Statistical analysis supports pervasive RNA subcellular localization and alternative 3' UTR regulation
- Department of Biochemistry Stanford University, United States
- Department of Biomedical Data Science Stanford University, United States
- Departments of Medicine (Cardiology) and Genetics Yale University, United States
Figures
Figure 1 with 1 supplement
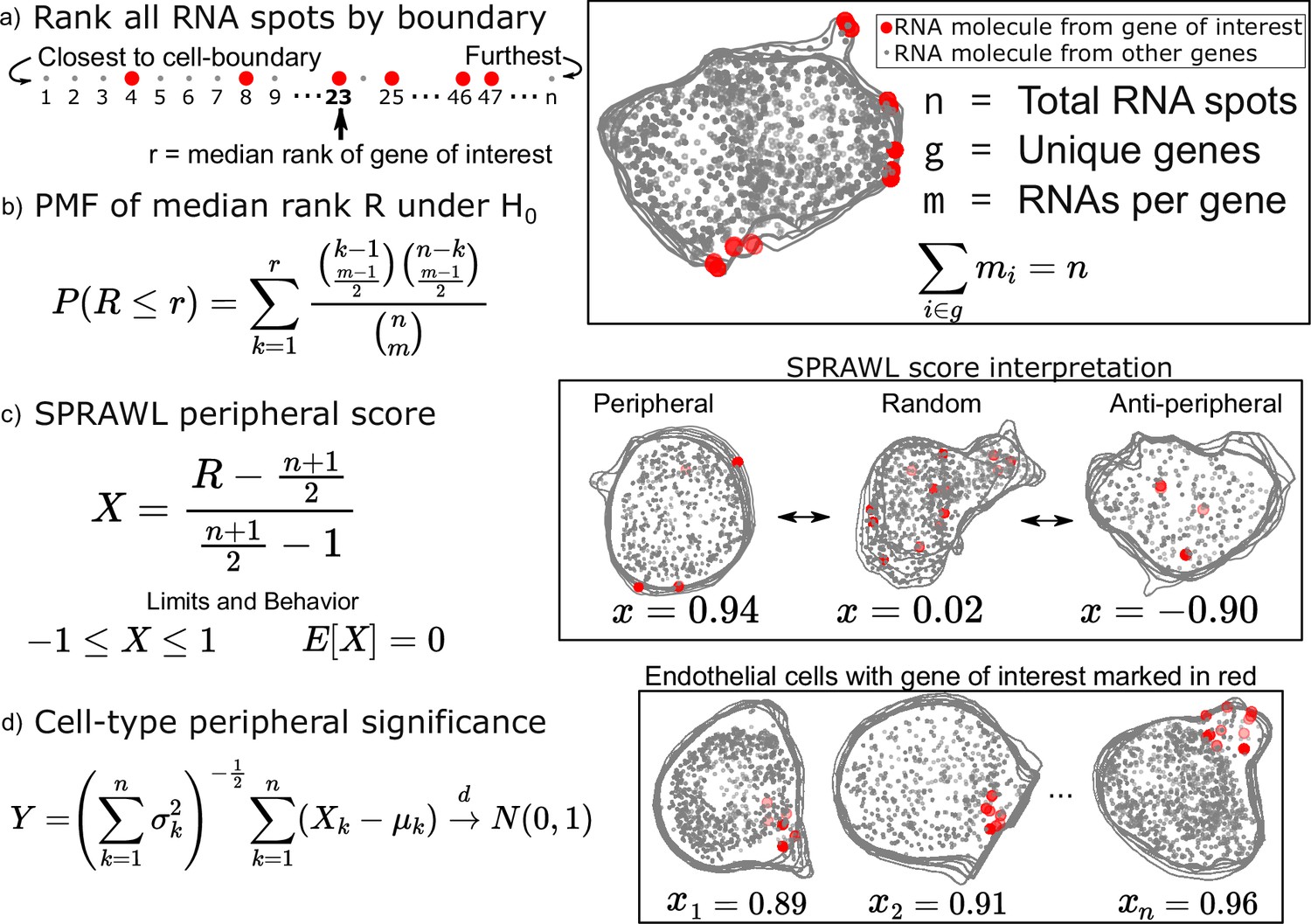
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) peripheral and central score workflow.
(a) RNAs are ranked from closest to furthest from the cell-boundary to calculate the median peripheral rank of the gene of interest. For the central metric, distances from the cell centroid are used for ranking instead. (b) Under the null hypothesis of each rank being equally likely, the probability mass function of the median is exactly calculable. (c) The intuitive SPRAWL score per gene per cell, X, will be near +1 for highly-peripheral patterns, near 0 for randomly-peripheral patterns, and near –1 for anti-peripheral patterns. (d) Peripheral significance of a gene within a cell-type is estimated from per cell SPRAWL scores using the Lyapunov Central Limit Theorem (CLT). Overlaying cell outlines are a result of viewing 3D slices from the top down.
Figure 1—figure supplement 1
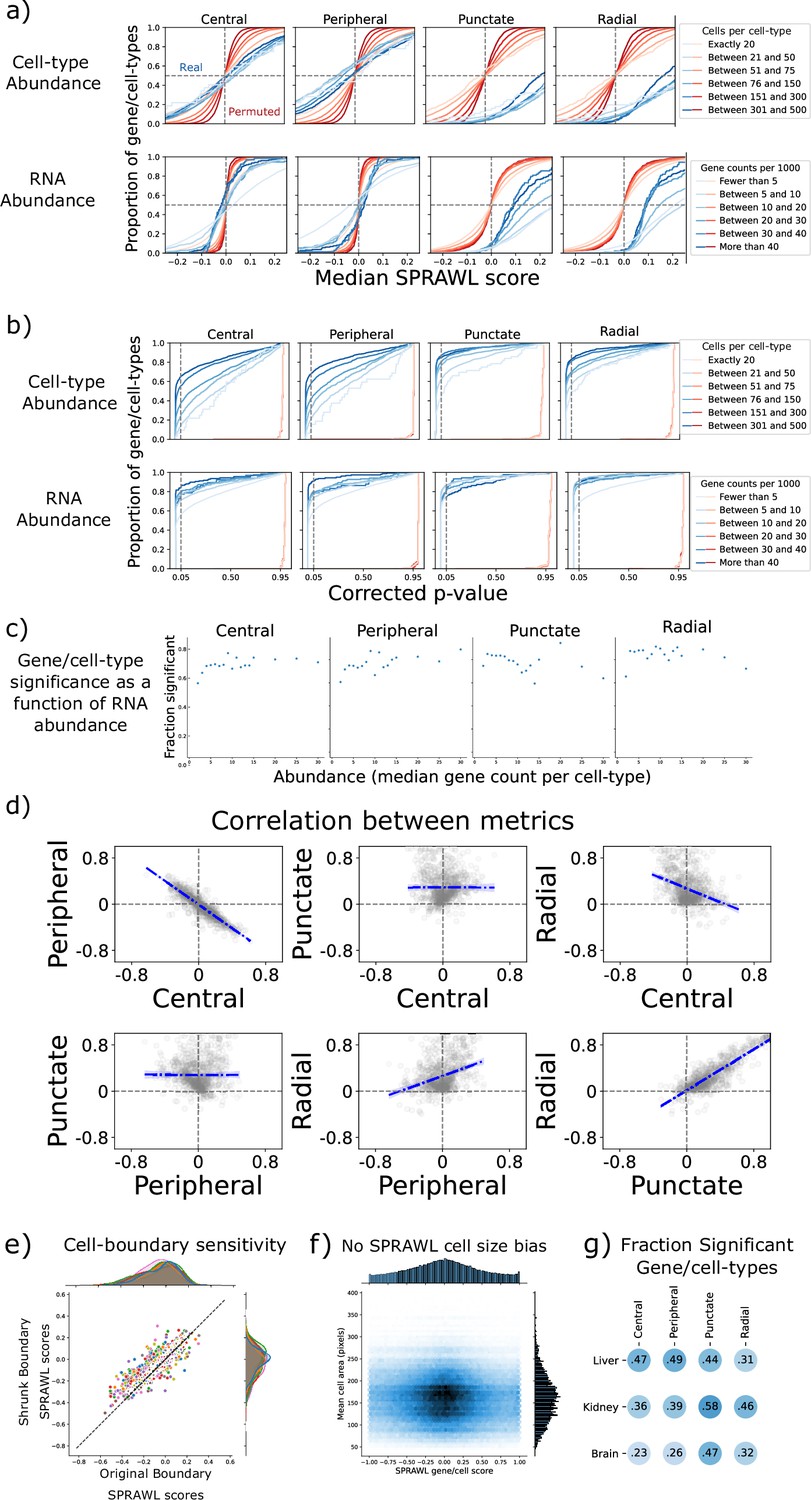
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) metrics have high specificity and lack bias.
