Spatial chromatin accessibility sequencing resolves high-order spatial interactions of epigenomic markers
- BGI Genomics, BGI-Shenzhen, China
- College of Life Sciences, University of Chinese Academy of Sciences, China
- Department of Biology, Cell Biology and Physiology, University of Copenhagen, Denmark
- Department of Breast Surgery, Harbin Medical University Cancer Hospital, China
Figures
Figure 1 with 1 supplement

Principle of SCA-seq.
(a) (1) After fixation, chromatin accessibility can be inferred from the extent of artificial methylation by methyltransferase (m6A or GpC, rare in native genomes). (2–4) Restriction enzymes to digest the labeled genome are selected. The interacted segments (>2) stay as a cluster of fragments (concatemer) due to fixation. Next, the spatially interacted segments in one concatemer are proximally ligated, along with the formation of chimeric long fragments. (5) These long chimeric fragments are sequenced using the Nanopore technology. The data-trained Nanopolish model helps calling the labeled artificial methylation marks (GpC or m6A), which reflect the regional chromatin accessibility and native CpG on the chimeric reads. Our algorithm analyzes the composition of the chimeric reads, indicating genome locations of these composed segments. Moreover, SCA-seq can simultaneously capture chromatin accessibility and native methylation information of these segments. (b) Comparison of advantages of SCA-seq with those of other similar technologies.
Figure 1—figure supplement 1
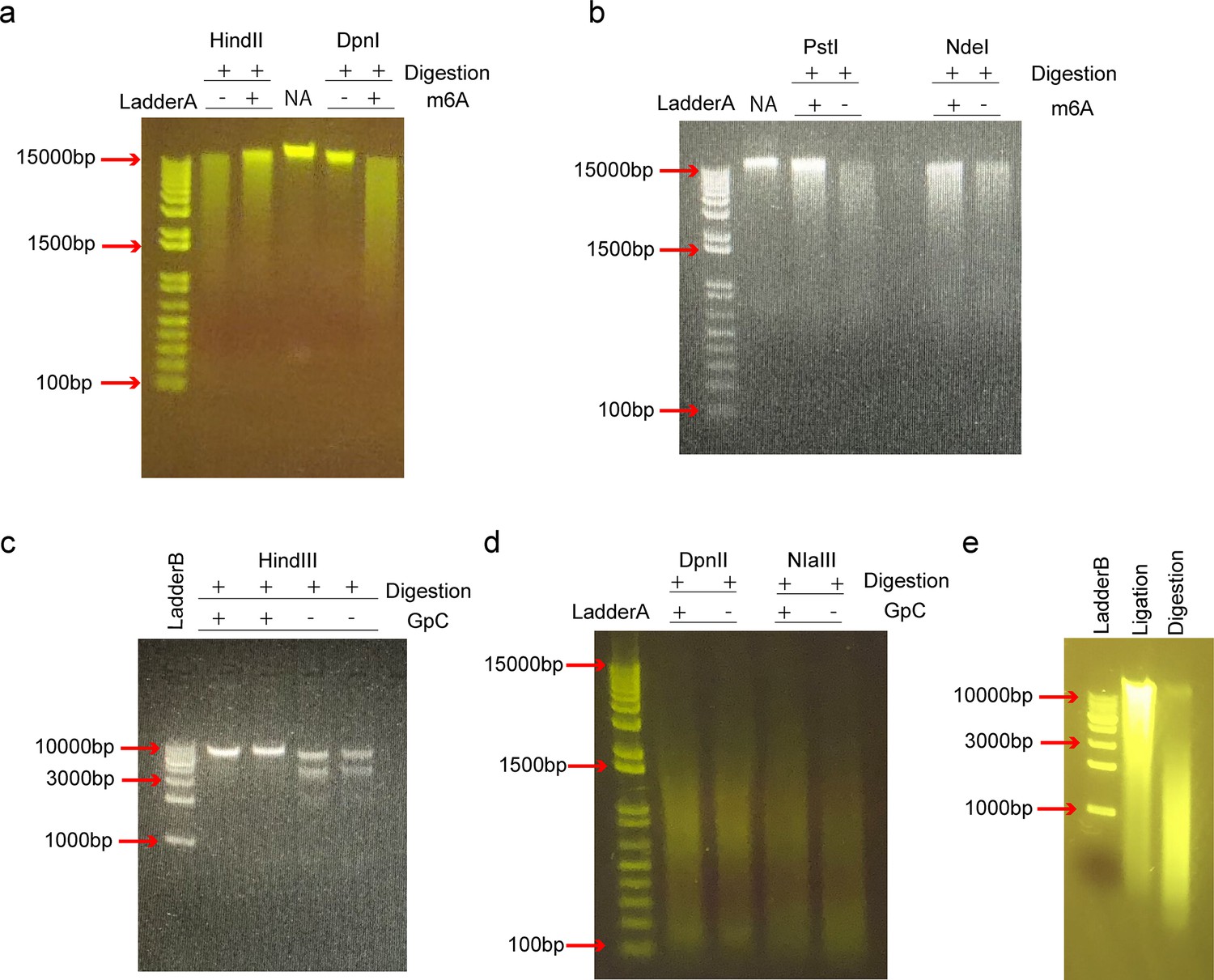
Selection of the compatible restriction enzymes.
In SCA-seq, we used methyltransferases (EcoGII or M.CviPI) to label the accessible chromatin and the restriction enzyme to digest the genome and prepare for the ligation step. Methyltransferase EcoGII transferred the methyl group to the 6′-carbon of adenosine (m6A modification). Due to the high density of adenosines in the genome (~25%), the abundant artificial m6A modification may impair restriction enzyme activity. We used EcoGII to treat the HEK293T genomic DNA (m6A+), whereas control DNA was left untreated (m6a−). We tested HindIII, DpnI (a), PstI and NdeI (b) on the methylated and unmethylated genomic DNAs, and all these restriction enzymes were inhibited by the abundant m6A modifications. DpnI is an m6A-dependent restriction enzyme that digests only genomic loci with the m6A modification. NA indicates the genomic DNA control. In a similar way, we tested the activities of HindIII (c) for lambda DNA; DpnII, and NIaIII (d) for HEK293T genomic DNA on the GpC methylated DNA preparations that were treated by methyltransferase M.CviPI. DpnII and NIaIII were not significantly inhibited by GpC methylation. (e) The HEK293T genomic DNA was digested by DpnII (Digestion) and then ligated by T4 DNA ligase (Ligation).
