4 mV; WT EQ 100 μM Mef (n=3): V1/2 = -80.3 mV, k=41.3 mV; K41C-EQ control (n=4): V1/2 = 15.2 mV, k=18.4 mV; K41C-EQ 100 μM Mef (n=4): V1/2 = 11.4 mV, k=19.4 mV; and K41C-EQ 1 mM Mef (n=3): V1/2 = 16.7 mV, k=19.8 mV. Error bars shown are SEM. (C) Summary plot of V1/2 change (ΔV1/2) for WT EQ in the presence of 100 µM mefenamic acid, WT EQ vs K41C-EQ in control and K41C-EQ in the presence of 100 µM and 1 mM mefenamic acid. Data are shown as mean ± SEM and unpaired t-test was used. **** denotes a significant ΔV1/2 compared to control where p<0.0001.
Figure 2 with 4 supplements
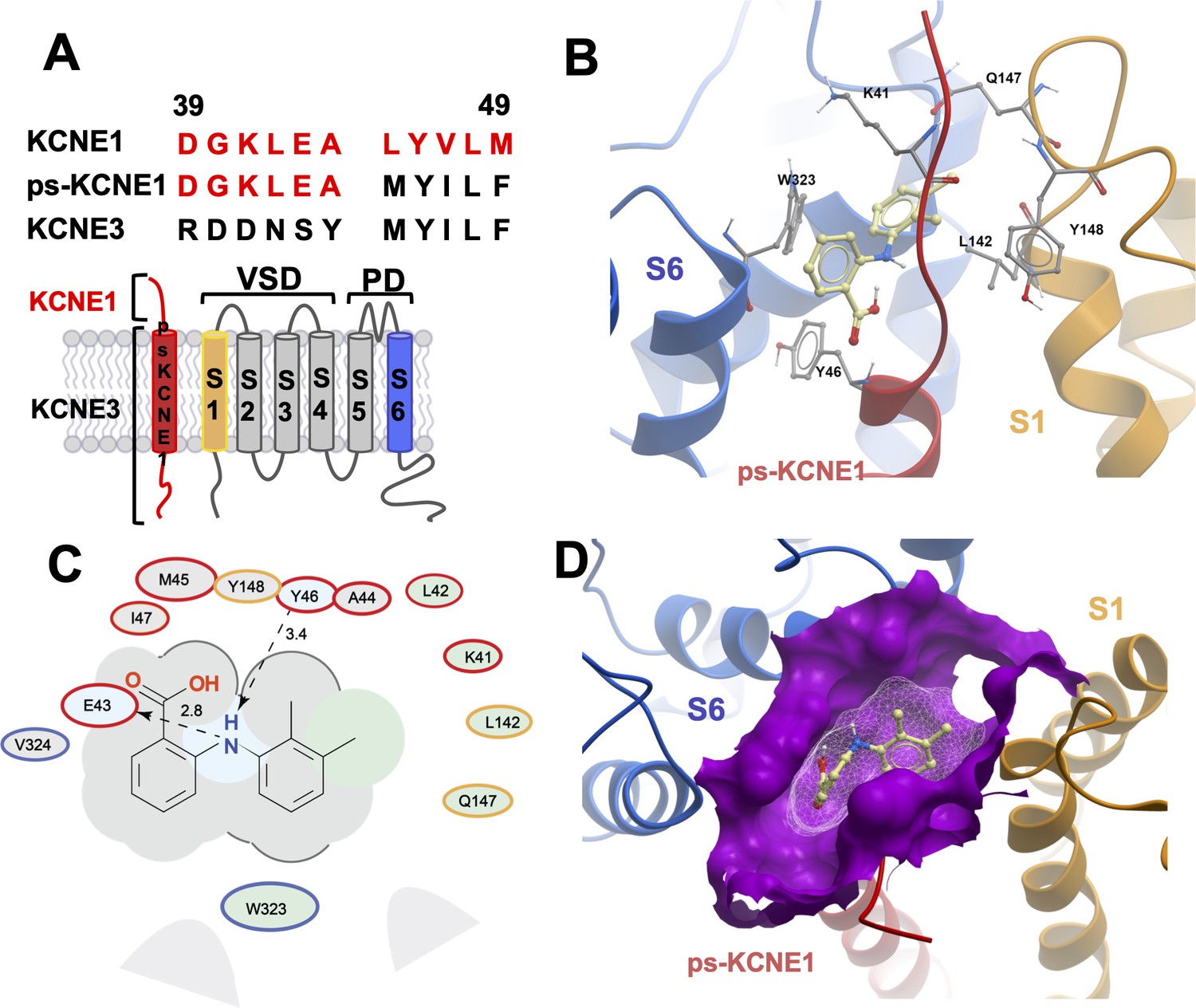
MD prediction of mefenamic acid binding site in the ps-IKs model.
(A) Pseudo-KCNE1 (ps-KCNE1) used to predict Mef binding site. Extracellular residues of KCNE1 (top), ps-KCNE1 (middle) and KCNE3 (bottom). Below, cartoon topology of the single transmembrane ps-KCNE1 β-subunit and the six transmembrane KCNQ1 α-subunit. S1-S4 transmembrane segments form the voltage sensor domain and S5-S6 form the pore domain. (B) Binding pose of Mef (yellow) in the external region of the ps-IKs channel complex obtained with docking (side view). Pore domain residues are blue, ps-KCNE1 subunit red, and the VSD of a neighbouring subunit is in yellow. (C) Ligand interaction map of Mef with ps-IKs from molecular docking. Size of residue ellipse is proportional to the strength of the contact. The distance between the residue label and ligand represents proximity. Grey parabolas represent accessible surface for large areas. Light grey ellipses indicate residues in van der Waals contacts, light green ellipses are hydrophobic contacts, and light blue are H-bond acceptors. Red borders indicate KCNE1, yellow are KCNQ1 VSD, and blue are pore residues. Dashed lines indicate H-bonds. The 2D diagram was generated by ICM pro software with cut-off values for hydrophobic contacts of 4.5 Å and hydrogen bond strength of 0.8. Further details in Materials and methods. (D) Mef binding pose observed in MD simulations in space-fill to highlight pocket formed by external S1 (yellow), S6 (blue) transmembrane domains of KCNQ1, and extracellular region of the ps-KCNE1 subunit (red). This binding conformation is the most frequent binding pose of Mef observed in ∼50% of frames and corresponds to the blue-framed conformation in Figure 2—figure supplement 3.
Figure 2—figure supplement 1
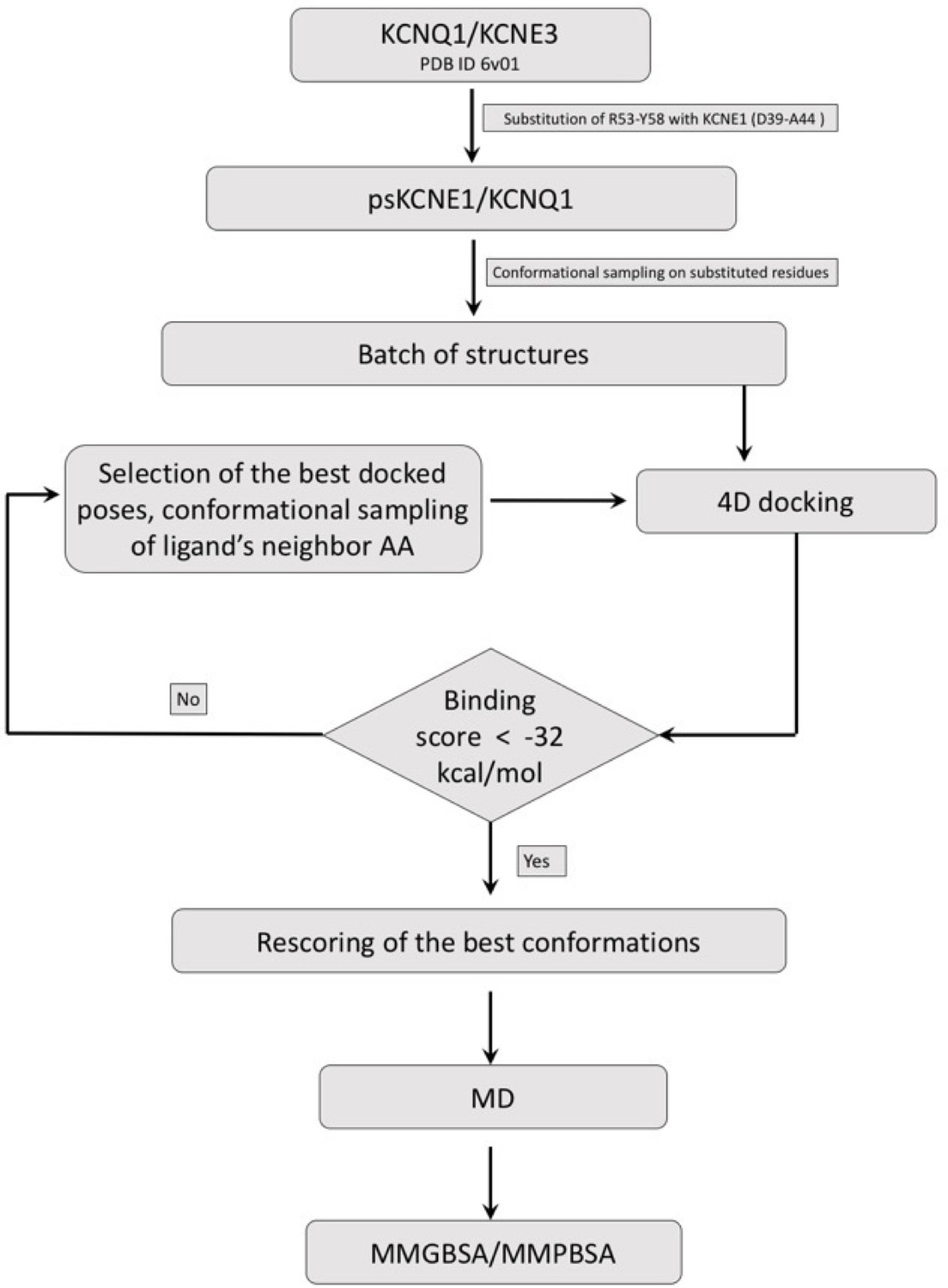
Drug docking and MD simulation workflow.
Schematic representation of the general workflow for construction of ps-IKs, Mef docking to the model ps-IKs channel complex, and MD simulations (See Materials and methods).
Figure 2—figure supplement 2
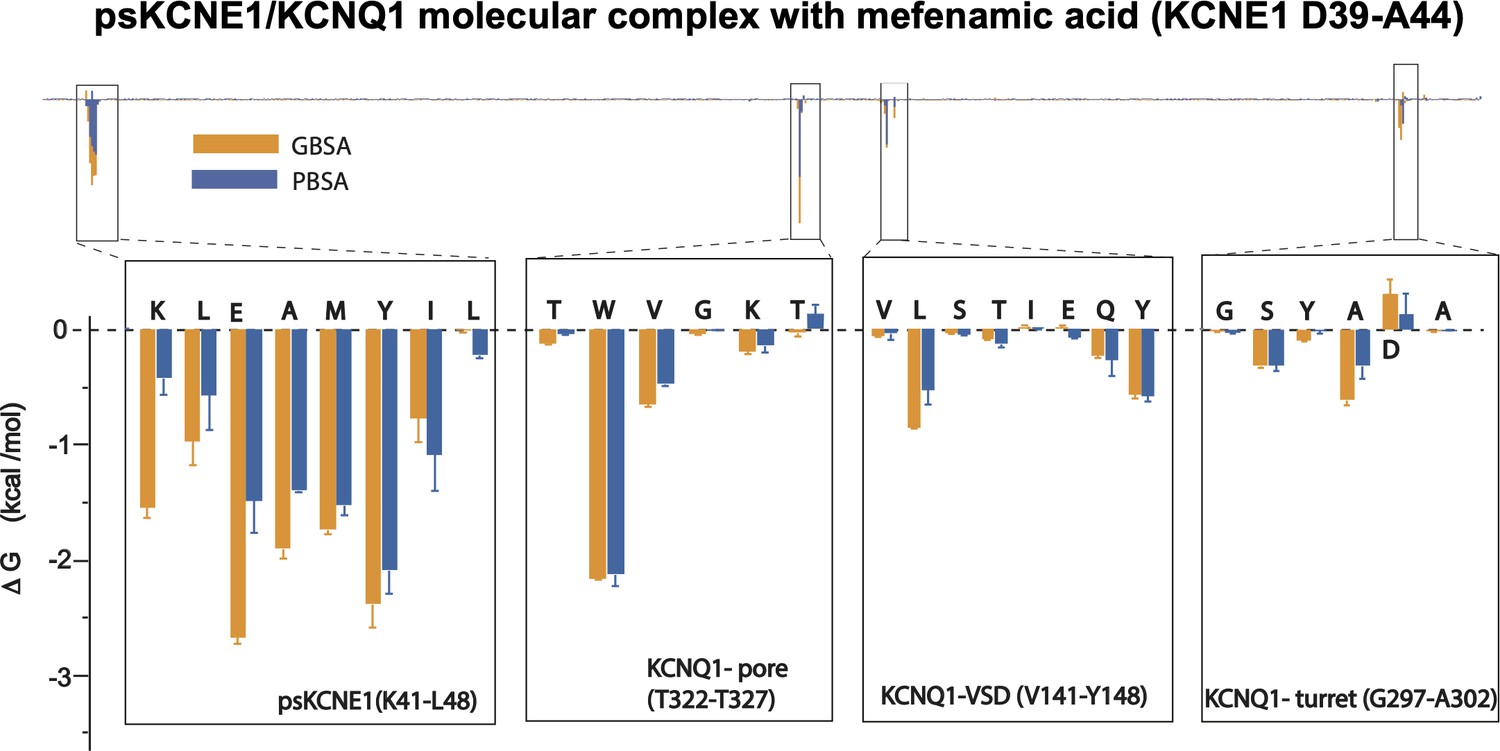
Energy decomposition per amino acid for mefenamic acid binding to ps-IKs.