(a) SPRAWL scores for permuted null datasets, reds, have expected mean values of zero regardless of either the number of cells per cell-type or the gene abundance. The permuted datasets have an expectedly lower variance for higher cells per cell-type and gene abundance. The real data, blue, shows expected means near 0 for the central and peripheral metrics, but higher scores for the punctate and radial metrics. (b) Under null simulations, red lines, all gene/cell-type pairs are deemed insignificant at an alpha level of 0.05 (vertical dashed line) for the four metrics. In the real data, blue lines, more gene/cell-type pairs are significant, after Benjamini-Hochberg correction, with higher cell-type and RNA abundance. (c) The fraction of significant gene/cell-type pairs in the BICCN samples are consistent across abundance levels measured as gene/cell-type median spot counts. (d) Peripheral and central scores are strongly anti-correlated for gene/cell-type scores while the radial and punctate scores are positively correlated. (e) To test whether peripheral localization patterns were driven artifactually by incorrect cell boundary calling, the cell boundary locations were computationally shrunk by a factor of 0.8 in the x and y direction, discarding spots that fell outside the new boundaries. In both the BICCN MOp and Vizgen Brainmap datasets, a Pearson correlation coefficient of greater than 0.85 was observed between the shrunk and original median gene/cell-type periphery scores. (f) SPRAWL scores are not conflated with cell size (g) Similar fractions of gene/cell-types are significant between the different datasets and metrics.
Figure 2
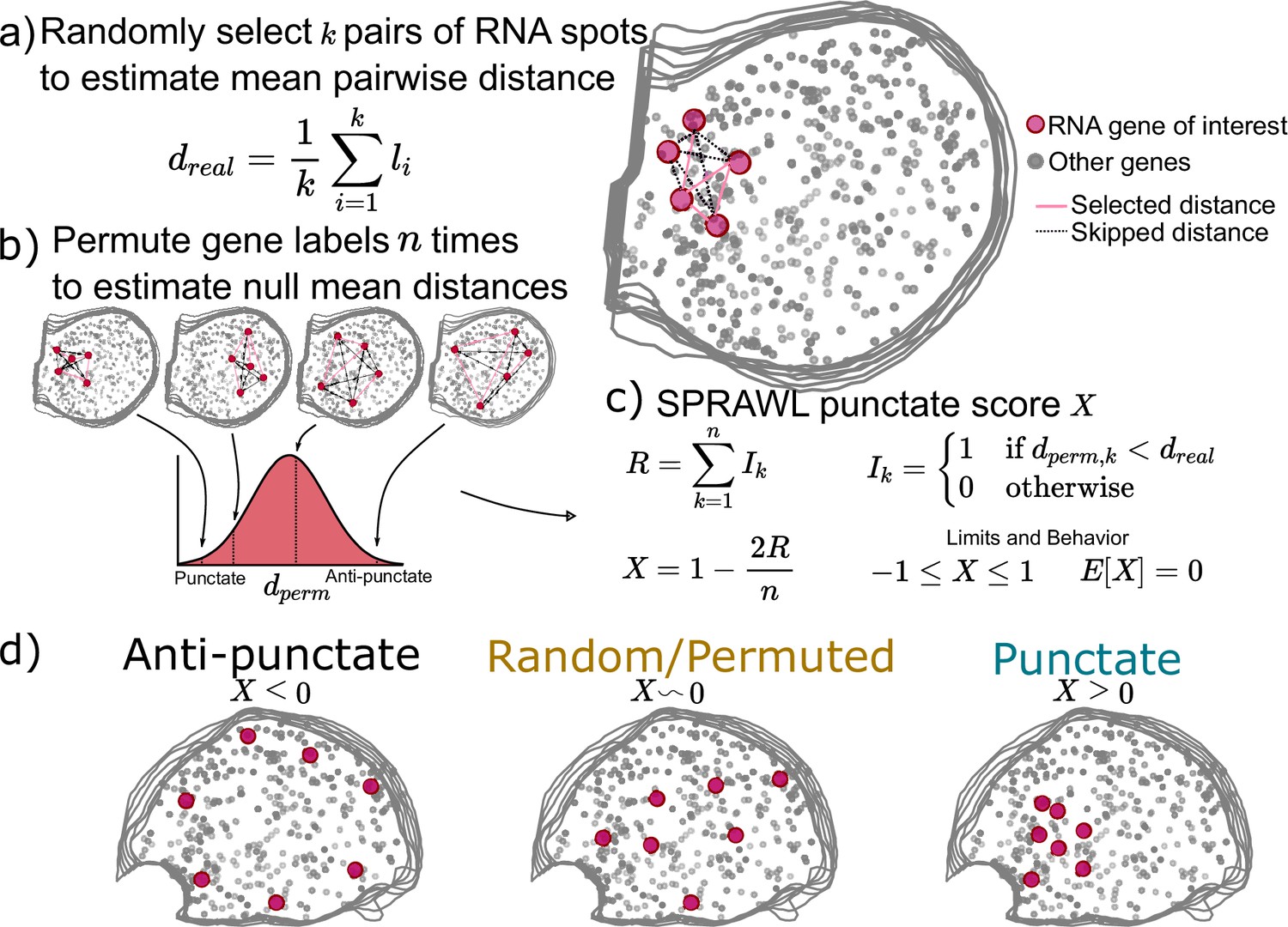
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) punctate and radial scores workflow.
(a) The SPRAWL punctate metric relies on (b) permutation testing to create a score (c) that represents whether RNA molecules from the gene of interest are closer together than expected by chance. The radial metric is identically calculated, except using average angle instead of distance. The significance of gene-cell-type punctate patterns is calculated using the Lyapunov Central Limit Theorem (CLT) as in the peripheral metric. (d) Depictions and interpretation of the SPRAWL punctate metric.