-
Figure 1—figure supplement 1—source data 1
Original gel images in Figure 1—figure supplement 1.
- https://cdn.elifesciences.org/articles/87868/elife-87868-fig1-figsupp1-data1-v1.zip
Figure 2 with 4 supplements
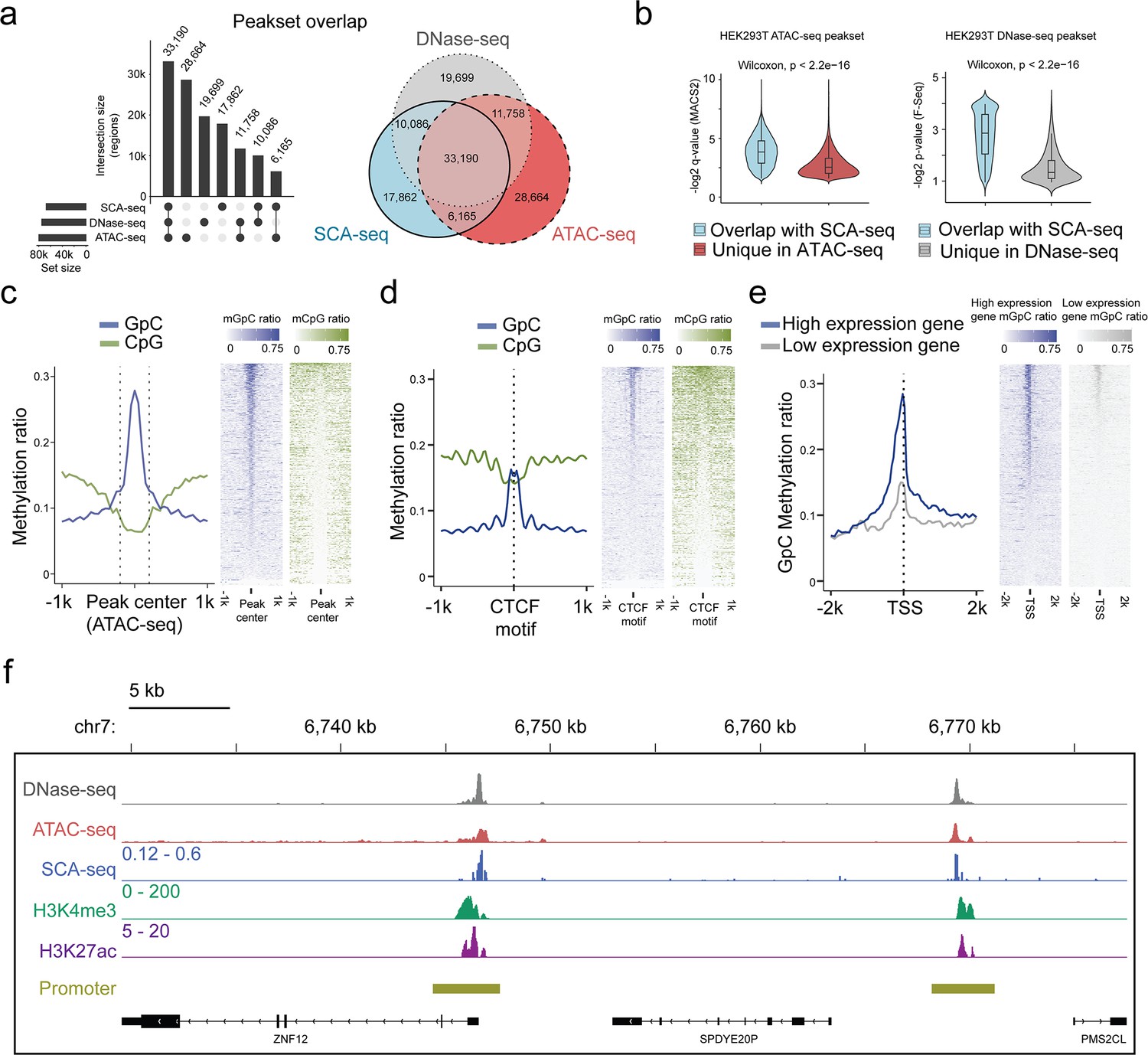
Feasibility of accessible chromatin labeling using SCA-seq.
(a) Venn diagram of the peak overlap between SCA-seq, ATAC-seq, and DNase-seq. The peak-calling algorithms were adapted from nanoNOME-seq. There were 80% peaks identified in SCA-seq overlapped with those detected by either ATAC-seq or DNase-seq. The upset plot is shown on the left. (b) Overlapping peaks in the violin plot between SCA-seq and ATAC-seq show the higher MACS2 peak q-value than the non-overlapping peaks (p < 2.6 × 10−16, Wilcoxon rank sum test). Similarly, the overlapping peaks in the violin plot between SCA-seq and DNase-seq have the higher F-seq p-value than the non-overlapping peaks (p < 2.6 × 10−16, Wilcoxon rank sum test). (c) ATAC-seq peak centered plot. ATAC-seq peak regions were centered at 0 on the x-axis and are indicated by the dashed line. The y-axis indicates the average methylation ratio on each site (methylation ratio = methylated GpC/all GpC in a 50-bp bin). Blue and green lines indicate the GpC methylation ratio (accessibility) and CpG methylation ratio, respectively. GpC and CpG ratio heatmaps around ATAC-seq peak centers are shown on the right. Each row represents a region of genome with an ATAC-seq peak. (d) CTCF motif centered plot at 0 on the x-axis indicating the CTCF motif position. The y-axis indicates the average methylation ratio among all the molecules across CTCF motifs. SCA-seq demonstrates classic nucleosome depletion patterns around the CTCF motif. GpC and CpG methylation ratio heatmaps around CTCF sites are shown on the right. Each row represents a region of genome with an CTCF site. (e) TSS-centered plot. We classified genes with high expression (upper quantile) and low expression (lower quantile) by the expression ranks. GpC and CpG methylation ratio heatmaps around TSSs are shown on the right. Each row represents a region of genome with a TSS. (f) A representative genome browser view showing chromatin accessibility signal of DNase-seq, ATAC-seq, SCA-seq, as well as H3K4me3 and H3K27ac ChIP-seq signals in a genome browser (47 kb span). The SCA-seq track shows the GpC methylation ratio in each genomic locus.