Poisson-Boltzmann Surface Area (MM/PBSA; blue) and Generalized Born Surface Area (MM/GBSA; orange) methods were used to estimate the interaction free energy contribution of each residue in the Mef-bound ps-IKs complex. Free energy for residues located in ps-KCNE1, pore domain and voltage-sensor domain of KCNQ1, that stand above background, are shown as enlarged panels. Error bars represent ± SD. 1000 frames for each simulation (n=3) were analyzed.
Figure 2—figure supplement 3
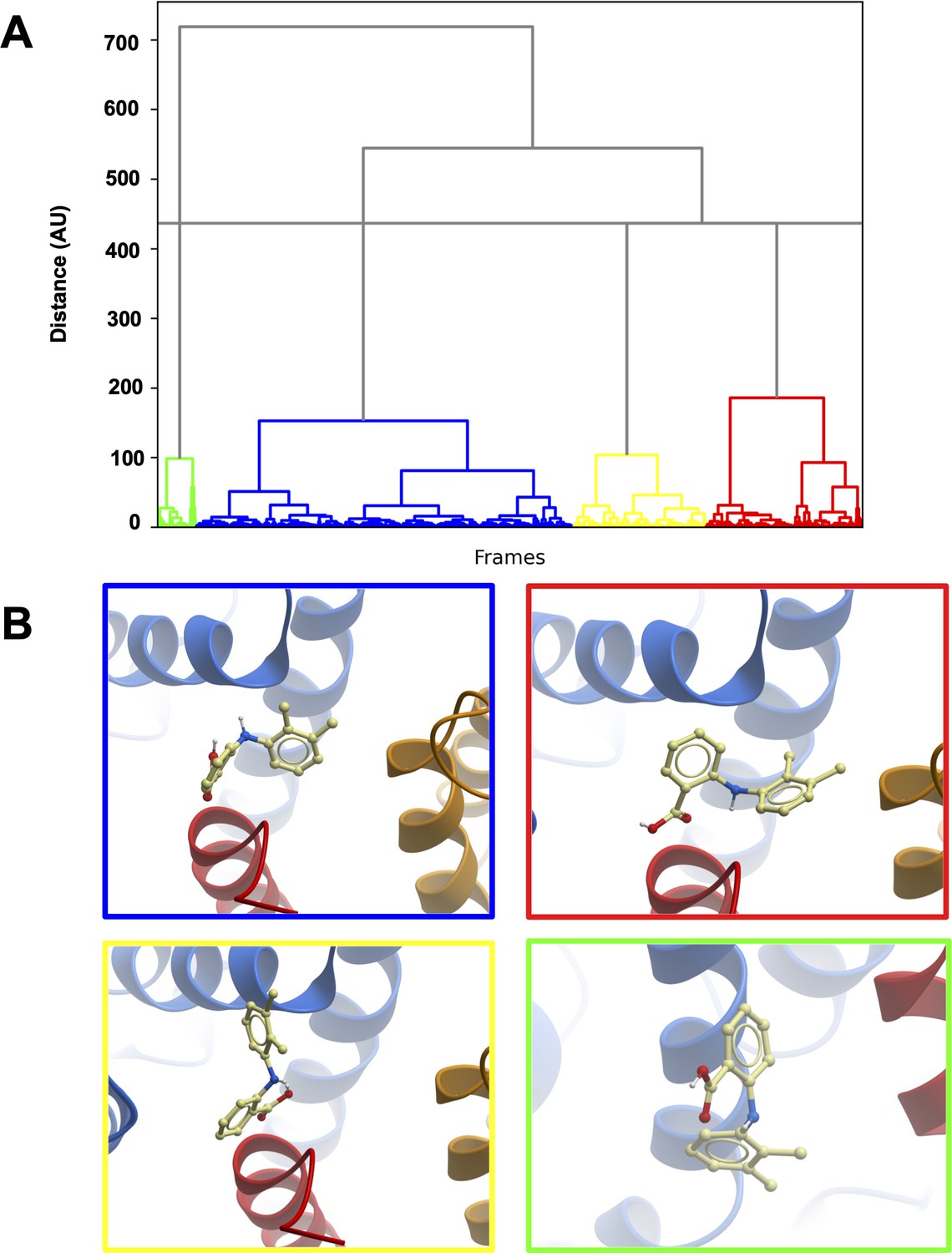
Clustering of MD trajectories based on mefenamic acid conformations.
(A) The clustering dendrogram based on Mef RMSD illustrates the similarity of Mef conformations in MD trajectories. (B) Centroids from each cluster with the lowest RMSD compared to all other conformations in the cluster. The color frame around the panels shows to which cluster the depicted conformation belongs.
Figure 2—figure supplement 4
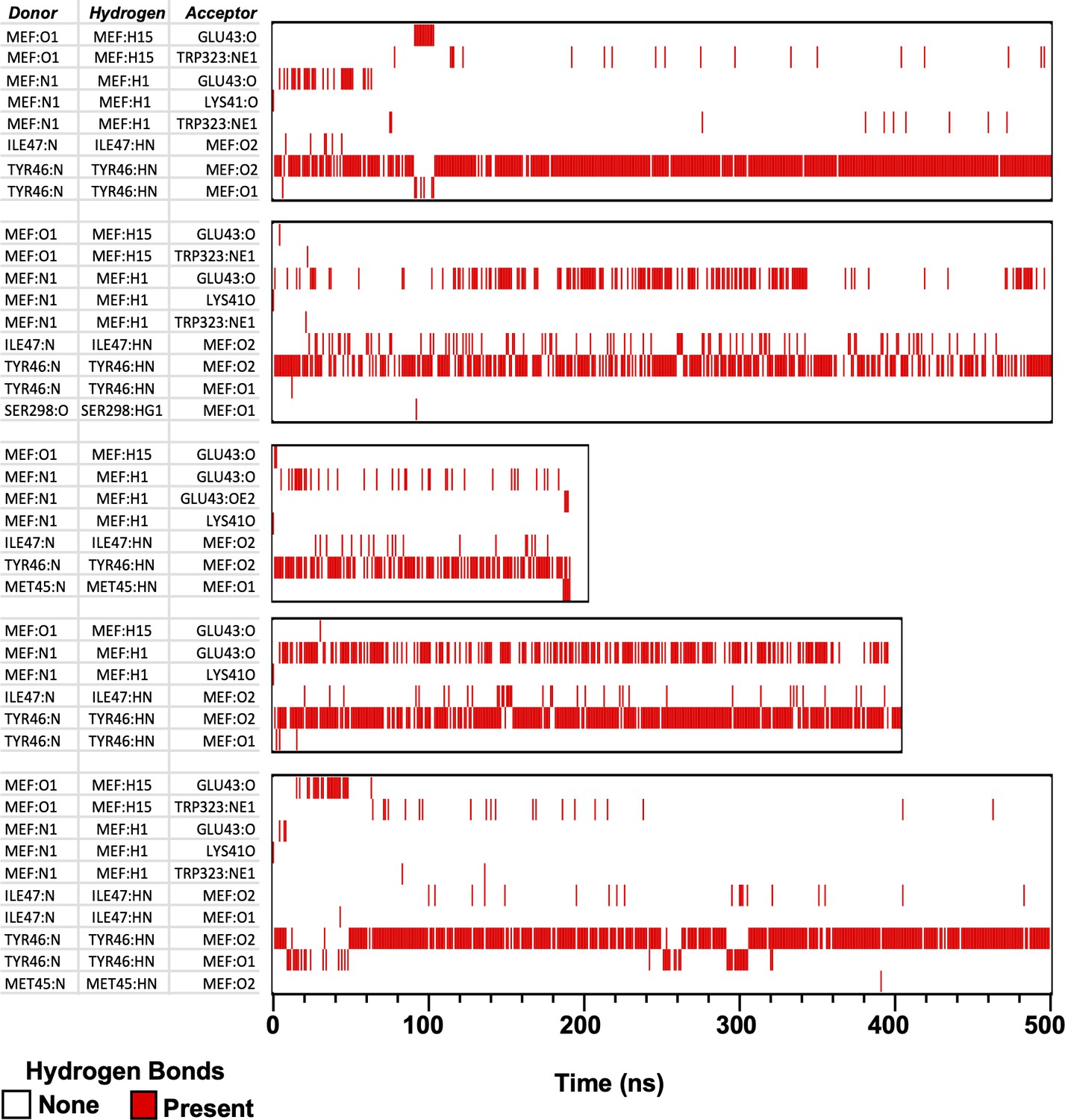
H-bonds formed between mefenamic acid and protein residues in its binding site during MD simulations.
MEF-N1 indicates the nitrogen of aminobenzoic acid; MEF:O1 and O2 are the oxygens of the aminobenzoic acid. Red lines indicate at which time point and between which atoms H-bond were formed.
Figure 3 with 2 supplements
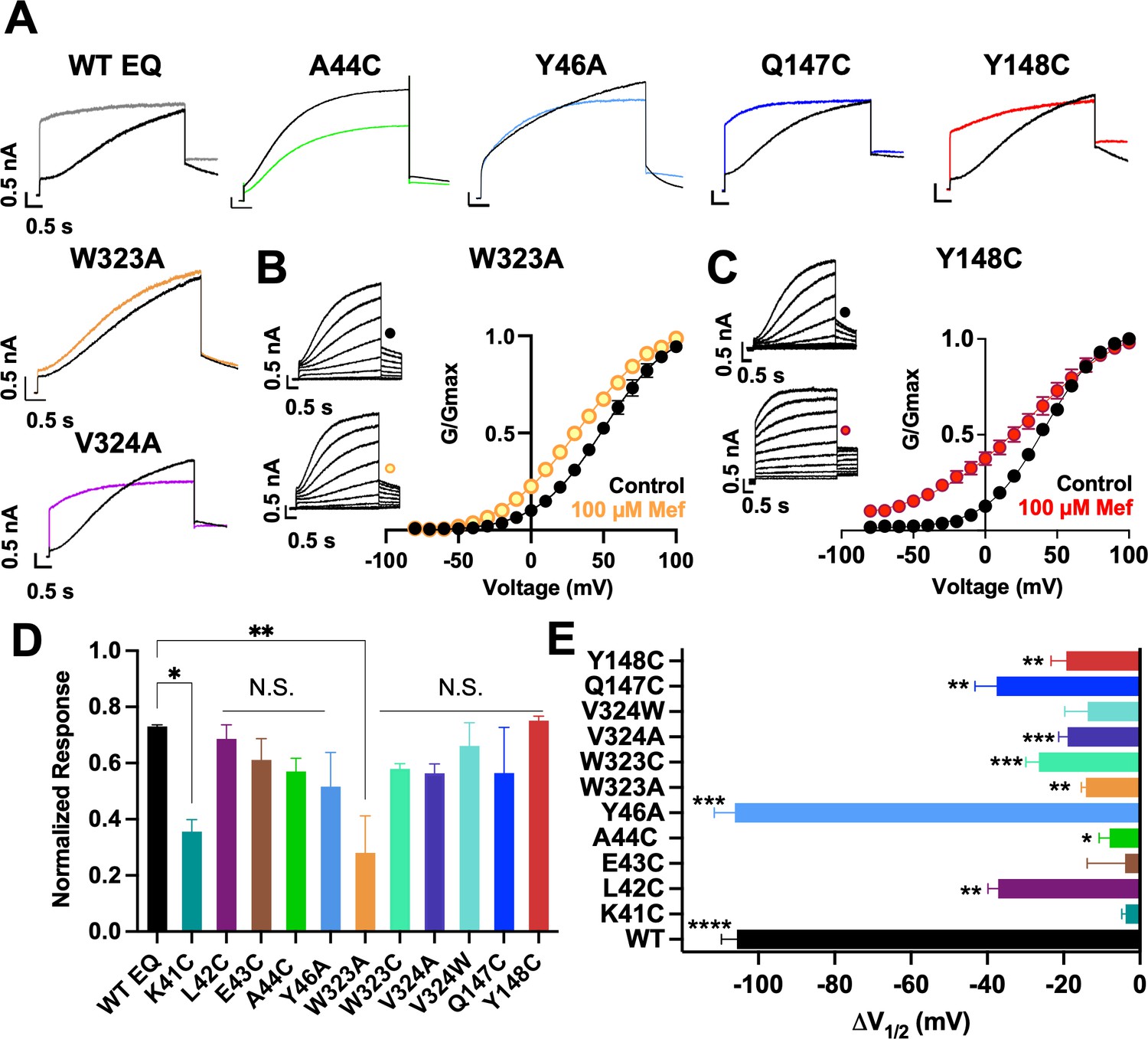
Current waveform and G-V changes induced by mefenamic acid in binding site mutants.