Figure 3 with 1 supplement
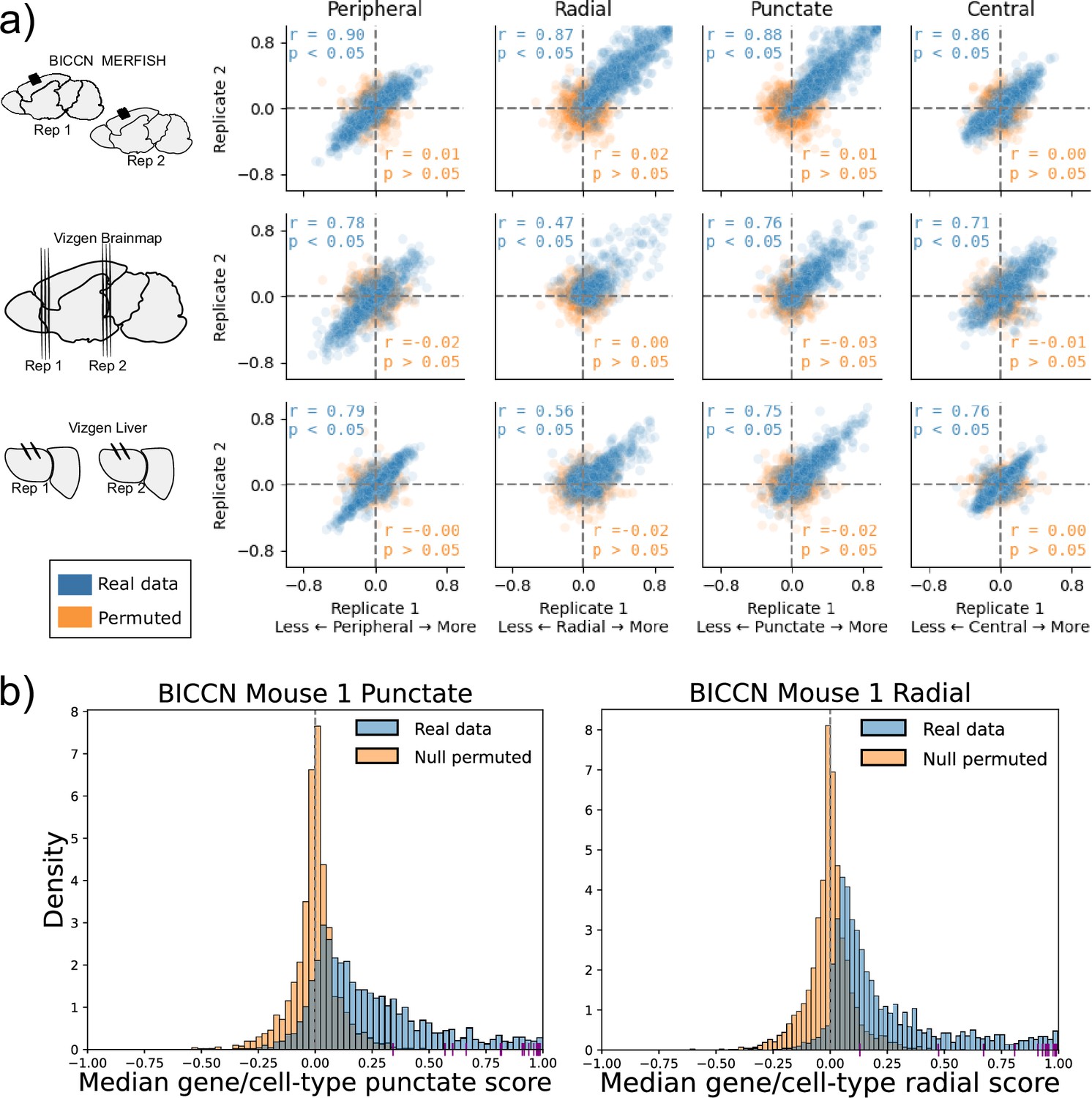
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) gene/cell-type scores are highly correlated between biological replicates.
(a) BICCN MERFISH, Vizgen Brainmap, and Vizgen Liver biological replicates (rows top to bottom) have Pearson correlation coefficients (blue) larger than 0.47 for SPRAWL peripheral, radial, punctate, and central metrics (columns left to right). Randomly permuting gene labels in these datasets eliminates underlying spatial patterning and yields insignificant Pearson correlation coefficients (orange) between biological replicates. Dotted lines indicate zero-valued SPRAWL gene-cell type scores. (b) In the motor cortex (MOp) BRAIN Initiative Cell Census Network (BICCN) dataset 87% of gene/cell-type pairs have positive punctate RNA patterning (blue), compared to 50% in the gene-label permuted data (orange). Similarly extreme trends of 95% and 52% are observed for the radial metric. Cldn5 RNA is consistently highly punctate and radial in all cell-types that express it, depicted by purple x-axis ticks.
Figure 3—figure supplement 1
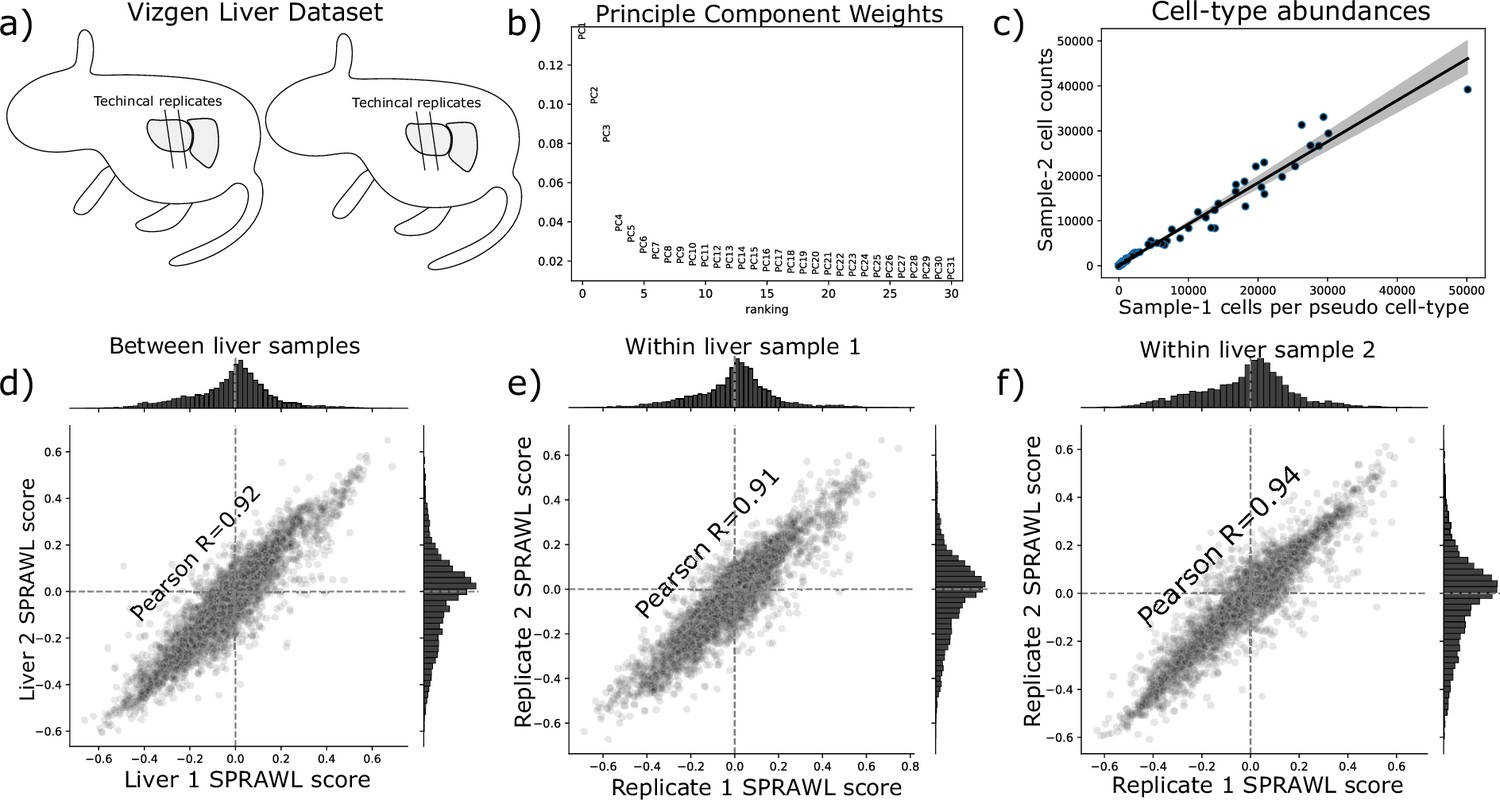
Vizgen Liver Showcase scores are highly correlated between replicates.