Figure 2—figure supplement 1
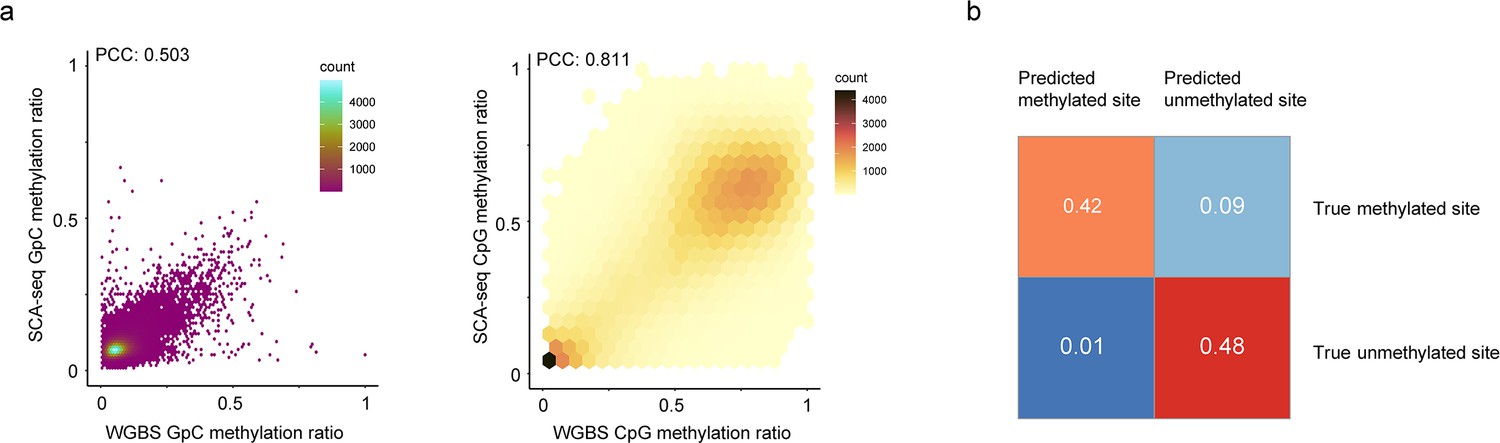
Feasibility of methylation calling.
(a) The similarity between the whole-genome bisulfite sequencing (WGBS) GpC methylation calls and SCA-seq GpC methylation calls is illustrated. The methylation ratios in 200 bp bins of SCA-seq were scatter plotted against WGBS methylation ratios (Pearson’s correlation, p < 2.6 × 10−16). A similar plot of the CpG methylation ratio is on the right (Pearson’s correlation, p < 2.6 × 10−16). (b) Confusion matrix of GpC methylation calling. Subsets of 100,000 sites in unmethylated pore-C HEK293T samples and methylated lambda DNA samples were methylation called by the default threshold of the Nanopolish cpggpc model. The results are classified into four categories: false positive and true negative from the unmethylated pore-C run and true positive and false negative from the GpC-labeled lambda DNA sample.
Figure 2—figure supplement 2
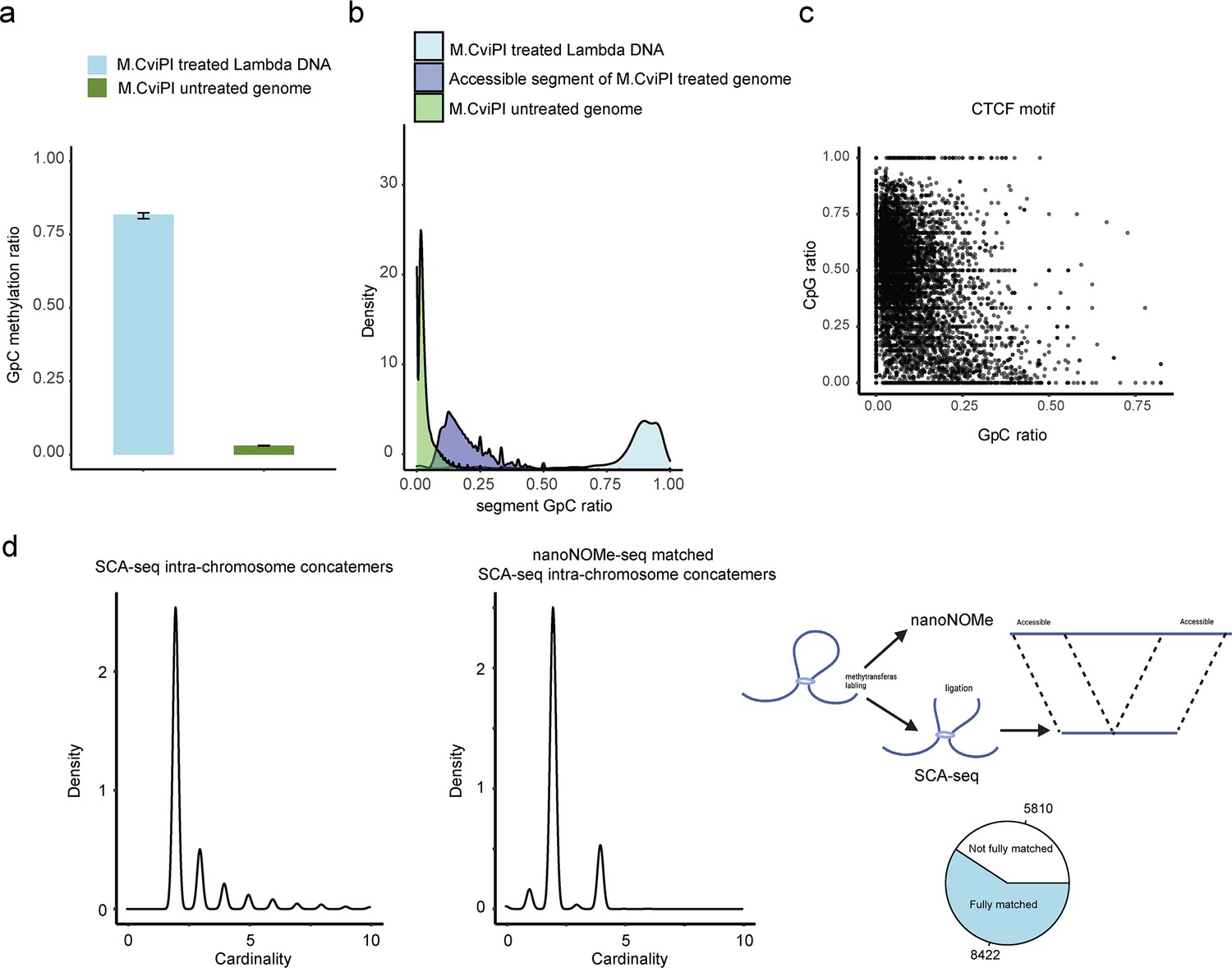
GpC labeling efficiency in vitro and in vivo.