(A) Current traces from WT EQ and key residue mutants in control (black) and 100 µM Mef (colors). (B) EQ-W323A and (C) EQ-Y148C current traces in control (top) and presence of 100 μM Mef (below). G-V plots in control (black) and presence of 100 μM Mef (colors). Boltzmann fits were: EQ-W323A control (n=4): V1/2 = 47.8 mV, k=23.7 mV; EQ-W323A Mef (n=3): V1/2 = 33.7 mV, k=28.5 mV; EQ-Y148C control (n=4): V1/2 = 36.8 mV, k=20.3 mV; and EQ-Y148C Mef (n=4): V1/2 = 17.5 mV, k=36.7 mV. Voltage steps were from –80 mV to +100 mV for 4 s, followed by a 1 s repolarization to –40 mV. Interpulse interval was 15 s. Error bars shown are ± SEM. (D) Summary plot of the normalized response to 100 µM Mef (see Materials and methods). Data are shown as mean + SEM and one-way ANOVA statistical test was used. **p<0.01 and *p<0.05 denote a significantly reduced response compared to WT EQ. N.S. denotes not significant. (E) Change in V1/2 (ΔV1/2) for WT EQ and each IKs mutant in control versus mefenamic acid. Data are shown as mean -SEM and unpaired t-test was used, where *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 indicate a significant change in V1/2 comparing control to the presence of the drug. n-values for mutants in D and E are stated in Table 1.
Figure 3—figure supplement 1
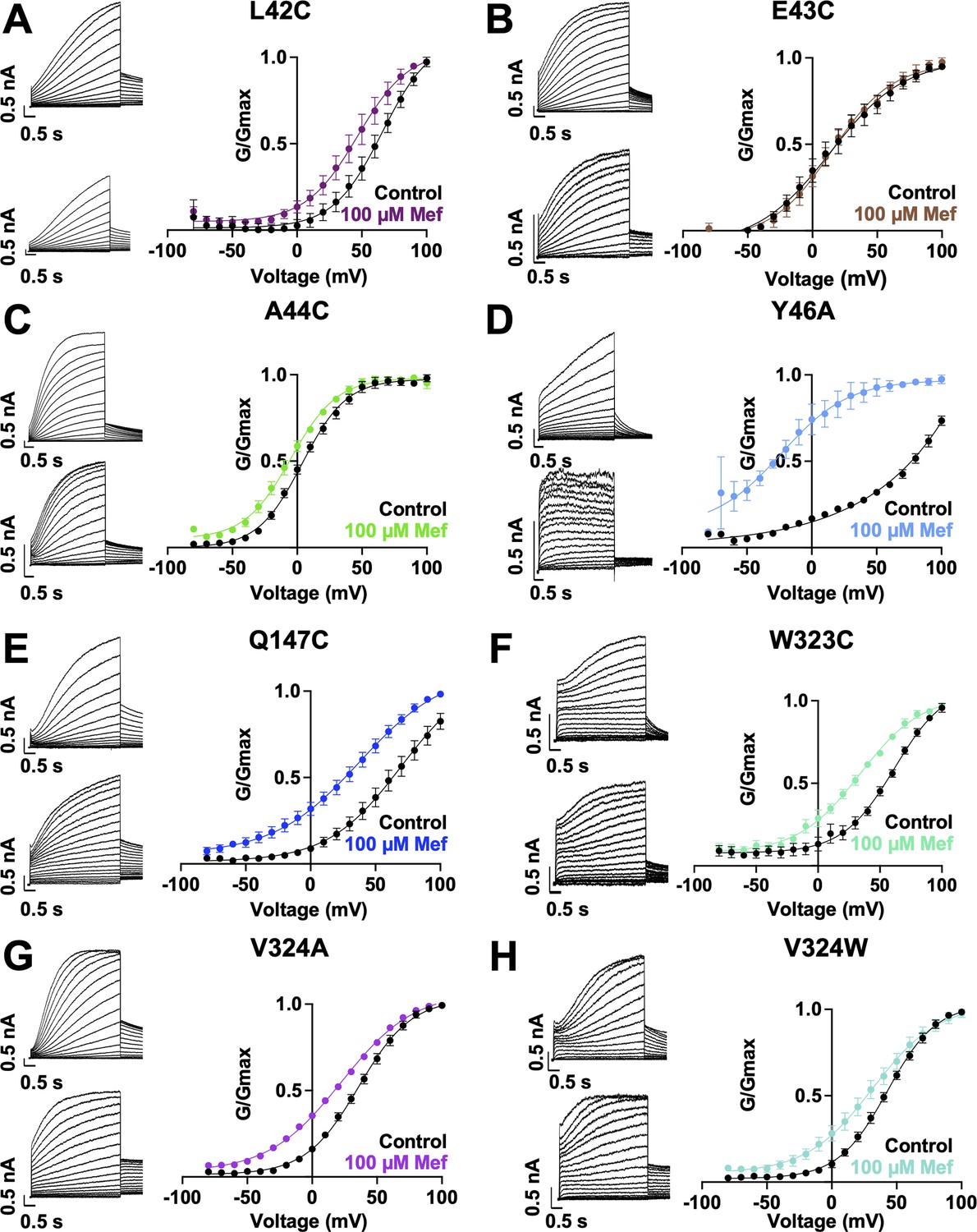
Current waveform and G-V changes induced by mefenamic acid for all binding site mutants.
(A)-(H) current traces in control (above) and presence of 100 μM Mef (below) and G-V plots for binding site mutants, as indicated. Cells were held at –80 mV, then pulsed from –90 mV to +100 mV for 4 s followed by –40 mV for 2 s. Error bars shown are ± SEM. Refer to Table 1 for Boltzmann fit values.
Figure 3—figure supplement 2
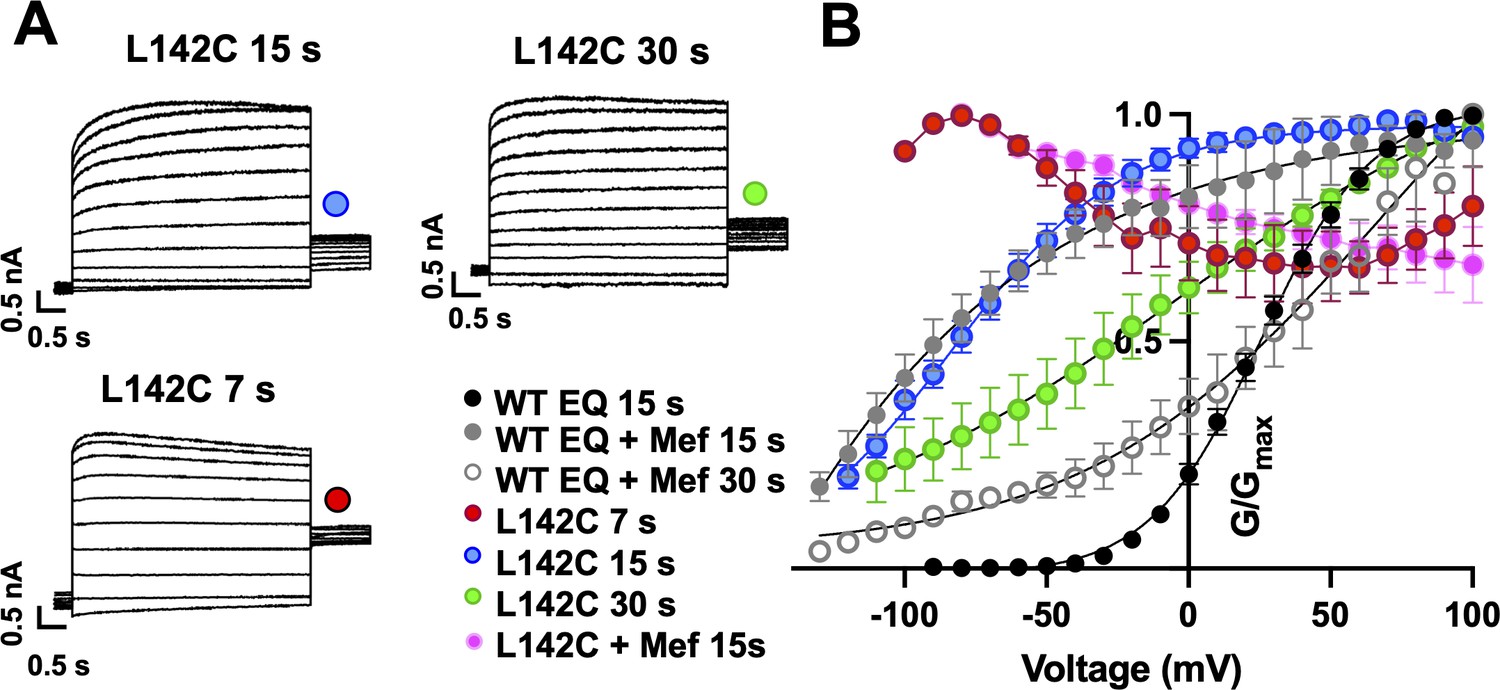
Augmented activation of EQ-L142C in the absence of mefenamic acid.
(A) EQ-L142C current traces in control solutions. Voltage steps were from –130 mV or higher in 10 mV steps to +100 mV for 4 s, followed by a repolarization step to –40 mV for 1 s. Holding potential was –80 mV. Interpulse interval was as indicated above, 15 s, 30 s and 7 s. (B) G-V plot of WT EQ in control (interpulse interval 15 s: black) and in the presence of 100 µM Mef (interpulse interval 15 s: grey closed circle; 30 s: grey open circle) and EQ-L142C in control solutions (interpulse interval 7 s: red; 15 s: blue; 30 s: green). Boltzmann fits were: WT EQ 15 s control (n=6): V1/2 = 25.4 mV, k=19.4 mV; WT EQ 15 s Mef (n=3): V1/2 = -80.3 mV, k=41.3 mV; WT EQ 30 s Mef (n=8); V1/2 = 26.7 mV, k=66.6 mV; EQ-L142C 15 s control (n=6) V1/2 = -80.3 mV, k=30.0 mV; EQ-L142C 30 s control (n=3) V1/2 = -28.7 mV, k=62.5. Due to the dramatic change in G-V plot shape, Boltzmann fits were not possible for EQ-L142C with an interpulse interval of 7 s. All mutations are forced saturated IKs channel complexes. Error bars shown are ± SEM.
Figure 4 with 2 supplements
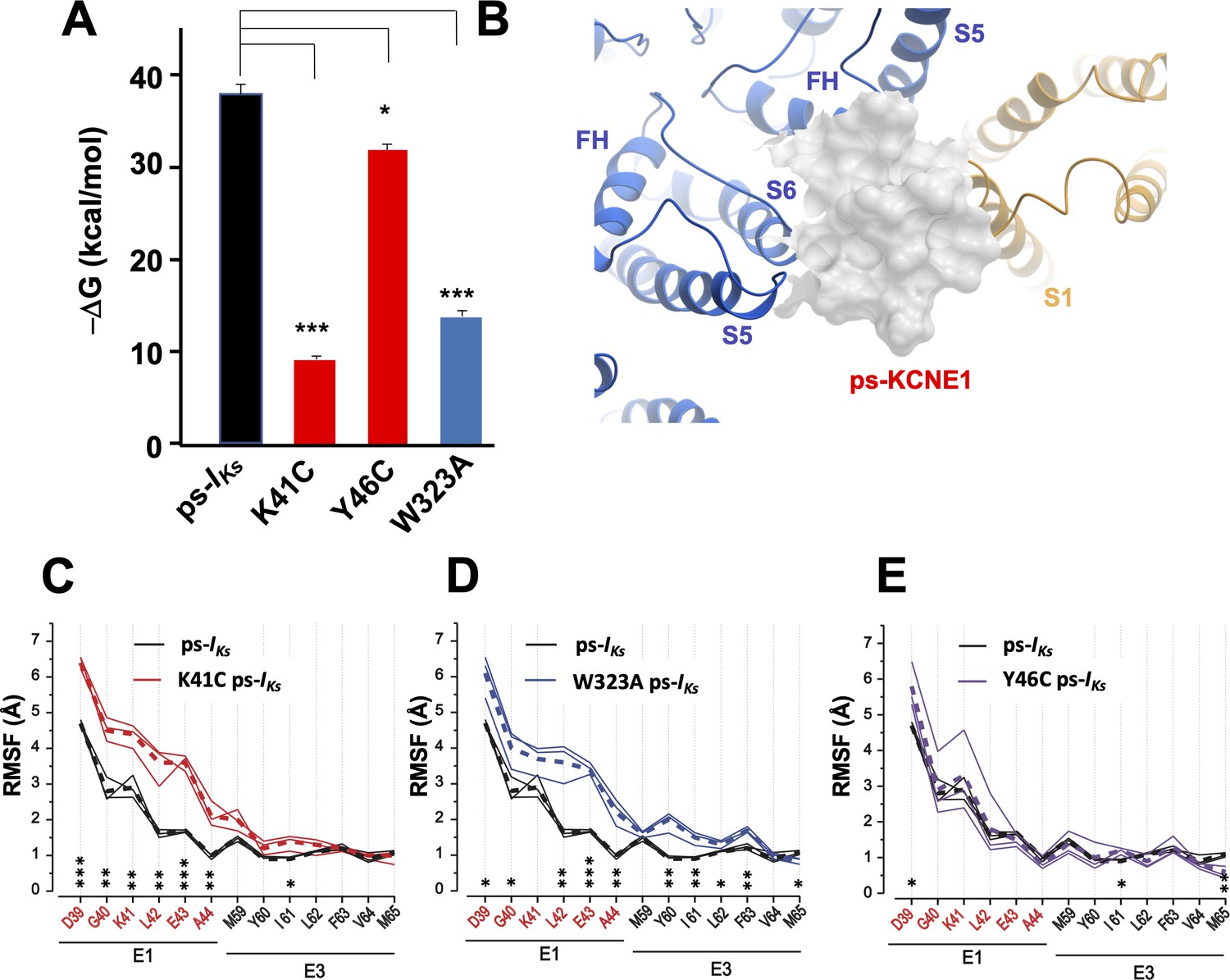
K41C, Y46C, and W323A mutant impact on mefenamic acid binding energy and flexibility of external KCNE1 residues.