(a) The Vizgen Liver showcase dataset provides spatial information for 2 mouse livers with two slices each. Cell annotation data was not provided in the Vizgen Liver Showcase, instead, clusters produced from off-the-shelf Leiden clustering (python Scanpy package) were used as pseudo cell-types. All four datasets were combined without reference to biological or technical replicate by first normalizing the read counts per cell, identifying highly variable genes, reducing to the first 10 principle components (b), and then computing a neighbor graph with n=40 which resulted in 100 clusters which had a similar number of cells from each animal (c) As well as having a high Pearson correlation coefficient between mice (d), the technical replicates were highly correlated within both Liver 1 (e) and Liver 2 (f).
Figure 4 with 2 supplements
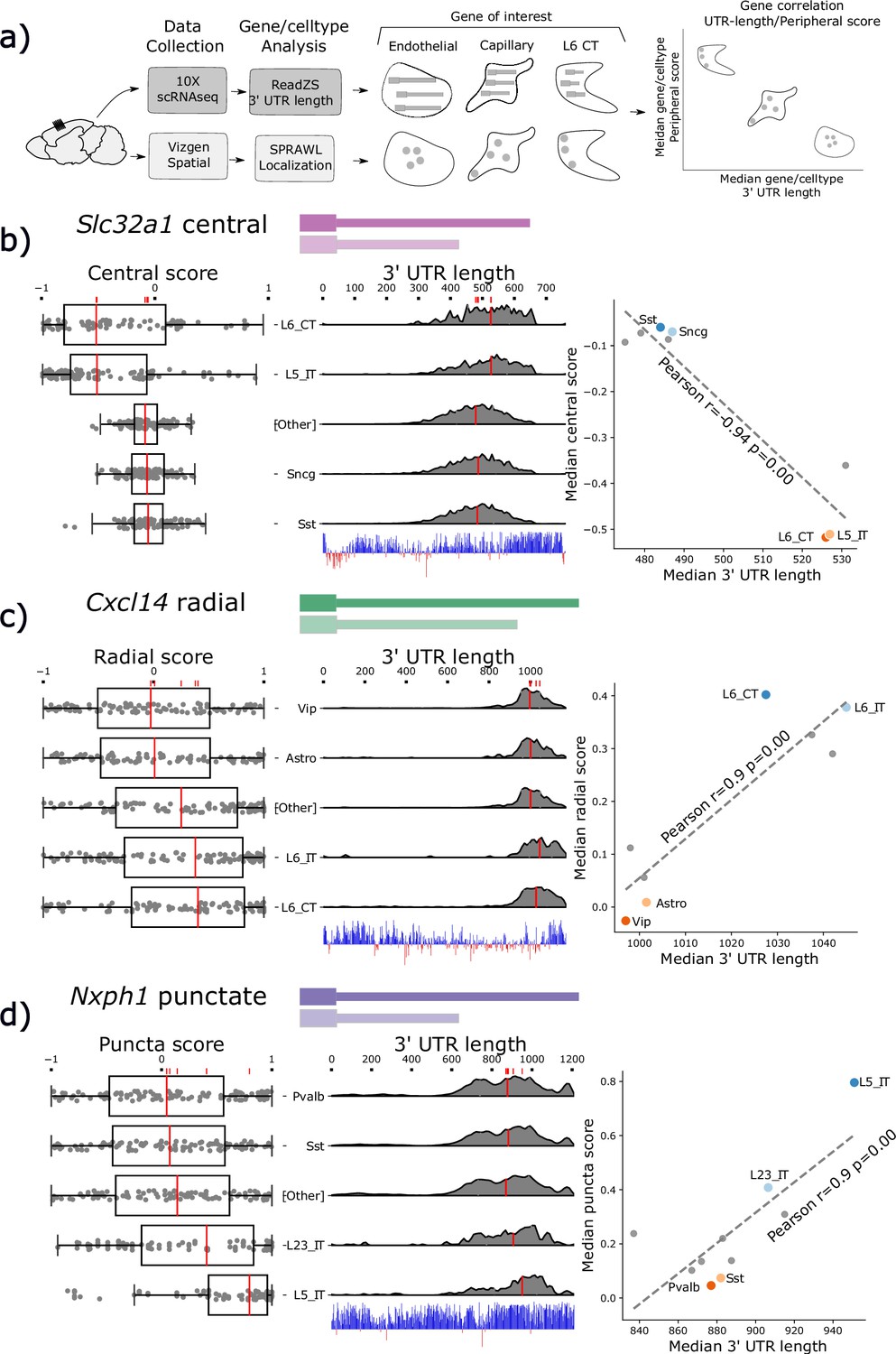
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) spatial scores and 3’ Untranslated Region (UTR) length are significantly correlated for a subset of genes.
(a) Workflow to calculate median 3’ UTR length and spatial score per gene/cell-type. (b) Slc32a1 median centrality, (c) Cxcl14 radial, and (d) Nxph1 punctate SPRAWL scores from the BRAIN Initiative Cell Census Network (BICCN) MERFISH dataset correlate significantly with 3’ UTR length determined from 10 X scRNA-seq data by ReadZS. The left-column boxplots show individual SPRAWL cell scores as overlaid dots. The cell-types are sorted by increasing median score marked in red. The two cell-types with the highest and lowest median SPRAWL scores are plotted individually while the remaining cell-types are collapsed into the ‘Other’ category. Gene/cell examples are shown to the left of the boxplots for each extreme cell-type group. The density plots in the middle column show estimated 3’ UTR lengths for each read mapping within the annotated 3’ UTR, stratified by cell-type. Lengths were approximated as the distance between the annotated start of the 3’ UTR and the median read-mapping position. Each density plot is normalized by cell-type to show relative shifts in 3’ UTR length with median lengths depicted with red lines. The scatterplots show the significant correlations between the median SPRAWL score and the median 3’ UTR length. The two cell-types with the highest, and the two with the lowest SPRAWL median scores are highlighted.