We used lambda DNA as the spiked-in DNA control to monitor M.CviPI efficiency. Genomic DNA without labeling was used as background control to calculate the background level of GpC. The labeled genomic DNA was processed using SCA-seq. We plotted the density plot of GpC ratios (methylated GpC/total GpC) in each segment for background genomic DNA, treated genomic DNA, and spike-in control DNA. The background GpC ratio in each segment was around 0.018 (a, b). Efficiency of spiked-in lambda DNA labeling by M.CviPI was around 0.88 in vitro. We then selected CTCF motifs as control to calculate in vivo labeling efficiency (c). We plotted the CpG methylation ratio (methylated CpG/total CpG) against the GpC methylation ratio in CTCF motifs. Previous publications showed that pre-existing methylation could antagonize CTCF binding to CTCF motifs (Bell and Felsenfeld, 2000; Hark et al., 2000; Kanduri et al., 2000). Therefore, we assumed that CTCF motifs with low methylation (CpG methylation <0.25) marked accessible chromatin regions with capacity for CTCF binding. The CTCF motif labeling efficiency was calculated as NCpGmethylation<0.25&GpCmethylation>0.07/NGpCmethylation<0.25 = 0.79. However, the highly methylated CTCF motifs might also bind CTCF to maintain chromatin accessibility. Therefore, the in vivo labeling efficiency could be estimated only roughly. We then used nanoNOME-seq to cross-validate SCA-seq labeling efficiency (d). Both nanoNOME-seq and SCA-seq were single-molecule assays. nanoNOME-seq could report chromatin accessibility in 2–20 kb single DNA molecules. Sixty-eight percent of SCA-seq concatemers (d, left panel) corresponded to similar DNAs identified by nanoNOME-seq (d, right panel). The most matched concatemers contained only two segments, representing the short-distance interaction. The concatemers with multiple segments, representing the long-distance interaction, may be out of the nanoNOME-seq detection range.
Figure 2—figure supplement 3
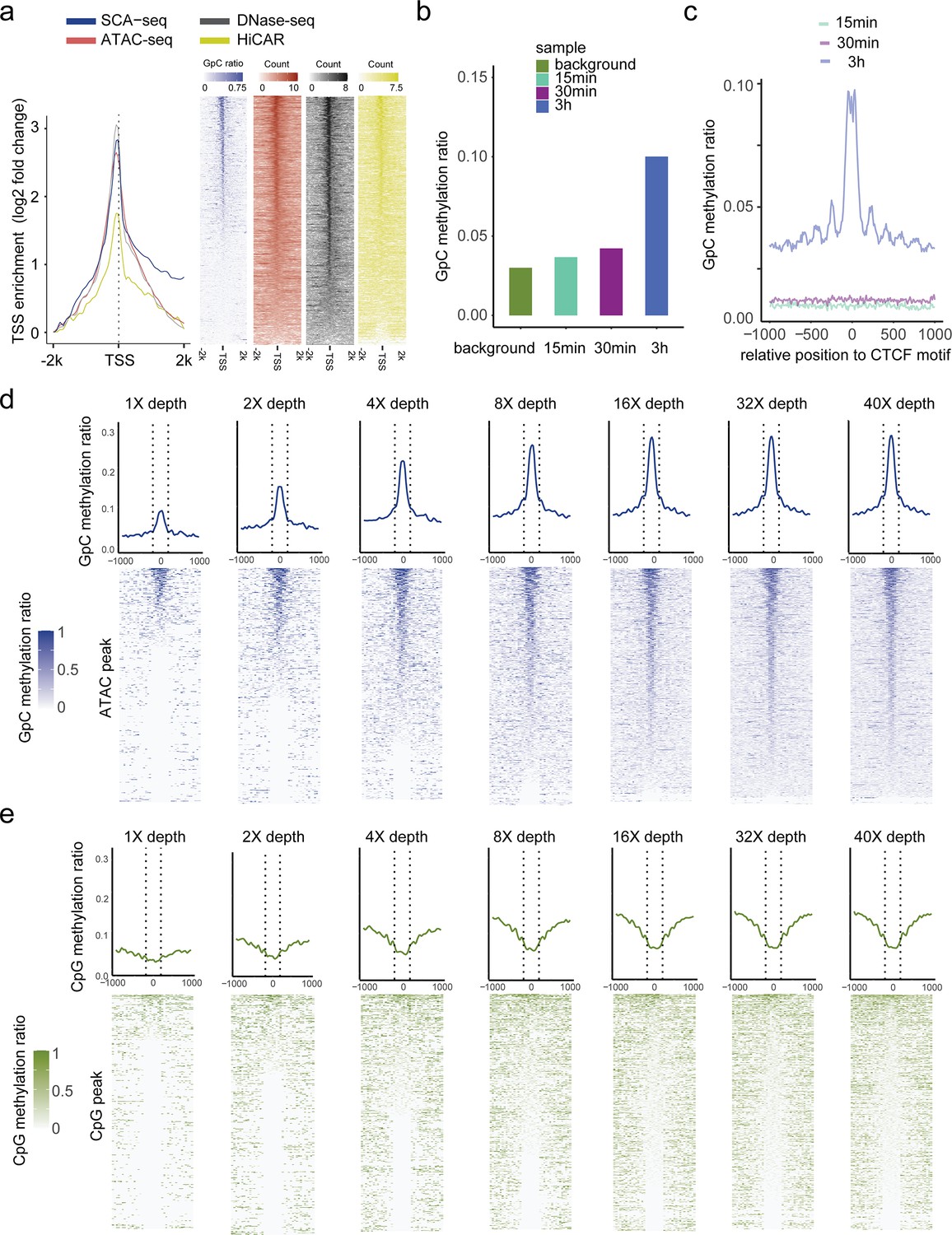
Dependence of labeling efficiency and signal strength from enzyme concentration and sequencing depth.