(A) Average free interaction energy of Mef-bound ps-IKs complexes calculated using MM/GBSA methods from three independent MD simulation runs. For K41C and W323A mutations, calculations correspond to interval of simulations before the detachment of ligand from the molecular complex. * and *** denote significant differences in average free interaction energy compared to ps-IKs. Student`s unpaired t-test was used for comparison of groups. (B) Surface representation of ps-IKs after removal of Mef. The pore residues are in blue and the VSD of a neighbouring subunit is yellow. (C–E) Root mean square fluctuations (RMSF) of ps-KCNE residues (Å) in the ps-IKs complex during the last 100 ns of simulations. Three separate MD simulations shown for the ps-IKs channel without Mef (black lines) and three for K41C (C, red), W323A (D, blue) after ligand detachment, and Y46C in absence of ligand (E, purple). Dashed lines show average values of RMSF calculated from three simulations. ***p<0.001; **p<0.01 *p<0.05 using an unpaired t-test. GROMACS software was used for RMSF analysis.
Figure 4—figure supplement 1
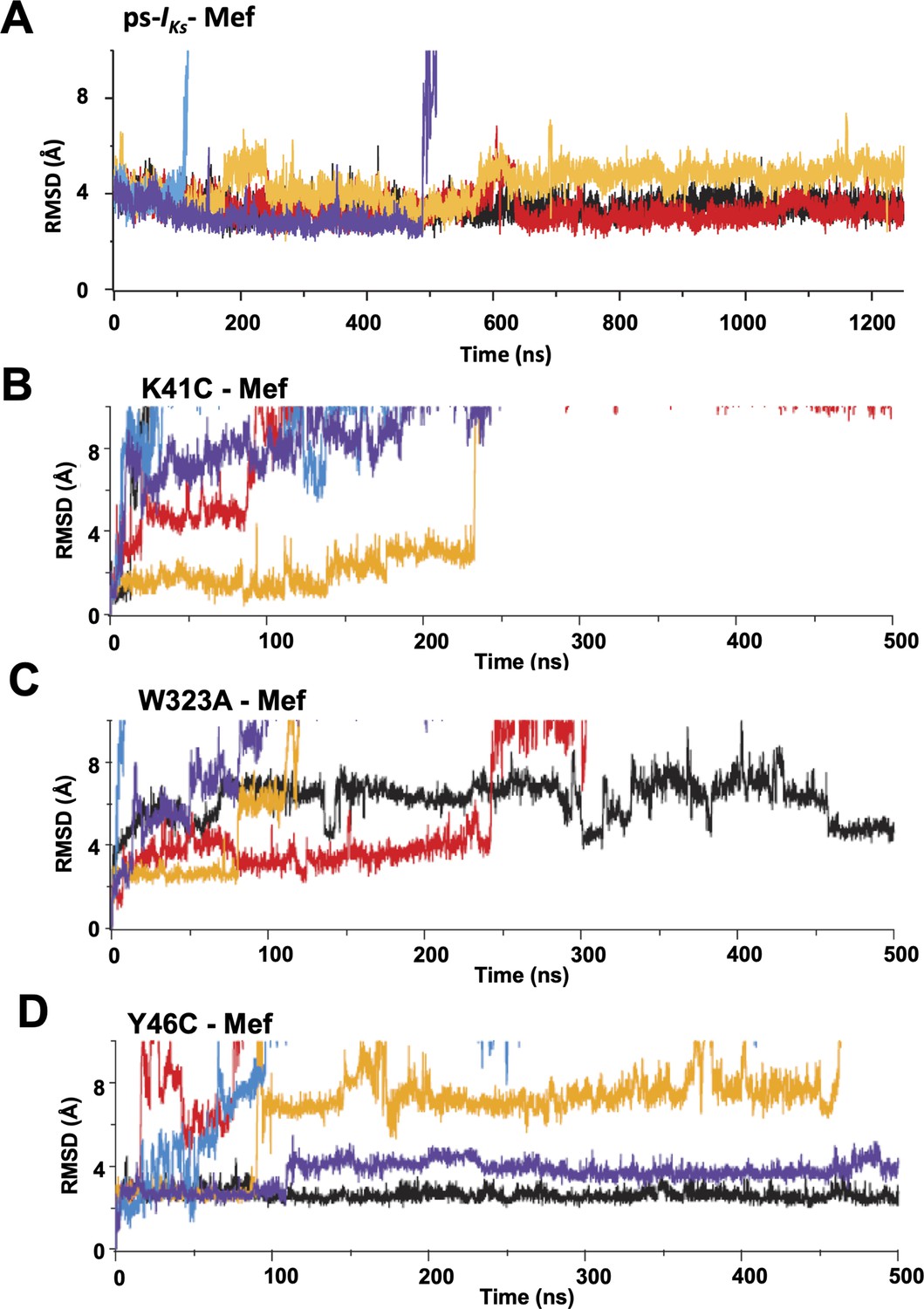
Root-mean-square deviations (RMSD) of mefenamic acid from its initial position during MD simulations.
(A) ps-IKs, (B) K41C, (C) W323A, and (D) Y46C-IKs mutants in the presence of mefenamic acid. Each color represents an individual simulation run, five in each case. Note that time scale in (A) is 2.5 x extended compared with (B–D).
Figure 4—figure supplement 2
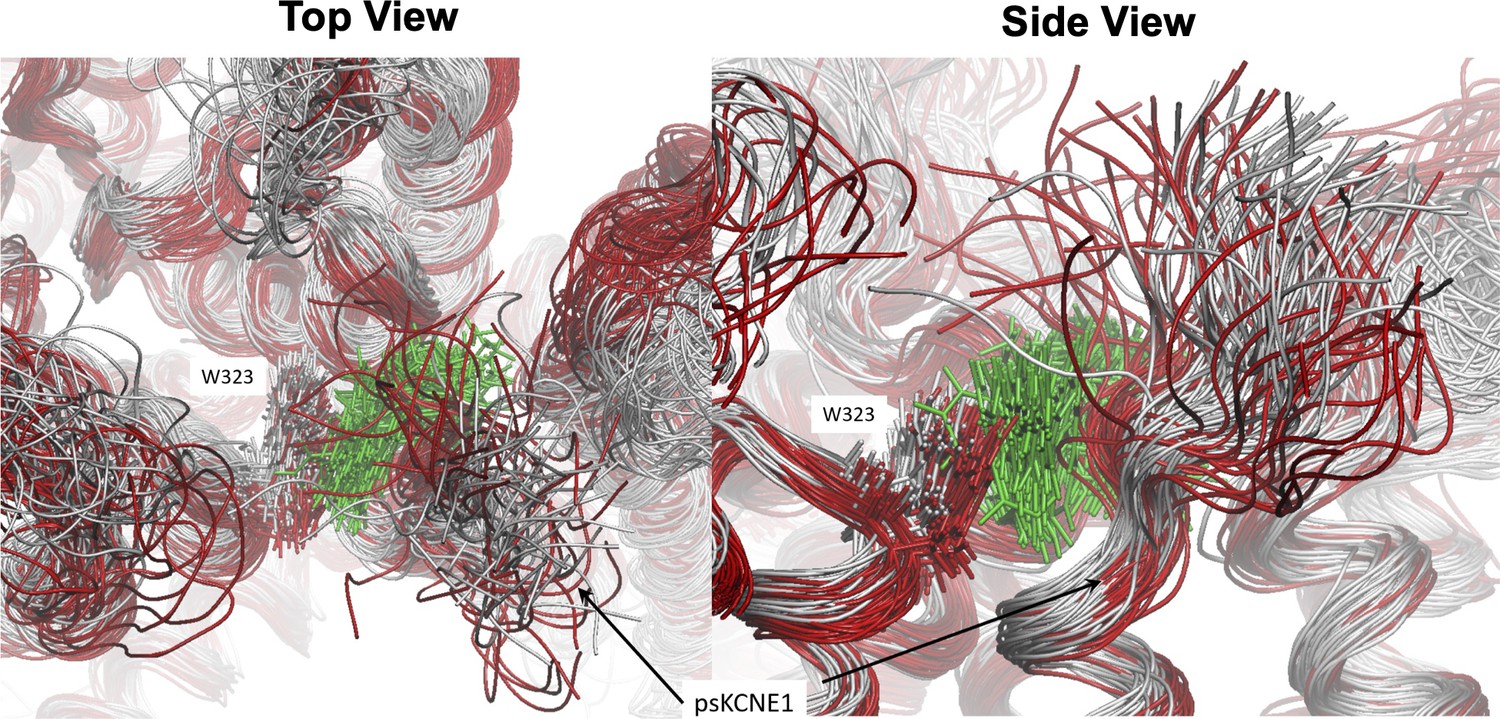
Result of loss of mefenamic acid from the binding site.
The binding pocket viewed from above (left) and the side (right) during a 1250 ns MD simulation before drug detachment (grey superimposed structure) and after (red superimposed structure). Mefenamic acid is represented as licorice and colored green. Snapshots for superimposition were collected every 10 ns. Visible from the snapshots is that when the drug leaves the binding site (after 500 ns, structures colored red), the N-terminal residues of ps-KCNE1, as well as W323 and other residues that form the pocket, shift toward the space that mefenamic acid previously occupied.
Figure 5
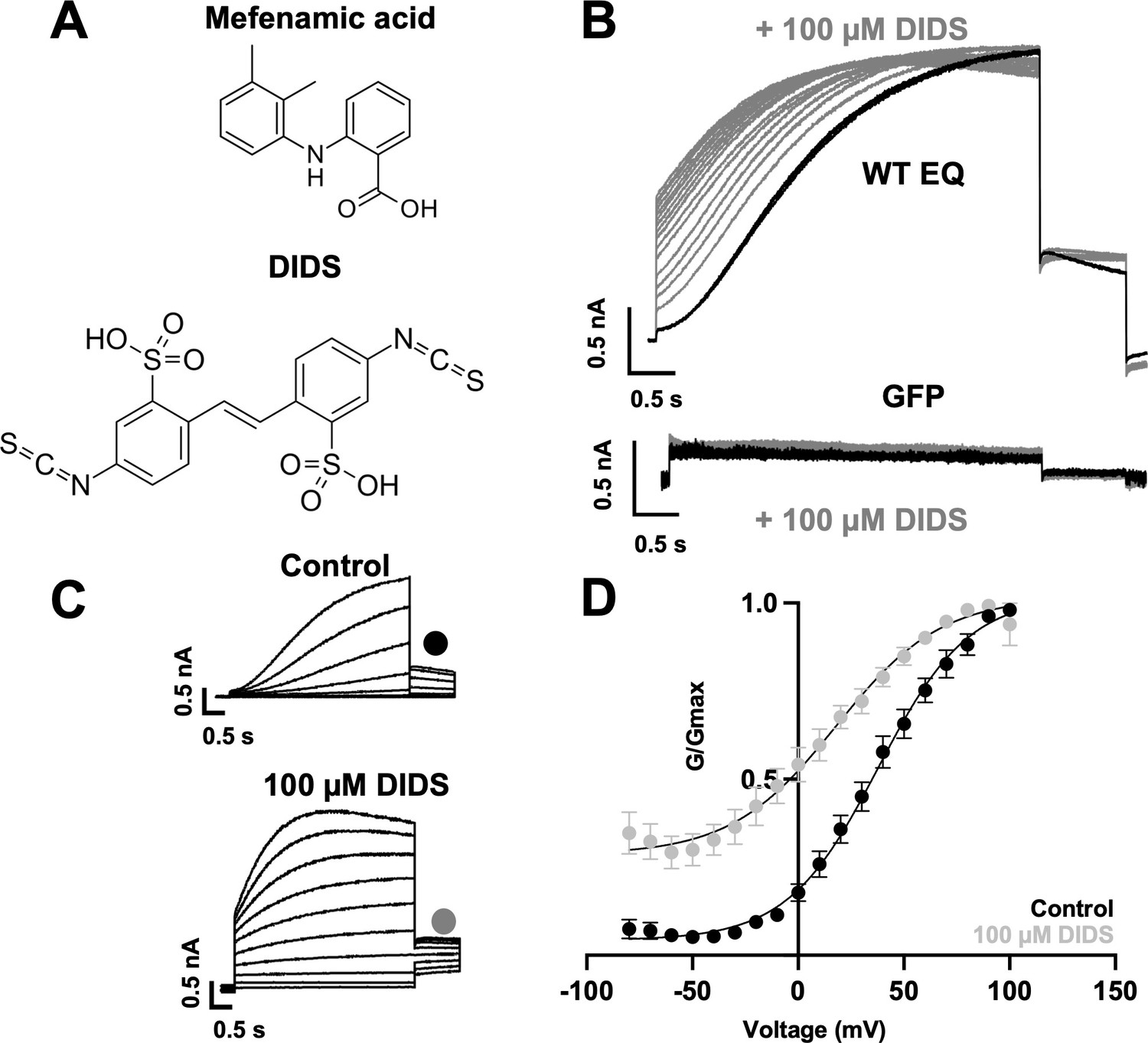
Effect of DIDS on IKs.