Figure 4—figure supplement 1
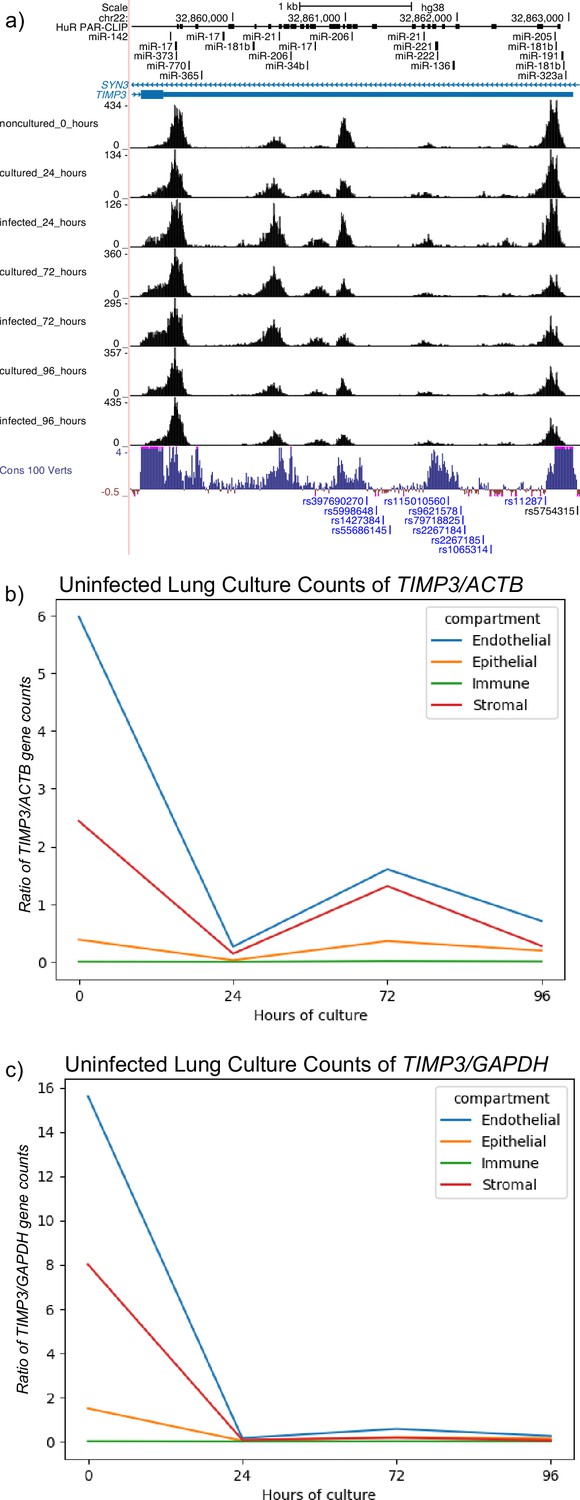
ReadZS detects Tabula Sapiens Lung differential 3’ Untranslated Region (UTR) length TIMP3 and decreases in Timp3 expression throughout culture.
(a) ReadZS detects statistically significant 3’ UTR length differences in the human TIMP3 3’ UTR in endothelial cell-types from the Tabula Sapiens consortium datasets across conditions. HuR binding sites from PAR-CLIP are shown above the Timp3 gene structure diagram. The last track shows high vertebrate sequence conservation throughout the UTR. Normalized expression of TIMP3 against (b) ACTIN and (c) GAPDH shows decreasing expression of TIMP3 throughout increasing culture direction in all tissue compartments.
Figure 4—figure supplement 2
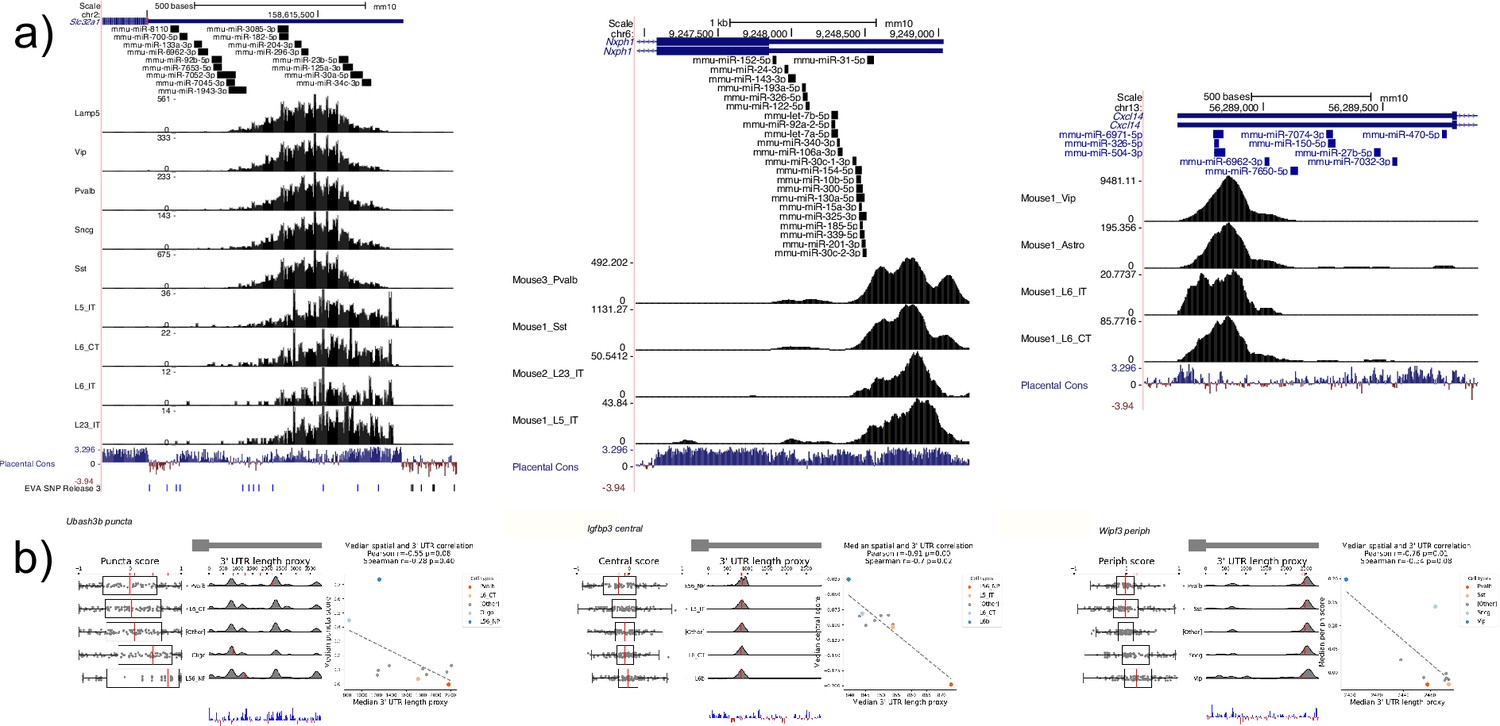
Computationally predicted miRNA binding sites in the 3’ Untranslated Regions (UTRs) of Slc32a1, Cxcl4, and Nxph1 and additional 3’ UTR correlated genes.