(a) TSS-centered plot illustrating the enrichment score (y-axis) for different methods around TSSs (0 on the x-axis), which shows a significant nucleosome depletion pattern. The enrichment score was calculated as log2(signal in each bin/lowest signal in scale). (b) Test of labeling efficiency at three different labeling times (15 min, twofold higher enzyme concentration; 30 min, twofold higher enzyme concentration; 3 hr, threefold higher enzyme concentration). GpC labeling efficiency was the highest after the 3 hr treatment, and the labeled GpC signal accurately showed the CTCF motif nucleosome pattern (c). The results of the 3 hr treatment were also similar to those obtained for nanoNOME-seq tissue sample preparation (Lee et al., 2020). (d, e) ATAC-seq peaks were identified first, and methylation ratios obtained using SCA-seq are plotted around the identified peaks (center to 0) with different sequencing depths. The line panels show a single-molecular resolution.
Figure 2—figure supplement 4
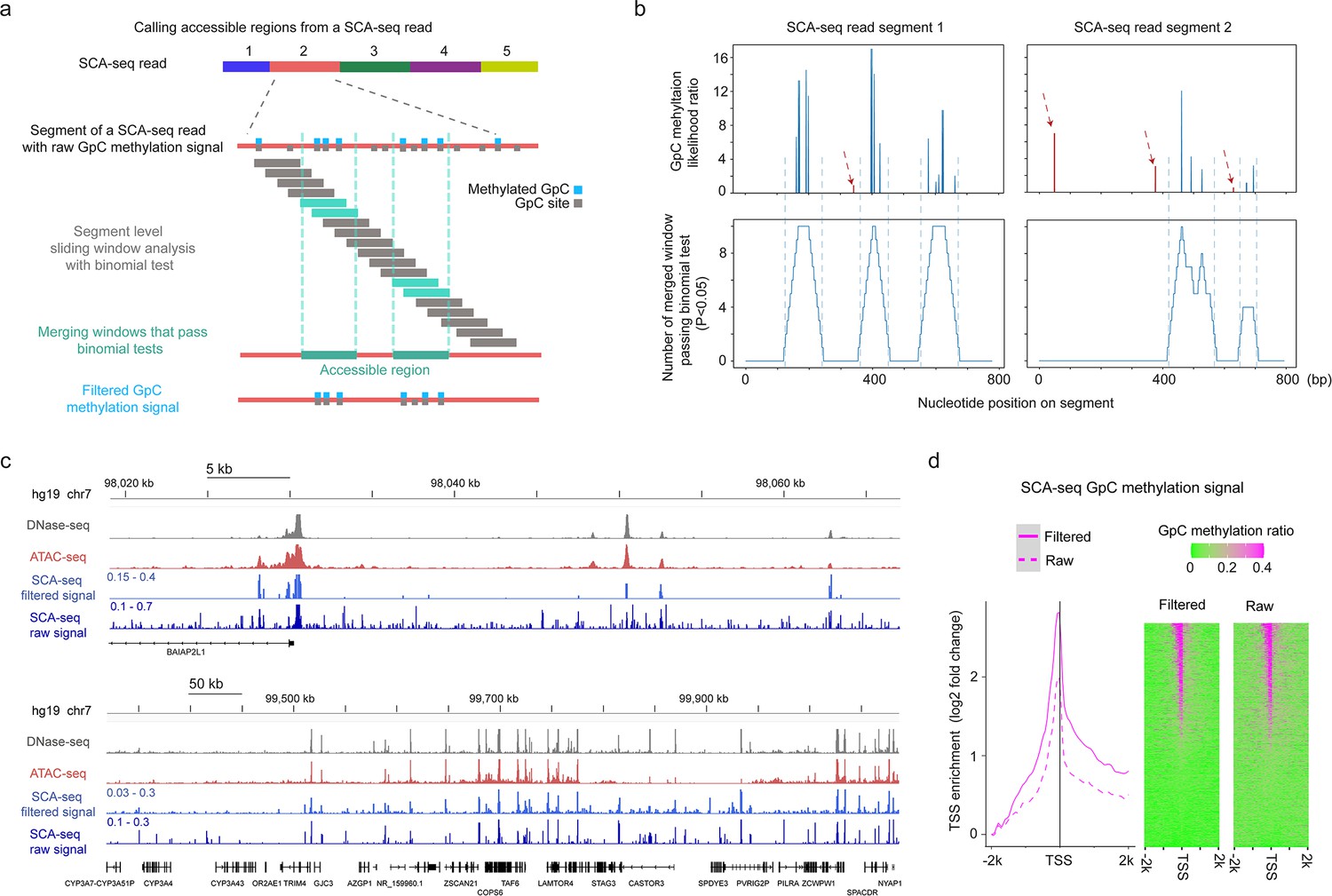
Reducing background noise by the detection of accessible regions at segment level.