(A) Molecular structure of mefenamic acid and DIDS. (B) WT EQ current in control (black) and exposed to 100 µM DIDS over time (grey). Pulses were from –80 to +60 mV every 15 s, and current traces are shown superimposed. Lower panel shows no effect on currents from GFP-only transfected cells exposed to 100 µM DIDS over time (grey). (C) Current traces from WT EQ in control and presence of 100 µM DIDS as indicated. Pulses were from –80 to +100 mV for 4 s, with a 1 s repolarization to –40 mV. Interpulse interval was 15 s. (D) Corresponding G-V plot in control (black) and DIDS (grey) from data as shown in panel C. Boltzmann fits were: WT EQ control (n=8): V1/2 = 30.5 mV, k=20.3 mV; WT EQ in the presence of DIDS (n=5): V1/2 = -16.1 mV, k=25.3 mV. Error bars shown are ± SEM.
Figure 6 with 3 supplements
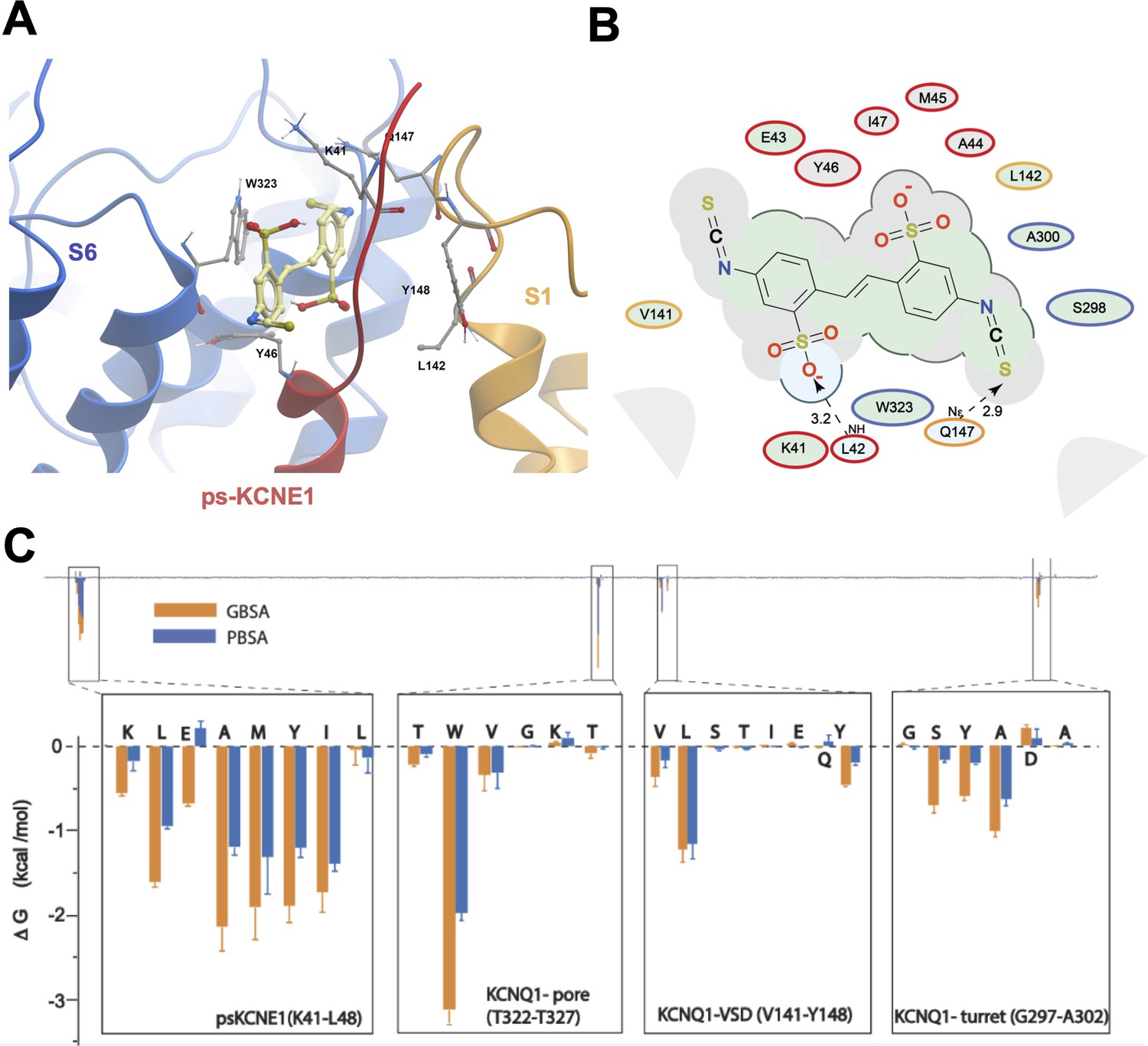
Ligand interaction and energy decomposition per amino acid for DIDS binding to ps-IKs.
(A) Binding pose of DIDS (yellow) in the external region of the ps-IKs channel complex obtained with molecular docking (side view). The residues of the pore domain are colored in blue, ps-KCNE1 subunit in red, and the voltage-sensor domain of a neighbouring subunit is presented in yellow. (B) Ligand interaction map of DIDS with ps-IKs from molecular docking. Size of residue ellipse is proportional to the strength of the contact. Light grey indicates residues in van der Waals contacts, light green hydrophobic contacts, and light blue are hydrogen bond acceptors. Red borders indicate KCNE1 residues, yellow are KCNQ1 VSD residues, and blue are pore residues. Dashed lines indicate H-bonds. The distance between the residue label and ligand represents proximity. Grey parabolas represent accessible surface for large areas. The 2D diagram was generated by ICM pro software with a cut-off value for hydrophobic contacts 4.5 Å and hydrogen bond strength 0.8. (C) Energy decomposition per amino acid for DIDS binding to ps-IKs. Generalized Born Surface Area (MM/GBSA; orange) and Poisson-Boltzmann Surface Area (MM/PBSA; blue) methods were used to estimate the interaction free energy contribution of each residue in the DIDS-bound ps-IKs complex. The lowest interaction free energy for residues in ps-KCNE1 and selected KCNQ1 domains are shown as enlarged panels (n=3 for each point). Error bars indicate ± SD.
Figure 6—figure supplement 1

H-bonds formed between DIDS and protein residues in its binding site during MD simulations.
DDS:O1-O6 are the oxygens of the sulfonic acids; DDS:N1 and N2 are nitrogens of the isothiocyanates. Red lines indicate at which time point and between which atom H-bonds were formed.
Figure 6—figure supplement 2
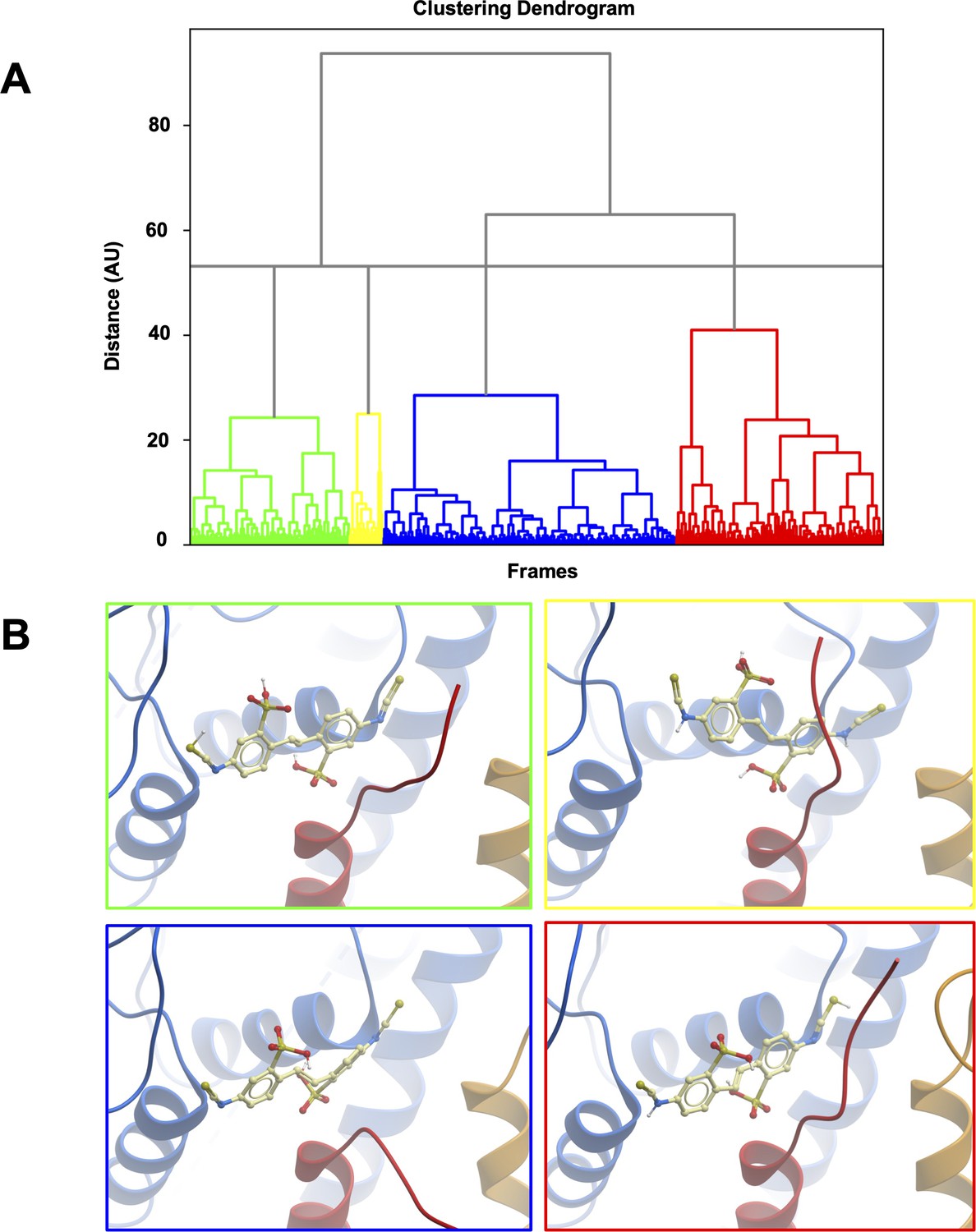
Clustering of MD trajectories based on DIDS conformations.
(A) The clustering dendrogram based on RMSD illustrates the similarity of DIDS conformations in MD trajectories. (B) Centroids from each cluster with the lowest RMSD compared to all other conformations in the cluster were selected and overlayed for comparison. The color of the frame around the panel shows to which cluster the depicted conformation belongs.