(a) Subset of computationally predicted 3’ UTRs from the miRWalk database for Slc32a1, Cxcl4, and Nxph1 indicate a potential mechanism of regulation for 3’ UTRs of different lengths. (b) Three genes Ubash3b, Igfbp4, and Wipf3 show significant negative correlation between various Subcellular Patterning Ranked Analysis With Labels (SPRAWL) metrics and estimated 3’ UTR length.
Figure 5 with 1 supplement
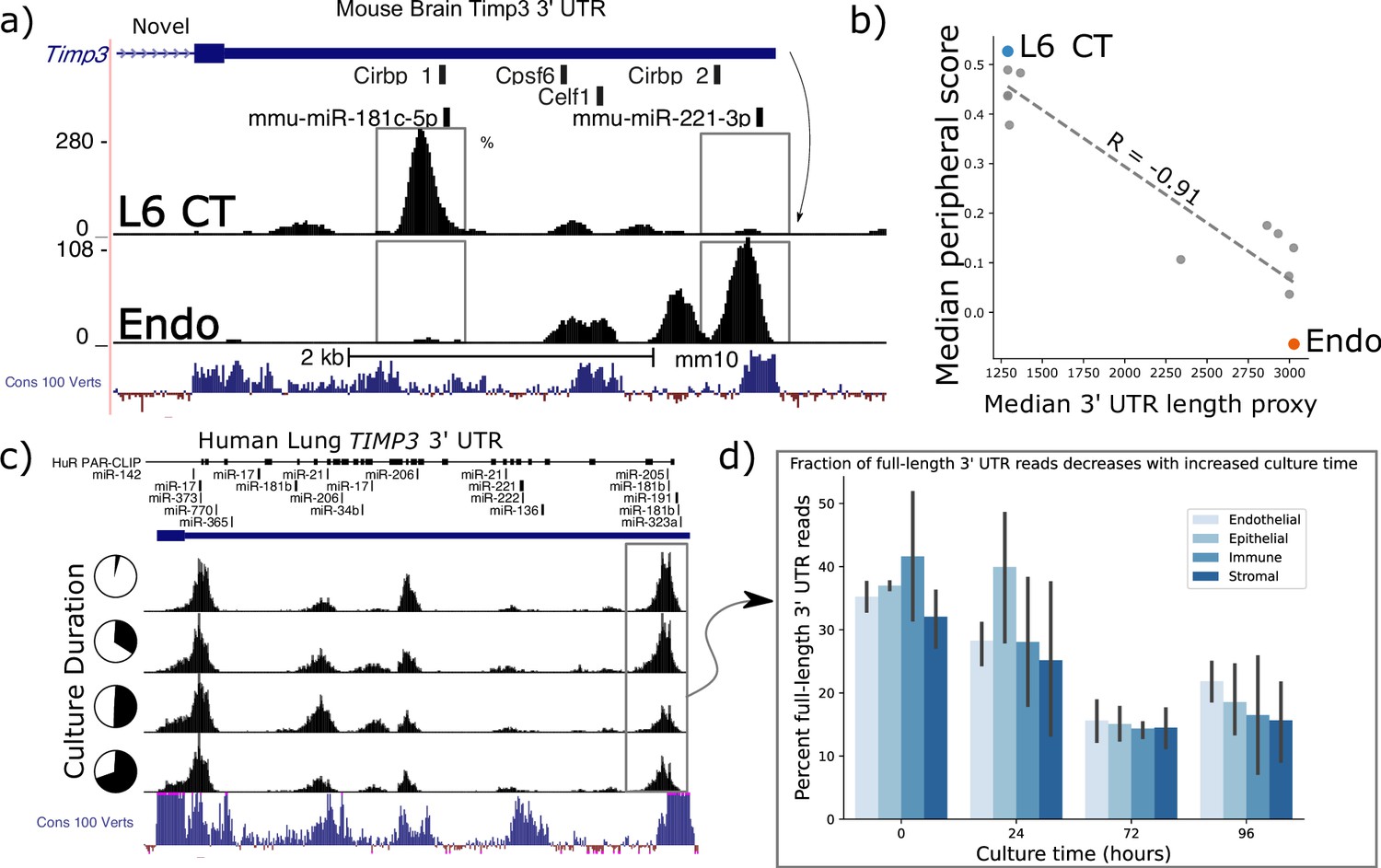
Timp3 alternative peripheral localization across motor cortex (MOp) cell types is statistically correlated with ReadZs differences in 3’ Untranslated Region (UTR) length.
(a) ReadZs detects two major alternative 3’ UTRs in mouse Timp3 from 10 X scRNA-seq which correspond to miR-181c-5p and miR-221–3 p binding sites. Reads from L6 critical threshold (CT) cells predominantly map to a novel upstream shortened 3’ UTR while endothelial cells primarily express the longer annotated 3’ UTR. The UCSC genome browser placental animal sequence conservation shows highly conserved regions in blue. Fisher’s exact test was highly significant between the two peaks denoted by the dotted lines between the two cell types. (b) Timp3 mean periphery score is significantly correlated with Timp3 median ReadZs score across MOp cell-types with Pearson correlation coefficient of –0.91 and p<<0.05. (c) Fraction of TIMP3 RNA full-length 3’ UTR reads, gray box, and (d) bar plots, decreases during human lung tissue culture.