(a) Schematic of calling accessible regions on an SCA-seq read. The accessible region of each segment of a read was called independently. The raw GpC methylation signals were divided into sliding windows, and binomial test was performed to determine the significant window (cyan box) where the GpC methylation ratio deviated from the background ratio. The overlapping significant windows were then merged to determine accessible regions (green box). GpC methylation signals outside the accessible regions were filtered out to reduce the background noise of SCA-seq accessibility signal. (b) Examples of calling accessible region on two SCA-seq segments. The top two plots show the raw GpC methylation likelihood ratio (y-axis) on the segment nucleotide position (x-axis). The bottom two plots show the number of merged significant windows (y-axis) on the segment nucleotide position (x-axis). The vertical dash lines aligned the called accessible region with the raw cluster of GpC methylation signal. The random GpC methylation signals (dark red) are highlighted in dark red dashed arrows. (c) Chromatin accessibility signal at various resolutions. The panels show DNase-seq, ATAC-seq, and SCA-seq filtered signal and raw signal in the genome browser. DNase-seq and ATAC-seq results show relative level of read counts in each genomic locus, whereas SCA-seq data represent GpC methylation ratio in each genomic locus. (d) TSS-centered plot demonstrating the filtered and raw SCA-seq GpC methylation signal at up- and downstream 2 k bp of TSS sites genome wide. The enrichment score was calculated as log2(TSS site signal/lowest signal in the 50 bp bin). The TSS-centered GpC methylation ratio heatmaps are shown on the right. Each row represents a TSS-centered region on the genome.
Figure 3 with 2 supplements
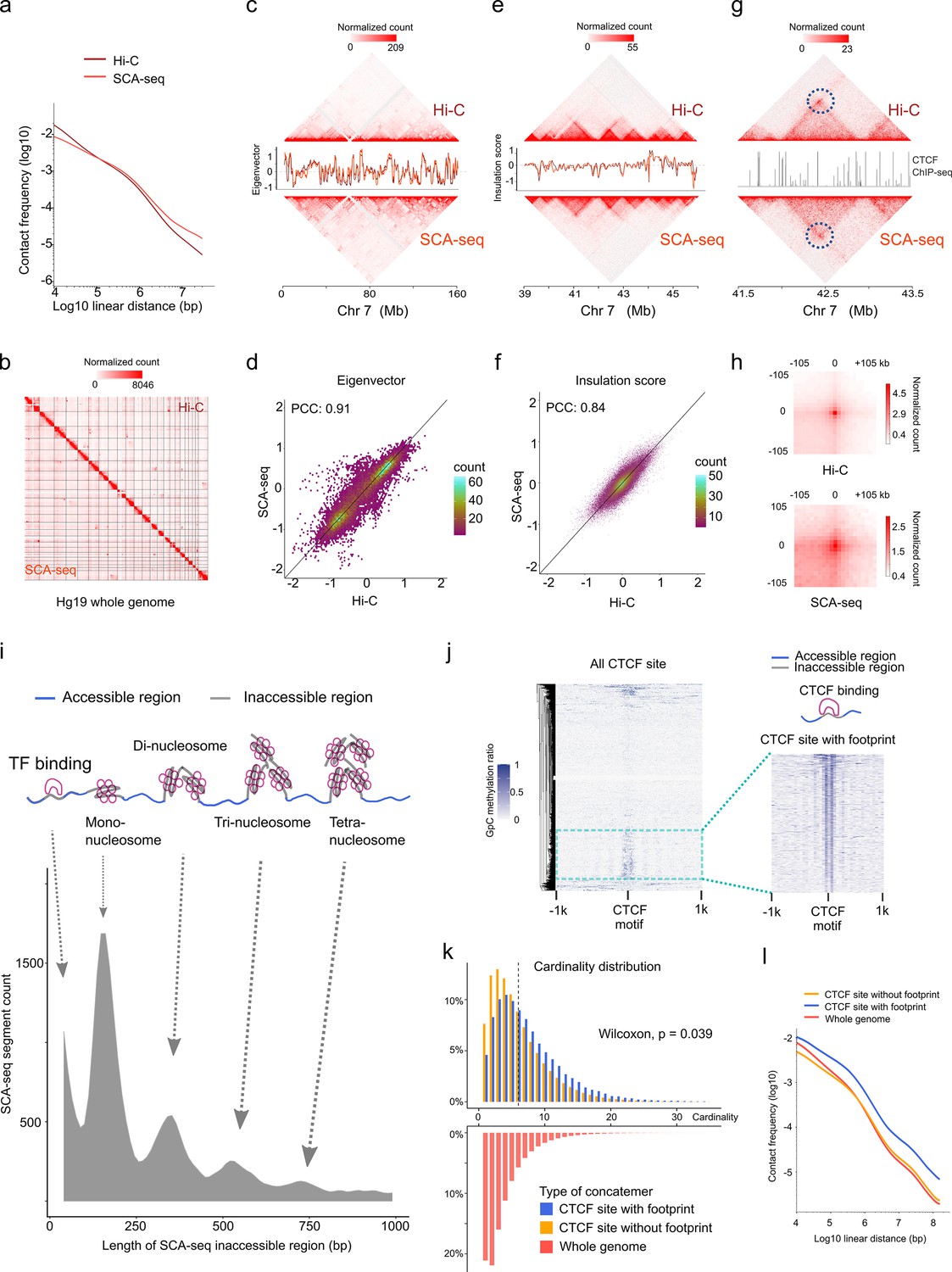
SCA-seq captures higher-order chromatin structure and CTCF footprints.