Figure 6—figure supplement 3
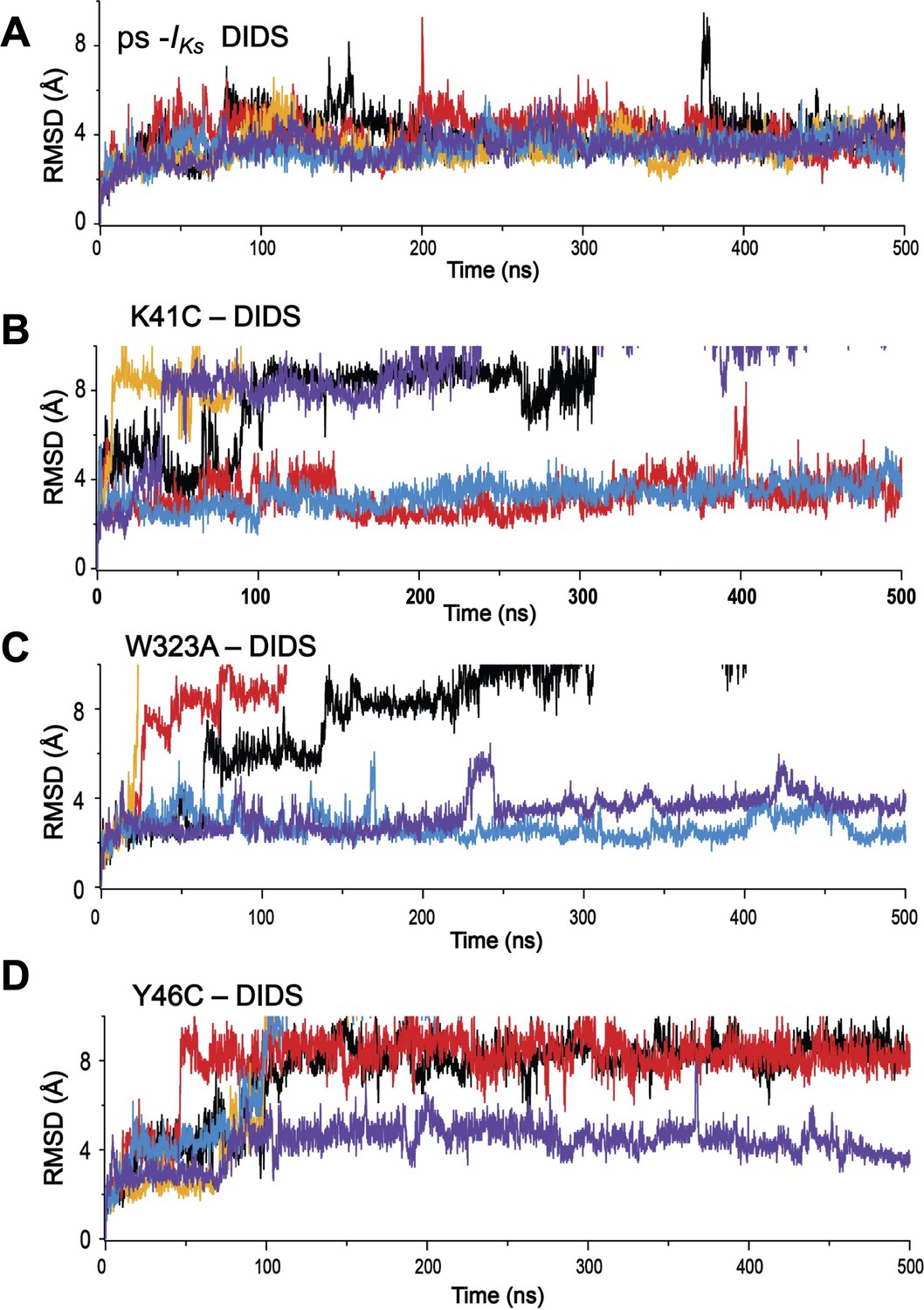
Root-mean-square deviations (RMSD) of DIDS from its initial position during MD simulations.
(A) ps-IKs, and mutations, (B) K41C, (C) W323A, and (D) Y46C complexes. Individual runs are shown in separate colors, n=5 for each.
Figure 7 with 1 supplement
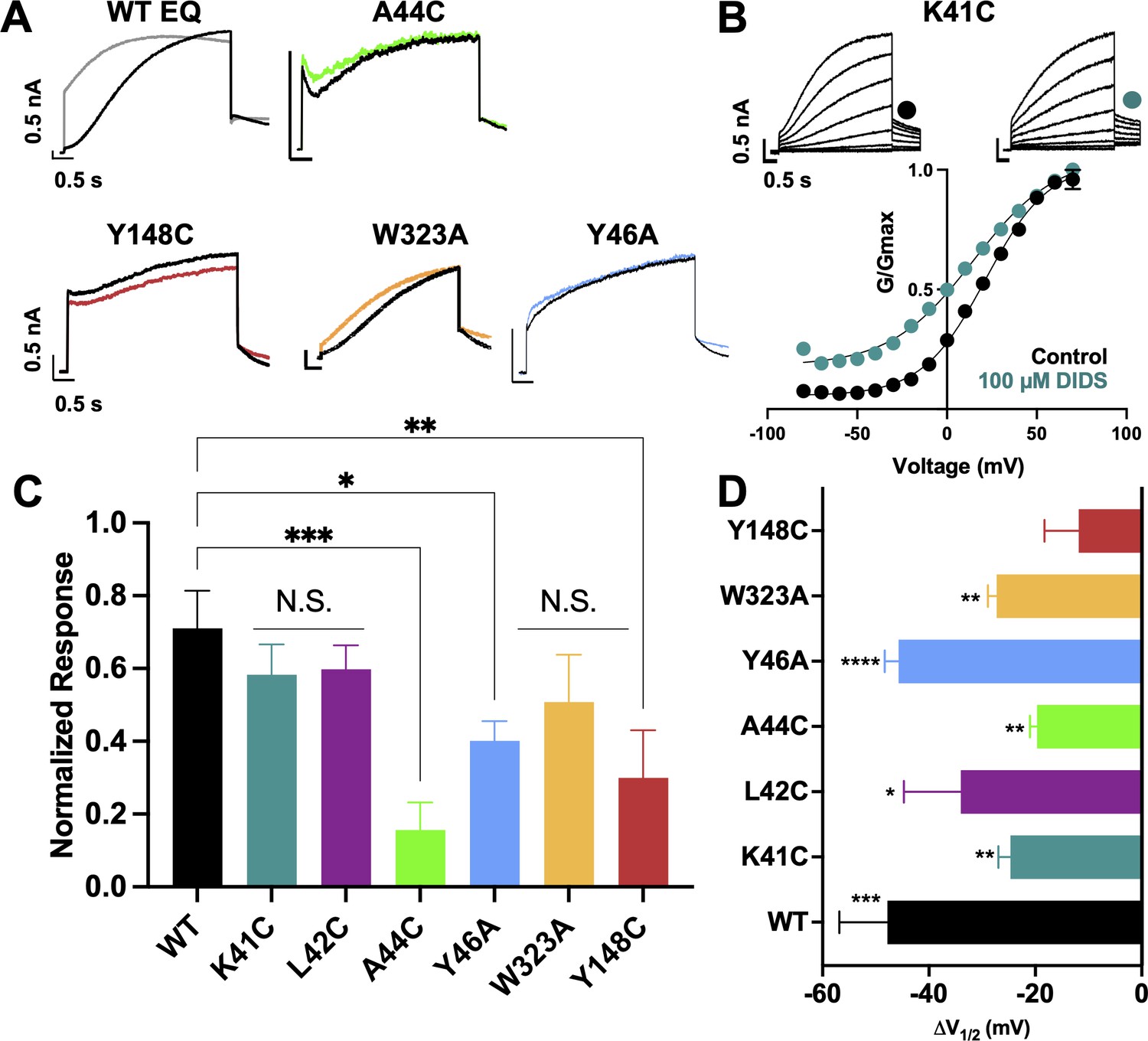
Binding site mutants important for the action of DIDS.
(A) Current traces from WT EQ and key mutants in the absence (control; black) and presence (colors) of 100 µM DIDS. (B) Data and G-V plots in control (black) and DIDS (teal). Boltzmann fits were: K41C-EQ control (n=3): V1/2 = 23.1 mV, k=20.2 mV; K41C-EQ DIDS (n=5): V1/2 = -1.6 mV, k=24.0 mV. Voltage steps from a holding potential of –80 mV to +70 mV for 4 s, followed by repolarization to –40 mV for 1 s. Interpulse interval was 15 s. Error bars shown are ± SEM. All calibration bars denote 0.5 nA/0.5 s. (C) Summary plot of the normalized response to 100 µM DIDS. Data are shown as mean + SEM and *p<0.05, **p<0.01, ***P<0.001 denote significant change in mutant versus WT currents (one-way ANOVA, see Materials and methods). For calculation, see Materials and methods. (D) Change in V1/2 (ΔV1/2) for WT EQ and each IKs mutant in control versus DIDS. Data are shown as mean - SEM and unpaired t-test was used, where *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 indicate a significant change in V1/2 comparing control to the presence of drug. n-values for mutants in C and D are stated in Table 5.
Figure 7—figure supplement 1
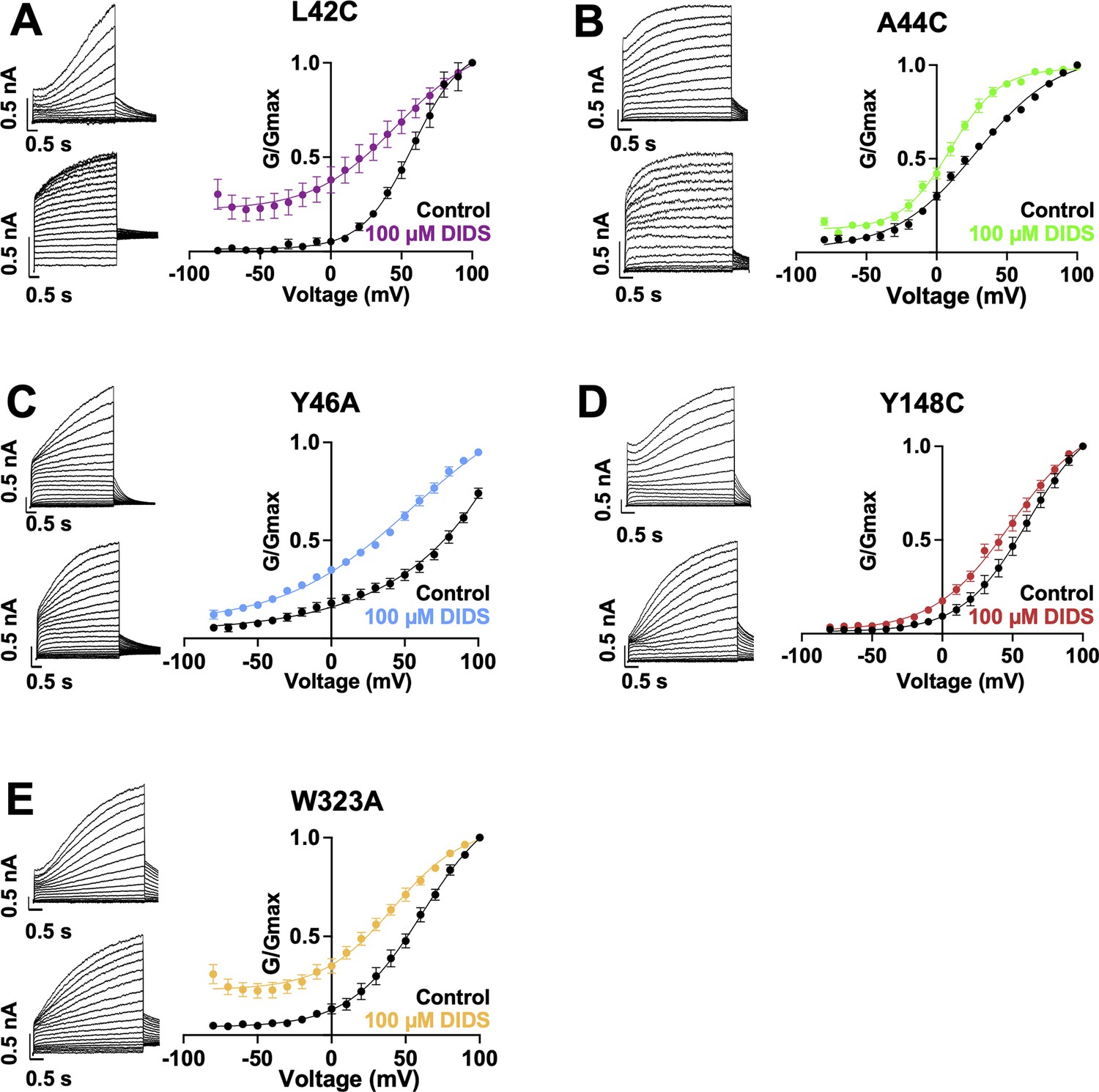
Current waveform and G-V changes induced by DIDS for all binding site mutants.
(A-E) current traces in control (top) and presence of 100 μM DIDS (bottom). For binding site mutants, cells were held at –80 mV, then pulsed to between –90 mV and +100 mV for 4 s followed by –40 mV for 2 s. G-V plots in control (black) and presence of 100 μM Mef (colors). Error bars are ± SEM. Refer to Table 5 for Boltzmann fit values.