Figure 5—figure supplement 1
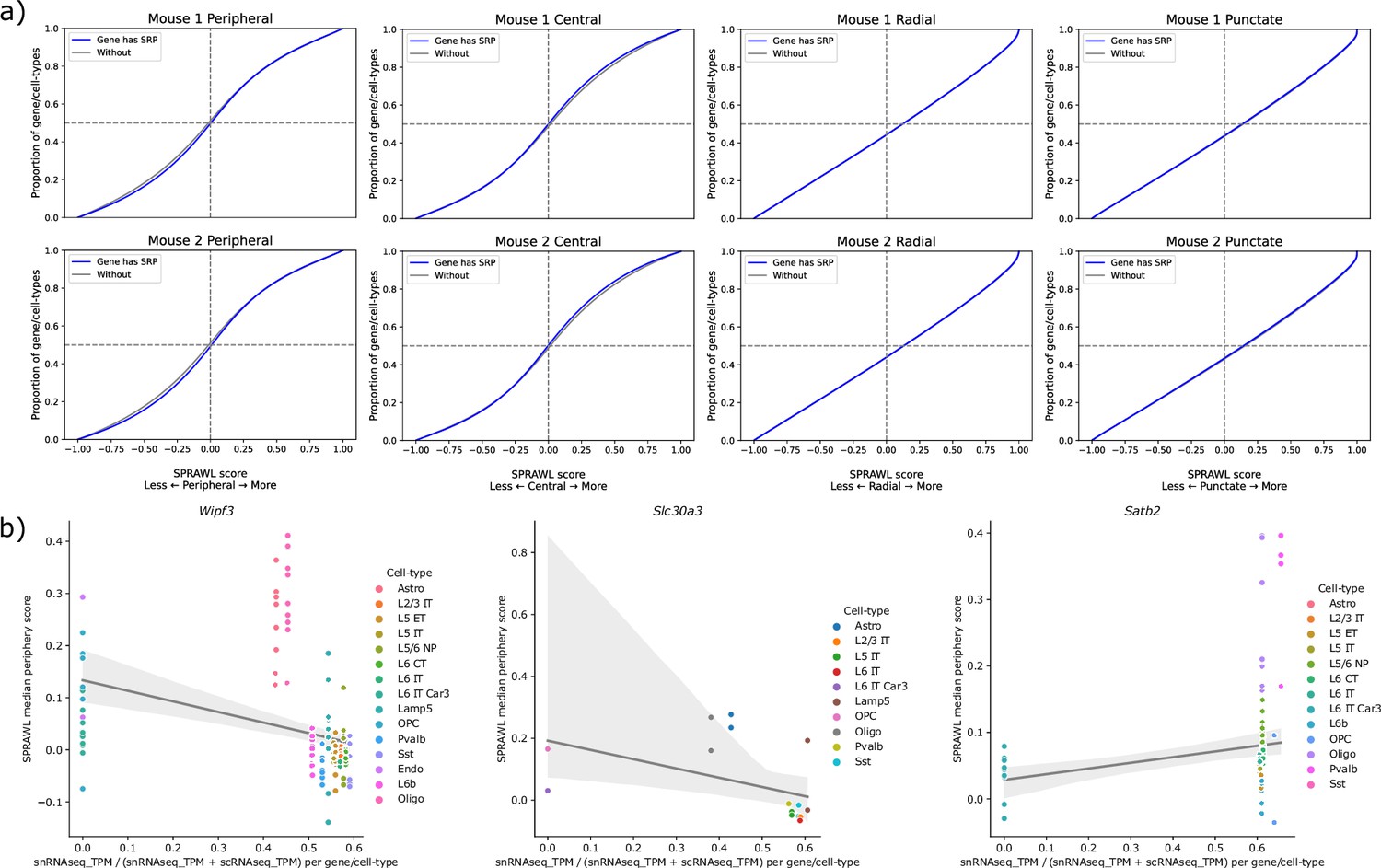
Subcellular Patterning Ranked Analysis With Labels (SPRAWL) scores do not correlate with the presence of signal recognition peptide, but do correlate with nuclear enrichment.
(a) Genes encoding signal recognition peptides do not have significantly differential SPRAWL scores while (b) genes such as Wipf3 and Slc30a3 have significantly lower peripheral scores in cell-types with higher nuclear expression. Satb2 shows the opposite unexpected correlation.
Figure 6 with 1 supplement
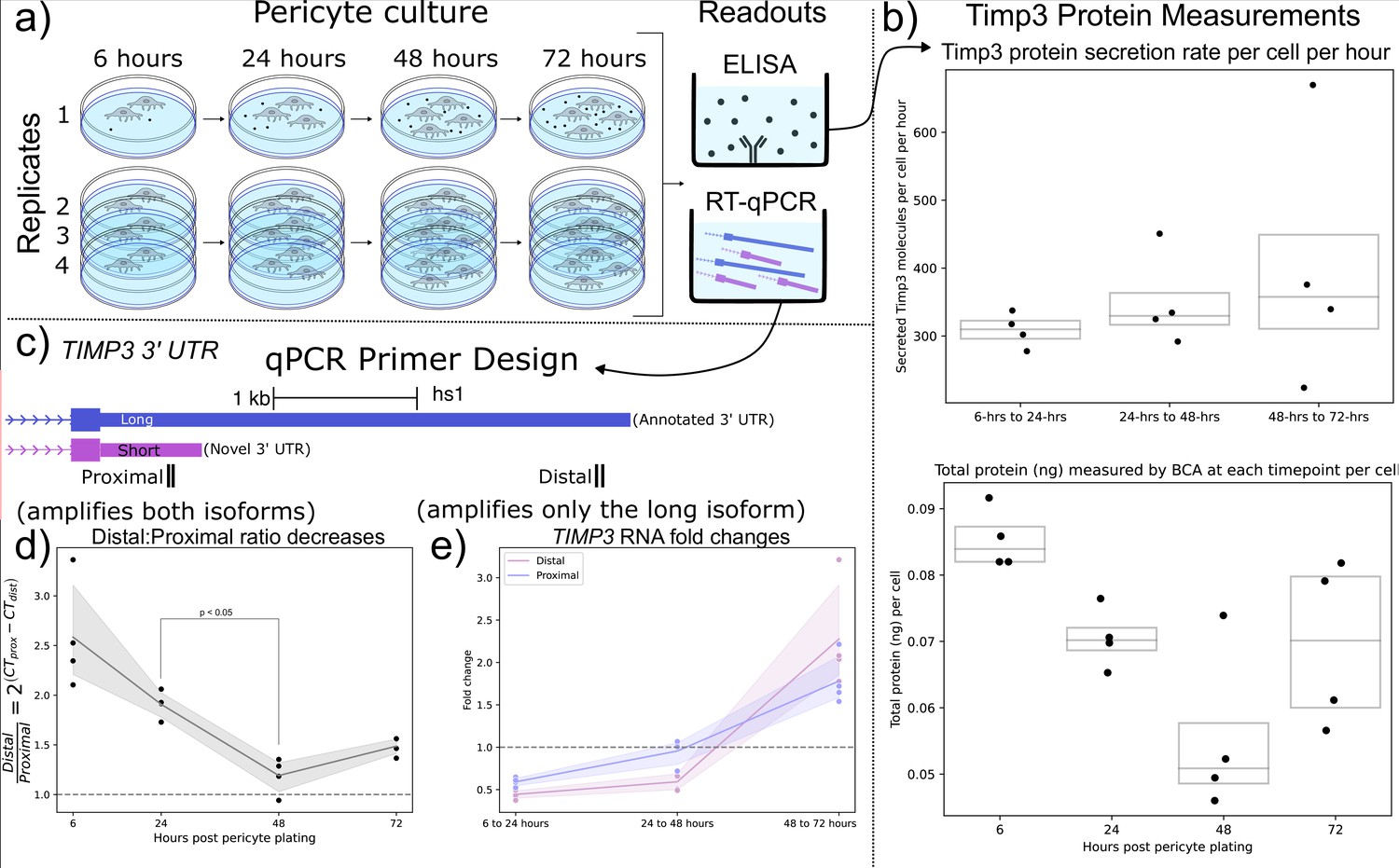
Shorter TIMP3 3’ Untranslated Regions (UTRs) become relatively more abundant in pericyte cell culture while TIMP3 protein production remains stable.