(a) Contact frequency (y-axis) as a function of linear genomic distance (x-axis) was plotted across all hg19 chromosomes for SCA-seq (red) and Hi-C (brown). (b) Comparison of 1.1 M SCA-seq virtual pairwise contacts (lower triangle) and 1.8 M Hi-C contacts (upper triangle) for chromosomes 1–22 and X (hg19) in HEK293T cells. Comparison of SCA and Hi-C 250 kb (c) and 25 kb (e) contact maps for chromosome 7. The corresponding eigenvector (c) and insulation score (e) indicating visual consistency between SCA-seq (red) and Hi-C (brown) signal patterns. Color scale bar: log normalized read counts. Scatterplots comparing eigenvector (d) and insulation score (f) between SCA-seq and Hi-C. PCC: Pearson’s correlation coefficient (p < 2.6 × 10−16). (g) Contact map showing an example of CTCF peaks at 10 kb resolution with a loop anchor signal indicated by black circles. Color scale bar: log normalized read counts. The CTCF ChIP-seq tract is plotted in between. (h) Aggregate peak analysis showing visual correspondence of enrichment patterns between Hi-C and SCA-seq within 100 kb of loop anchors at 10 kb resolution. Color scale bar: sum of contacts detected across the entire loop sets at CTCF sites in a coordinate system centered around each loop anchor. (i) The lower density plot shows the inaccessible region length distribution of SCA-seq. The upper panel shows the schematic diagram of the transcription factor binding and nucleosome footprint patterns. (j) The heatmap of GpC methylation level shows chromatin accessibility at the CTCF motif at a single-molecule resolution. A subset of CTCF sites with CTCF-binding footprint (50 bp accessible–50 bp inaccessible–50 bp accessible) is shown in a magnified view. (k) Cardinality distribution of three sets of concatemers. Concatemers with a CTCF footprint (blue) and without the footprint (yellow) are determined by the CTCF-binding footprint. The genome-wide concatemers are shown in red. Distributions of concatemers with and without CTCF footprint were compared by the Wilcoxon test. (l) Multiplex concatemers were converted to pairwise contacts. The linear distances between pairwise contacts are plotted as density distributions.
Figure 3—figure supplement 1
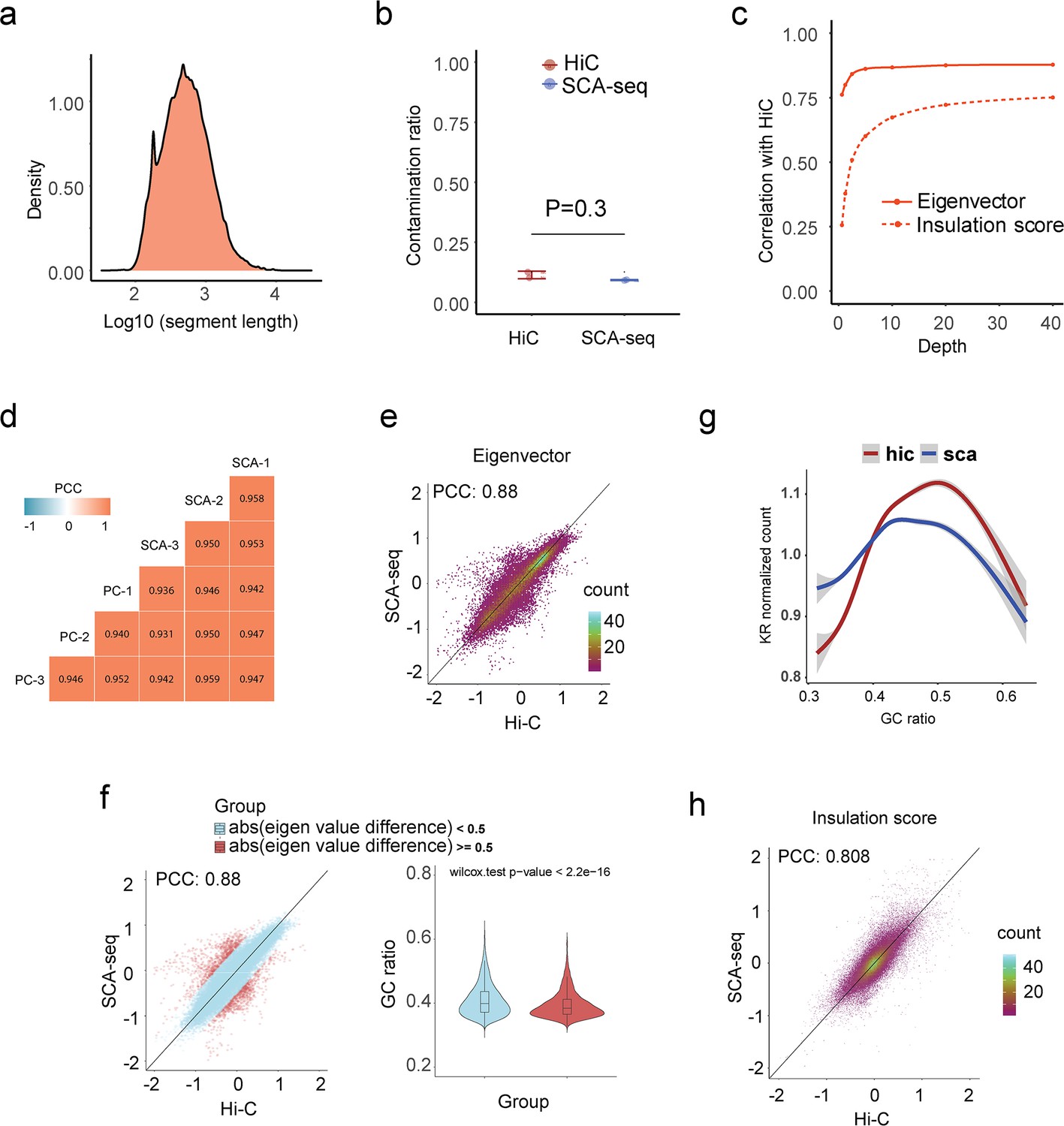
Quality parameters of SCA-seq for resolving the genome structure.