Figure 8
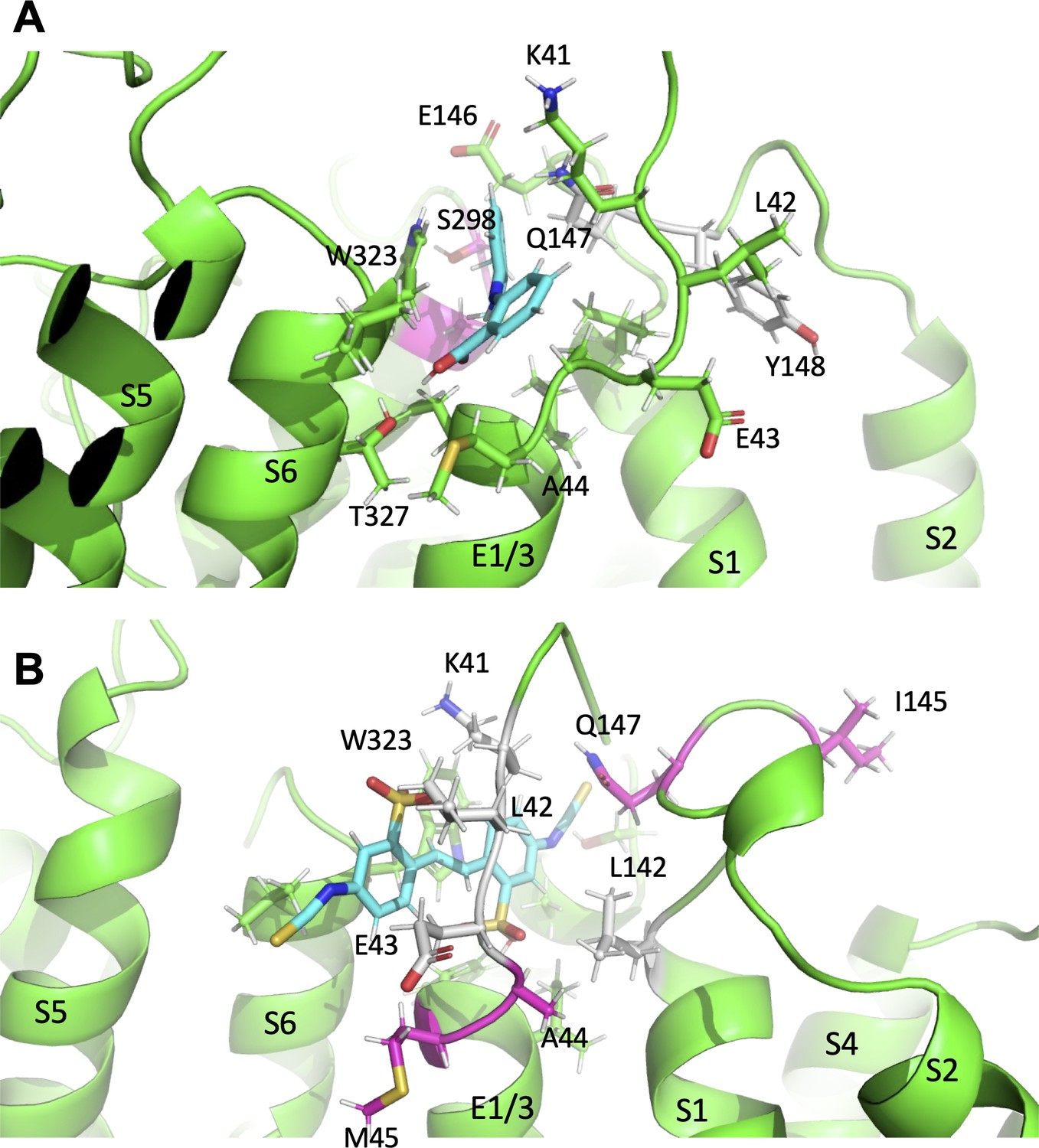
Subtle differences in activator binding to Y46C- and A44C-ps-IKs mutant channels.
(A) Mefenamic acid bound to the ps-IKs channel. Residues that are part of the binding site are shown in stick format colored green, except those residues that were important in the WT channel that had reduced ΔG in Y46C (in grey). Residues that had slight increases in ΔG values in Y46C are shown in magenta (S298, A300). Mefenamic acid is in cyan. See Figure 8—source data 1. (B) DIDS bound to the ps-IKs channel. Residues that are part of the binding site are shown in stick format colored green except those residues that were important in the WT channel that had reduced ΔG in the A44C mutant (in grey). Residues that had slight increases in ΔG values in A44C are shown in magenta. DIDS is in cyan. W323 may be seen behind DIDS. See Figure 8—source data 2. Images were made with the PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
-
Figure 8—source data 1
Source data for Figure 8A.
- https://cdn.elifesciences.org/articles/87038/elife-87038-fig8-data1-v1.zip
-
Figure 8—source data 2
Source data for Figure 8B.
- https://cdn.elifesciences.org/articles/87038/elife-87038-fig8-data2-v1.zip
Author response image 1

Videos
Video 1
MD simulations at the molecular level of binding of mefenamic acid to ps-IKs, and K41C-, W323A-, Y46C- ps-IKs mutants.
Note that videos may be shorter than the actual 500 ns simulations if drugs do not remain bound. Mefenamic acid binding to ps-IKs. W323, and K41 side chains are shown.
Video 2
Mefenamic acid binding to K41C ps-IKs.
K41C, W323, and Y46 side chains are shown.
Video 3
Mefenamic acid binding to W323A ps-IKs.
K41, W323A, and Y46 side chains are shown.
Video 4
Mefenamic acid binding to Y46C ps-IKs.
K41, W323, and Y46C side chains are shown.
Video 5
MD simulations at the molecular level of binding of DIDS to ps-IKs, and K41C-, W323A-, Y46C- ps-IKs mutants.
DIDS binding to ps-IKs. K41, W323, and Y46 side chains are shown.
Video 6
DIDS binding to K41C ps-IKs.
K41C, W323, and Y46 side chains are shown.
Video 7
DIDS binding to W323A ps-IKs.
K41, W323A, and Y46 side chains are shown.
Video 8
DIDS binding to Y46C ps-IKs.
K41, W323, and Y46C side chains are shown.
Video 9
Binding pocket fluctuations before and after exit of mefenamic acid.
W323A and the backbone of ps-KCNE1 (residues 41–44) gradually appear ~100 ns. Frames before mefenamic acid detachment are white, and after detachment red.
Tables
Table 1
Mean V1/2 of activation (mV) and slope values (k-factor, mV) in the absence and presence of mefenamic acid for fully saturated IKs channel complexes.
A statistical difference in V1/2 compared to control is shown as p-value, determined using an unpaired t-test. NS denotes not significant. Values are shown ± SEM.
| Control | 100 µM or 1 mM mefenamic Acid* | p-value | |||||
|---|---|---|---|---|---|---|---|
| V1/2 | k-factor | n | V1/2 | k-factor | n | ||
| WT EQ | 25.4±2.4 | 19.4±1.2 | 6 | -80.3±4.1 | 41.3±8.4 | 3 | <0.0001 |
| K41C-EQ | 15.2±1.1 | 18.4±1.7 | 4 | 11.4±1.0 | 19.4±0.8 | 4 | <0.05 |
| 16.7±2.0 | 19.8±1.4 | 3 | NS | ||||
| L42C-EQ | 68.9±1.5 | 21.5±3.7 | 3 | 31.8±0.4 | 14.8±4.27 | 3 | <0.01 |
| E43C-EQ | 18.3±10.0 | 25.7±1.5 | 6 | 14.4±5.6 | 22.8±1.3 | 6 | NS |
| A44C-EQ | 4.1±1.8 | 17.6±1.4 | 4 | -5.6±2.7 | 18.3±2.0 | 4 | <0.05 |
| Y46A-EQ† | 76.4±1.5 | 57.3±1.6 | 3 | -29.8±4.1 | 18.9±3.1 | 3 | <0.05 |
| EQ-W323A | 47.8±2.7 | 23.7±2.2 | 4 | 33.7±1.2 | 28.5±3.1 | 3 | <0.05 |
| EQ-W323C | 54.0±2.1 | 22.2±2.1 | 4 | 27.5±3.3 | 25.6±1.8 | 4 | <0.05 |
| EQ-V324A | 34.4±2.3 | 20.7±1.1 | 6 | 15.5±2.3 | 27.9±1.6 | 5 | <0.05 |
| EQ-V324W | 41.0±2.4 | 18.4±0.3 | 4 | 27.3±6.0 | 25.5±1.3 | 4 | NS |
| EQ-Q147C | 63.9±5.8 | 25.4±2.0 | 4 | 26.3±5.7 | 32.7±1.7 | 4 | <0.05 |
| EQ-Y148C | 36.8±0.3 | 20.3±0.4 | 4 | 17.5±4.0 | 36.7±3.7 | 4 | <0.05 |
-
*
For K41C-EQ, the concentration of mefenamic acid used was either 100 µM (upper row values) or 1 mM (lower row values). For all other constructs, 100 µM mefenamic acid was used.
-
†
An estimation of the activation V1/2 was calculated from a right shifted, non-saturating GV curve.
Table 2
Mean frequency of H-bonds between MEF or DIDS and residues in ps- IKs during 500 ns MD simulations or until drug unbound.
MEF357-N1 indicates the nitrogen of aminobenzoic acid; MEF357:O1 and O2 are the oxygens of the aminobenzoic acid; DDS357:O1-O6 are the oxygens of the sulfonic acids; DDS357:N1 and N2 are nitrogens of the isothiocyanates. n=5 for each.
| Donor | Hydrogen | Acceptor | % Frames with H-Bonds | SEM | |
|---|---|---|---|---|---|
| MEF | MEF-N1 | MEF-H1 | GLU43-O | 22.5 | 10.2 |
| TYR46-N | TYR46-HN | MEF-O2 | 79.7 | 4.2 | |
| ILE47-N | ILE47-HN | MEF-O2 | 7.9 | 2.7 | |
| DIDS | DIDS-O2 | DDS357-H10 | TRP323-NE1 | 12.9 | 2.1 |
| DIDS-O1 | DDS357-H9 | GLU43-OE2 | 25.1 | 4.0 | |
| DIDS-O1 | DDS357-H9 | GLU43-OE1 | 25.2 | 4.6 | |
| ILE47N | ILE47-HN | DIDS-O5 | 22.0 | 3.2 | |
| TYR46-N | TYR46-HN | DIDS-O6 | 56.8 | 10.4 | |
| TYR46-N | TYR46-HN | DIDS-O5 | 7.1 | 1.8 | |
| TYR299N | TYR299-HN | DIDS-N2 | 11.9 | 0.6 | |
| SER298-OG | SER298-HG1 | DIDS-N2 | 26.0 | 10.6 |
Table 3
Mean V1/2 of activation (mV) and slope values (k-factor, mV) for WT EQ treated with 100 µM mefenamic acid, and untreated mutant EQ-L142C.
Interpulse interval used is as indicated. Values are shown ± SEM. An interpulse interval of 7 s created such a dramatic change in the shape of the EQ-L142C G-V plot (see Figure 3—figure supplement 2) that a Boltzmann curve could not be fit and V1/2 and k-values are therefore not available.
| V1/2 | k-factor | n | |
|---|---|---|---|
| WT EQ +100 µM mefenamic acid: Interpulse interval 15 s | -80.3±4.1 | 41.3±8.4 | 3 |
| WT EQ +100 µM mefenamic acid: Interpulse interval 30 s | 26.7±10.5 | 66.6±28 | 8 |
| EQ-L142C control: Interpulse interval 15 s | -80.3±4.5 | 30.0±2.9 | 6 |
| EQ-L142C control: Interpulse interval 30 s | -28.7±19 | 62.5±9.6 | 3 |
Table 4
Average free interaction energies of MEF-bound ps-IKs complexes calculated according to MM/GBSA methods from three independent MD simulation runs using the AMBER force field.
Mean values are in kcal/mol, ± SD. For W323A and K41C mutations, calculations correspond to interval of simulations before the detachment of ligand from the molecular complex. Note that unbinding occurred in K41C and W323A in all 3 simulations with 300 ns duration.
| Run | Method | ps-IKs | W323A | Y46C | K41C |
|---|---|---|---|---|---|
| I | GBSA | -39.3±4.1 | -13.0±5.1 | -31.2±2.9 | -10.5±3.8 |
| - | Unbinding after ~75 ns | Unbinding after ~25 ns | |||
| II | GBSA | -39.0±3.3 | -23.1±3.1 | -32.2±3.8 | -17.0±2.6 |
| - | Unbinding after ~120 ns | - | Unbinding after ~70 ns | ||
| III | GBSA | -35.5±2.6 | -22.6±4.1 | -28.8±5.7 | -17.2±2.5 |
| - | Unbinding after ~85 ns | - | Unbinding after ~20 ns |
Table 5
Mean V1/2 of activation (mV) and slope values (k-factor, mV) in the absence and presence of 100 µM DIDS for fully saturated IKs channel complexes.
A statistical difference in V1/2 compared to control is shown as p-value determined using an unpaired t-test. NS denotes not significant. Values are shown ± SEM.
| Control | 100 µM DIDS | p-value | |||||
|---|---|---|---|---|---|---|---|
| V1/2 | k-factor | n | V1/2 | k-factor | n | ||
| WT EQ | 30.5±4.3 | 20.3±0.9 | 8 | -16.1±2.8 | 25.3±1.9 | 5 | <0.001 |
| K41C-EQ | 23.1±3.0 | 20.2±1.8 | 3 | -1.6±3.7 | 24.0±3.6 | 3 | <0.01 |
| L42C-EQ | 55.4±3.3 | 19.9±1.5 | 3 | 21.4±10.7 | 28.5±2.6 | 4 | <0.05 |
| A44C-EQ | 24.1±1.7 | 29.6±3.3 | 4 | 5.6±2.4 | 17.6±1.8 | 3 | <0.05 |
| *Y46A-EQ | 75.2±2.1 | 59.1±5.3 | 5 | 25.8±0.9 | 39.2±1.4 | 5 | <0.0001 |
| EQ-Y148C | 51.8±4.6 | 23.5±1.7 | 4 | 40.0±3.1 | 26.3±1.5 | 5 | NS |
| EQ-W323A | 50.7±3.6 | 23.9±1.6 | 4 | 23.4±4.9 | 24.1±1.3 | 4 | <0.05 |
-
*
An estimation of the activation V1/2 was calculated from a right shifted, non-saturating GV curve.
Table 6
Average free interaction energies of DIDS-bound ps-IKs complexes calculated according to MM/PBSA and MM/GBSA methods from three independent MD simulation runs of 300 ns duration using the AMBER force field.