(a) Experimental setup for human pericyte cell culture with reverse-transcriptase quantitative PCR (RT-qPCR) and extracellular TIMP3 protein ELISA readouts at four-timepoints. (b) TIMP3 protein secretion per cell per hour does not significantly change throughout culture time, even though the total protein measured by BCA does change. (c) qPCR experiment design with proximal and distal qPCR primers to distinguish long and short 3’ UTR isoforms. The proximal qPCR primer can detect both long and short isoforms while the distal primer can only amplify the long 3’ UTR. (d) The ratio of distal to proximal primer-template abundances significantly decreases throughout culture time, implying increased usage of the short TIMP3 3’ UTR compared to the long isoform. (e) TIMP3 3’ UTR abundance, normalized by 18 s housekeeper abundance, fluctuates from halving to doubling between culture timepoints for both distal and proximal primers.
Figure 6—figure supplement 1
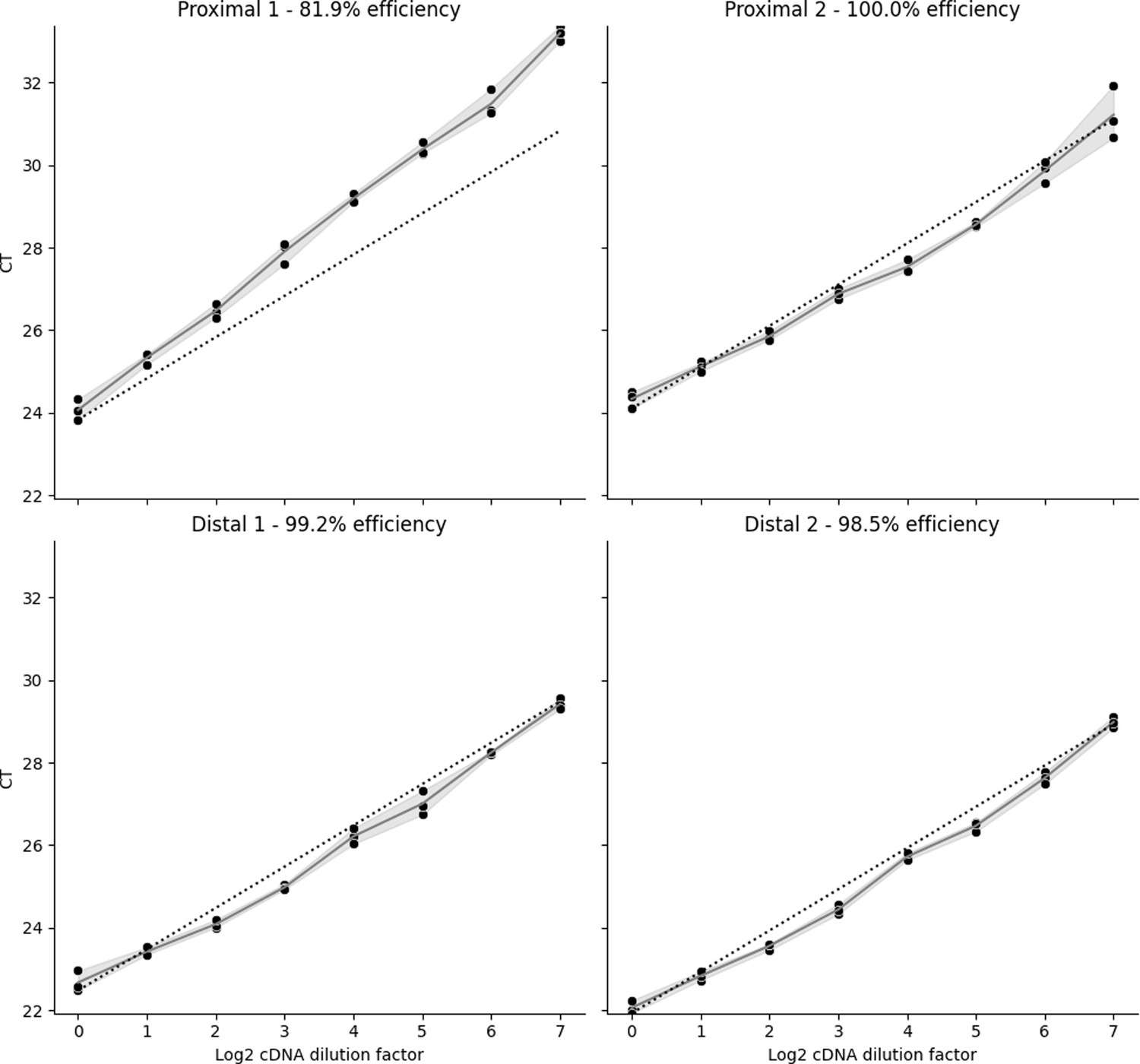
qPCR primer efficiencies for Timp3 3’ Untranslated Region (UTR) were estimated by using twofold cDNA dilutions of the same 6 hr timepoint sample.
Of the two proximal and two distal primer pairs, only proximal primer 1 had low efficiency at 81.9%. The remaining three primers showed nearly perfect 100% efficiency, where a cDNA dilution of 2 X resulted in a critical threshold value increase of 1. Dots indicate critical threshold (CT) readings of technical replicates done in triplicate with shaded regions between them. Dashed lines indicate 100% efficiency curves.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Software, algorithm | SPRAWL | This paper, Bierman, 2024 | https://github.com/salzman-lab/SPRAWL | |
| Cell line (Homo-sapiens) | Human brain vascular pericytes | Sciencell | #1200 | |
| Sequence-based reagent | Proximal_primer_1_fwd | This paper | Timp3 qPCR primer | GGGAACTATCCTCCTGGCCC |
| Sequence-based reagent | Proximal_primer_1_rev | This paper | Timp3 qPCR primer | TTCTGGCATGGCACCAGAAAT |
| Sequence-based reagent | Proximal_primer_2_fwd | This paper | Timp3 qPCR primer | AGGTCTATGCTGTCATATGGGGT |
| Sequence-based reagent | Proximal_primer_2_rev | This paper | Timp3 qPCR primer | TGGGGCCAGGAGGATAGTTC |
| Sequence-based reagent | Distall_primer_1_fwd | This paper | Timp3 qPCR primer | AATTGGCTCTTTGGAGGCGA |
| Sequence-based reagent | Distal_primer_1_rev | This paper | Timp3 qPCR primer | GCGGATGCTGGGAGAATCTA |
| Sequence-based reagent | Distal_primer_2_fwd | This paper | Timp3 qPCR primer | TAGCCAGTCTGCTGTCCTGA |
| Sequence-based reagent | Distal_primer_2_rev | This paper | Timp3 qPCR primer | GGGTTCGAGATCTCTTGTTGG |
| Commercial assay or kit | qPCR Kit | BioRad | SsoAdvanced Universal supermix | |
| Commercial assay or kit | Human TIMP-3 ELISA Kit | Invitrogen | # EH458RB |
Additional files
-
Supplementary file 1
Counts of unique, significant, and opposite-effect genes in each experiment/metric combination.
Genes are defined as significant if they are observed to be significant in at least cell-type in any replicate. Opposite-effect genes are those observed to have at least one significantly positive Subcellular Patterning Ranked Analysis With Labels (SPRAWL) gene/cell-type score, and one significantly negative SPRAWL gene/cell-type score.
- https://cdn.elifesciences.org/articles/87517/elife-87517-supp1-v1.docx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/87517/elife-87517-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Statistical analysis supports pervasive RNA subcellular localization and alternative 3' UTR regulation
eLife 12:RP87517.
https://doi.org/10.7554/eLife.87517.2




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}