Mean values are in kcal/mol, ± SD. For W323A and Y46C mutations, calculations correspond to interval of simulations before the detachment of ligand from the molecular complex. Note that unbinding occurred in W323A in 2 of 3 simulations, and K41C did not unbind but the binding pose shifted.
| Run | Method | ps-IKs | W323A | Y46C | K41C | A44C | Y148C |
|---|---|---|---|---|---|---|---|
| I | PBSA | -32.3±6.7 | -25.7±4.9 | -35.5±6.3 | -33.5±4.4 | -29.2±4.2 | -34.1±4.6 |
| GBSA | -35.5±4.6 | -30.2±4.5 | -42.7±5.4 | -41.5±5.8 | -36.8±2.9 | -40.7±4.0 | |
| unbinding after 240 ns | changed binding pose after ~140 –150 ns | ||||||
| II | PBSA | -32.6±4.8 | -25.3±5.5 | -25.7±6.5 | -36.4±4.2 | -29.0±4.0 | -29.3±3.7 |
| GBSA | -36.0±4.8 | -30.0±4.8 | -32.4±6.4 | -31.6±4.0 | -34.0±5.7 | -31.8±4.7 | |
| unbinding after ~100 –132 ns | |||||||
| III | PBSA | -29.2±8.1 | -27.1±3.9 | -38.3±4.8 | -35.1±4.3 | -31.2±45.0 | -35.7±5.8 |
| GBSA | -34.0±8.9 | -35.1±4.8 | -42.5±4.7 | -30.8±3.9 | -35.6±5.6 | -40.3±5.9 | |
| unbinding after ~240 ns |
Table 7
Average free energy of mefenamic acid and DIDS-bound ps-IKs and mutant complexes.
Values are calculated according to the MM/GBSA method from three independent MD simulation runs using the AMBER force field, and further broken down by residue and channel region. For K41C and W323A, calculations correspond to the interval of simulations before detachment of the ligand from the complex. Values are in kcal/mol. Mutated residues are in bold italics.
| Residue | ps-IKs MEF | K41C MEF | Y46C MEF | W323A MEF | ps-IKs DIDS | A44C DIDS | Y46C DIDS | Y148C DIDS | W323A DIDS | |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | -38.02 | -16.39 | -31.40 | -20.31 | -34.29 | -35.40 | -39.16 | -37.60 | -34.33 | |
| Drug | -19.69 | -8.79 | -15.25 | -11.65 | -17.42 | -17.52 | -21.21 | -18.18 | -19.17 | |
| Channel | -17.91 | -6.52 | -16.61 | -7.55 | -19.21 | -18.40 | -16.94 | -19.21 | -15.10 | |
| Ch +Drug | -37.60 | -15.31 | -31.86 | -19.20 | -36.63 | -35.92 | -38.15 | -37.39 | -34.27 | |
| Difference | -0.42 | -1.08 | 0.46 | -1.11 | 2.34 | 0.52 | -1.01 | -0.21 | -0.06 | |
| E1/E3 | K41 | -1.548 | -0.349 | -1.411 | -0.954 | -0.639 | -0.439 | -1.148 | -0.283 | -0.083 |
| L42 | -0.970 | -0.895 | -0.943 | -1.193 | -1.061 | -0.798 | -0.165 | -0.320 | -0.587 | |
| E43 | -2.681 | -0.472 | -2.257 | -0.309 | -1.312 | -0.660 | -0.014 | -2.264 | -0.705 | |
| A44 | -1.906 | -0.886 | -2.035 | -1.102 | -1.778 | -2.062 | -1.027 | -1.061 | -1.780 | |
| M45 | -1.744 | -0.163 | -1.913 | -0.064 | -1.756 | -2.154 | -1.222 | -2.810 | -1.633 | |
| Y46 | -2.391 | -0.121 | -2.702 | -0.083 | -1.985 | -1.797 | -1.245 | -2.377 | -1.603 | |
| I47 | -0.771 | -0.136 | -0.412 | -0.203 | -2.036 | -1.993 | -1.004 | -0.621 | -1.528 | |
| L48 | -0.010 | -0.003 | -0.025 | 0.005 | -0.045 | -0.100 | -0.233 | -0.081 | -0.035 | |
| Sum | -12.02 | -3.03 | -11.70 | -3.90 | -10.61 | -10.00 | -6.06 | -9.82 | -7.79 | |
| Pore | T322 | -0.125 | -0.047 | -0.091 | -0.968 | -0.200 | -0.213 | -0.127 | -0.152 | -0.410 |
| W323 | -2.159 | -0.736 | -2.226 | -0.932 | -3.312 | -3.395 | -2.279 | -3.566 | -1.590 | |
| V324 | -0.644 | -0.037 | -0.123 | -0.047 | -0.172 | -0.210 | -0.111 | -2.016 | -0.063 | |
| G325 | -0.038 | -0.006 | -0.006 | 0.017 | -0.004 | 0.000 | 0.003 | -0.048 | 0.032 | |
| K326 | -0.185 | -0.015 | -0.024 | 0.008 | 0.038 | 0.058 | 0.091 | 0.000 | 0.118 | |
| T327 | -0.130 | -0.015 | 0.020 | 0.027 | -0.040 | -0.022 | -0.018 | -1.298 | -0.014 | |
| Sum | -3.28 | -0.86 | -2.49 | -1.89 | -3.69 | -3.78 | -2.44 | -7.08 | -1.93 | |
| S1 | V141 | -0.061 | -0.014 | -0.247 | -0.006 | -0.569 | -0.411 | -0.660 | -0.076 | -0.504 |
| L142 | -0.861 | -0.434 | -1.052 | -0.267 | -1.525 | -0.822 | -1.285 | -0.460 | -1.036 | |
| S143 | -0.028 | -0.004 | -0.014 | -0.063 | -0.008 | -0.006 | -0.104 | -0.026 | -0.017 | |
| T144 | -0.091 | -0.006 | -0.242 | -0.526 | -0.015 | -0.122 | -0.982 | -0.002 | -0.001 | |
| I145 | 0.004 | 0.005 | 0.002 | 0.009 | 0.003 | -0.329 | -0.067 | -0.009 | -0.001 | |
| E146 | 0.000 | 0.048 | 0.056 | 0.114 | 0.028 | -0.014 | -1.601 | 0.113 | 0.013 | |
| Q147 | -0.230 | -0.365 | 0.006 | -0.244 | 0.012 | -0.143 | -1.061 | -0.974 | -0.054 | |
| Y148 | -0.565 | -1.556 | 0.006 | -0.128 | -0.428 | -0.336 | 0.002 | -0.022 | -0.223 | |
| Sum | -1.83 | -2.32 | -1.49 | -1.11 | -2.50 | -2.18 | -5.76 | -1.40 | -1.82 | |
| Turret | G297 | -0.024 | -0.001 | -0.010 | -0.027 | -0.016 | -0.027 | -0.089 | -0.001 | -0.076 |
| S298 | -0.320 | -0.163 | -0.419 | -0.427 | -0.791 | -0.848 | -1.107 | -0.216 | -1.026 | |
| Y299 | -0.103 | -0.046 | -0.104 | -0.052 | 0.718 | -0.598 | -0.563 | -0.167 | -0.673 | |
| A300 | -0.618 | -0.183 | -0.636 | -0.431 | -1.130 | -1.112 | -0.751 | -0.652 | -1.093 | |
| D301 | 0.311 | 0.075 | 0.248 | 0.299 | 0.231 | 0.172 | -0.181 | -0.136 | -0.683 | |
| A302 | -0.024 | -0.003 | -0.011 | -0.007 | -0.011 | -0.020 | 0.009 | -0.008 | -0.015 | |
| Sum | -0.78 | -0.32 | -0.93 | -0.65 | -2.40 | -2.43 | -2.68 | -0.91 | -3.57 |
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Homo sapiens) | KCNQ1 | GenBank | HGNC:HGNC:6294 | Gene ID: 3784 |
| Gene (Homo sapiens) | KCNE1 | GenBank | HGNC:HGNC:6240 | Gene ID: 3753 |
| Strain, strain background (include species and sex here) | n/a | n/a | n/a | n/a |
| Genetic reagent (include species here) | n/a | n/a | n/a | n/a |
| Cell line (Homo-sapiens) | tsa201 | Sigma-Aldrich | Cat # CB_96121229 | Transformed human embryonic kidney 293 cells. The cells have been eradicated from mycoplasma at ECACC. The identity of tsA201 and 293 has been confirmed by STR profiling. |
| Transfected construct (synthetic) | KCNQ1 in pcDNA3 | This paper | KCNQ1 DNA in pcDNA3 vector. | |
| Transfected construct (synthetic) | KCNE1 in pcDNA3 | This paper | KCNE1 DNA in pcDNA3 vector. | |
| Biological sample (include species here) | n/a | n/a | n/a | n/a |
| Antibody | n/a | n/a | n/a | n/a |
| Recombinant DNA reagent | pcDNA3 | Invitrogen | Cat # V79020 | pcDNA3.1 (+) Mammalian Expression Vector |
| Sequence-based reagent | K41C_F | This paper | PCR primers | CCGCAGCGGTGACGGCTGCCTGGAGGC |
| Sequence-based reagent | K41C_R | This paper | PCR primers | GTAGAGGGCCTCCAGGCAGCCGTCACCG |
| Sequence-based reagent | L42C_F | This paper | PCR primers | CAGCGGTGACGGCAAGTGCGAGGCCCT |
| Sequence-based reagent | L42C_R | This paper | PCR primers | GACGTAGAGGGCCTCGCACTTGCCGTCA |
| Sequence-based reagent | E43C_F | This paper | PCR primers | GCGGTGACGGCAAGCTGTGCGCCCTCTA |
| Sequence-based reagent | E43C_R | This paper | PCR primers | GGACGTAGAGGGCGCACAGCTTGCCGTC |
| Sequence-based reagent | A44C_F | This paper | PCR primers | CGGCAAGCTGGAGTGCCTCTACGTCCTC |
| Sequence-based reagent | A44C_R | This paper | PCR primers | GAGGACGTAGAGGCACTCCAGCTTGCCG |
| Sequence-based reagent | Y46A_F | This paper | PCR primers | GGAGGCCCTCTGCGTCCTCATGGTAC |
| Sequence-based reagent | Y46A_R | This paper | PCR primers | GTACCATGAGGACGGCGAGGGCCTCC |
| Sequence-based reagent | W323A_F | This paper | PCR primers | GGTCTTCCCGACCGCCGTCTGGGGCAC |
| Sequence-based reagent | W323A_R | This paper | PCR primers | GTGCCCCAGACGGCGGTCGGGAAGACC |
| Sequence-based reagent | W323C_F | This paper | PCR primers | GGTCTTCCCGACACACGTCTGGGGCAC |
| Sequence-based reagent | W323C_R | This paper | PCR primers | GTGCCCCAGACGTGTGTCGGGAAGACC |
| Sequence-based reagent | V324A_F | This paper | PCR primers | CCCCAGACGTGGGCCGGGAAGACCATC |
| Sequence-based reagent | V324A_R | This paper | PCR primers | GATGGTCTTCCCGGCCCACGTCTGGGG |
| Sequence-based reagent | V324W_F | This paper | PCR primers | CCCCAGACGTGGTGGGGGAAGACCATC |
| Sequence-based reagent | V324W_R | This paper | PCR primers | GATGGTCTTCCCCCACCACGTCTGGGG |
| Sequence-based reagent | Q147C_F | This paper | PCR primers | CAGGGCGGCATAGCACTCGATGGTGGAC |
| Sequence-based reagent | Q147C_R | This paper | PCR primers | GTCCACCATCGAGTGCTATGCCGCCCTG |
| Sequence-based reagent | Y148C_F | This paper | PCR primers | GCCAGGGCGGCACACTGCTCGATGGTG |
| Sequence-based reagent | Y148C_R | This paper | PCR primers | CACCATCGAGCAGTGTGCCGCCCTGGC |
| Peptide, recombinant protein | eGFP in pcDNA3 | Gift | Enhanced Green Fluorescent Protein | |
| Commercial assay or kit | Midiprep | ThermoFisher Scientific | Cat# K210004 | DNA extraction kit |
| Chemical compound, drug | DIDS | Sigma-Aldrich | CAS # 53005-05-3 | 4,4'-diisothiocyano-2,2'-stilbenedisulfonic acid (stock 50 mM) |
| Chemical compound, drug | Mef | Sigma-Aldrich | CAS # 61-68-7 | mefenamic Acid (stock 50 mM) |
| Software, algorithm | pCLAMP 11 | Molecular Devices | pCLAMP 11 software | |
| Software, algorithm | GraphPad Prism 9 | GraphPad Software | GraphPad Prism 9 software | |
| Software, algorithm | ICM-pro | MolSoft LLC | ICM-pro 3.8 software | |
| Software, algorithm | GROMACS | Royal Institute of Technology and Uppsala University, Sweden | GROMACS 2021.4 | |
| Software, algorithm | TTClust | Thibault Tubiana, PhD | TTClust, a molecular simulation clustering program | |
| Other | Axopatch 200B amplifier | Molecular Devices | Axopatch 200B amplifier | |
| Other | Digidata 1440 A digitizer | Molecular Devices | Digidata 1440 A digitizer | |
| Other | Lipofectamine 2000 | ThermoFisher Scientific | Cat # 11668019 | Lipofectamine 2000 transfection reagent |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A generic binding pocket for small molecule IKs activators at the extracellular inter-subunit interface of KCNQ1 and KCNE1 channel complexes
eLife 12:RP87038.
https://doi.org/10.7554/eLife.87038.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}