(a) The medium length of fragments in concatemers was around 700 bp. (b) We further calculated the false positive contacts caused by the ligations (contamination). We used the contacts between the mitochondrial genome and nucleic genome/total contacts to calculate the contamination ratios. SCA-seq and Hi-C contamination ratios were 9% and 12%, respectively. (c) We then subsampled the reads to test the sequencing threshold to calculate the eigenvector (AB compartment) and insulation score (TAD). We needed over 5 and 30 million reads to accurately produce results similar to the Hi-C output for in the AB compartment and TAD, respectively. (d) We then tested the correlation (0.93–0.94) between SCA-seq and pore-C biological replicates (N = 3) (Ulahannan et al., 2019). SCA-seq demonstrated good consistency between samples. (e) Plot of global eigenvalue correlation (without quality control) between SCA-seq and Hi-C data. (f) The compartment correlations with bias were far from the diagonal line and are colored red. The bias regions had a significantly lower GC ratio than the unbiased region in the violin plot. (g) The GC ratio distribution in SCA-seq and Hi-C. (h) Plot of global insulation score correlation (without quality control) between SCA-seq and Hi-C data.
Figure 3—figure supplement 2
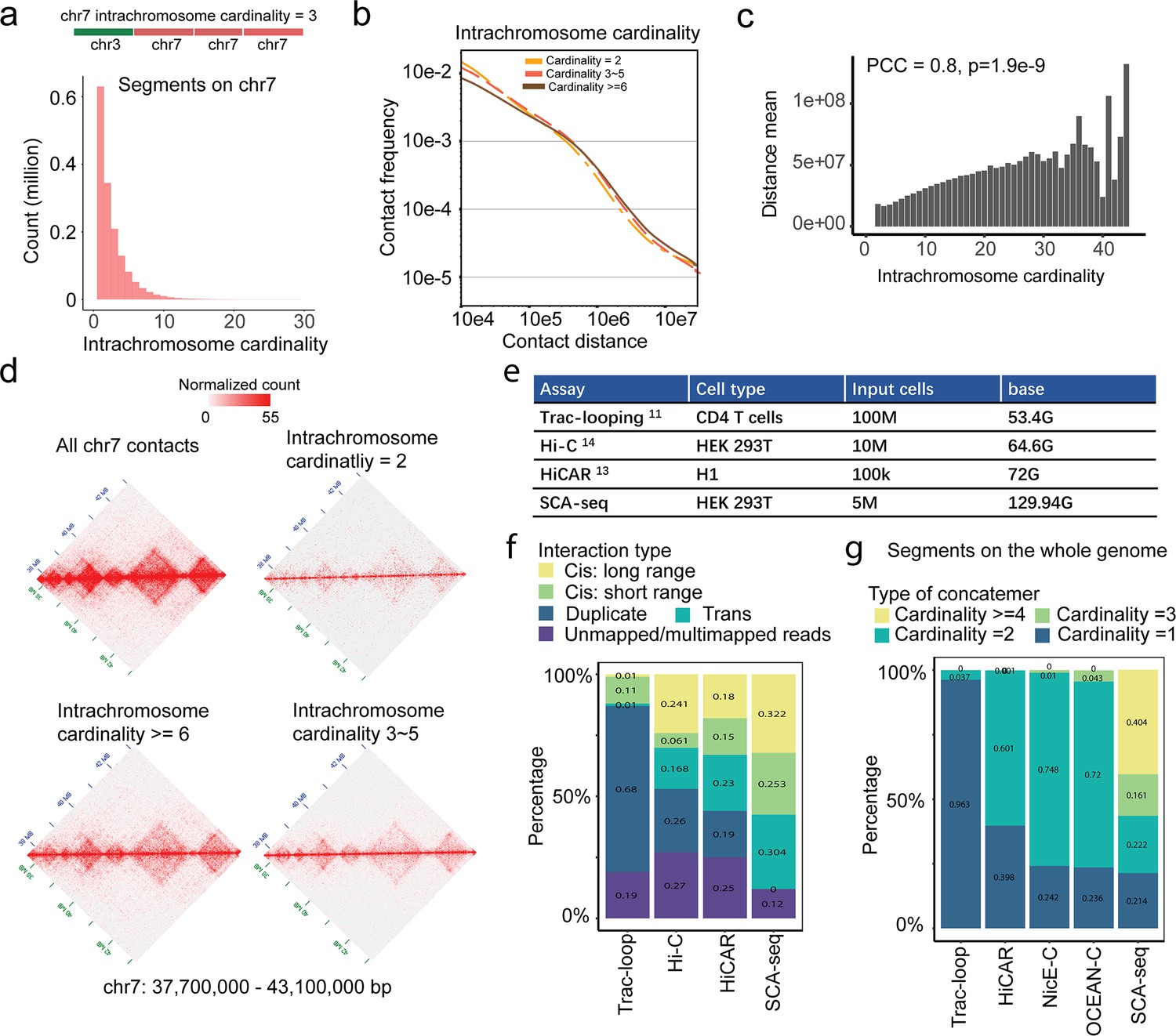
High-order contacts resolve the long-distance genome interaction.
(a) Intra-chromosome contacts on chromosome 7. The Chr7 cardinality only considers the chr7 intra-chromosome segments that are adjacent to each other on an SCA-seq concatemer. 65.4% of concatemers contain one segment without any intra-chromosome contacts, 14.7% of concatemers contain two segments, 14.5% contain three to five segments. 5.4% of concatemers have ≥ six segments. (b) Plot of the contact frequency against the contact distance for concatemers of segments on chromosome 7 (intra-chromosome concatemers) with different cardinality values. The high-cardinality concatemers had more distant contacts than the low-cardinality concatemers. (c) The maximum contact distance in each concatemer positively correlates with concatemer cardinality. (d) Concatemers of segments on chromosome 7 with different cardinality values were converted to pairwise contact maps and normalized by the JuicerTools KR method. The high-cardinality concatemers tend to maintain the TAD structures and indicate long-distance interaction. (e) The data output, cell type, and cell number of each method. (f) Genomic interactions at distances of ≥20 and <20 kb were classified as long- and short-range interactions. ‘Duplicate’ indicates duplicated reads. ‘Trans’ indicates interaction across chromosomes. (g) The cardinality represented the genome-wide segment number in one read, referring the conformation complexity.
Figure 4 with 1 supplement
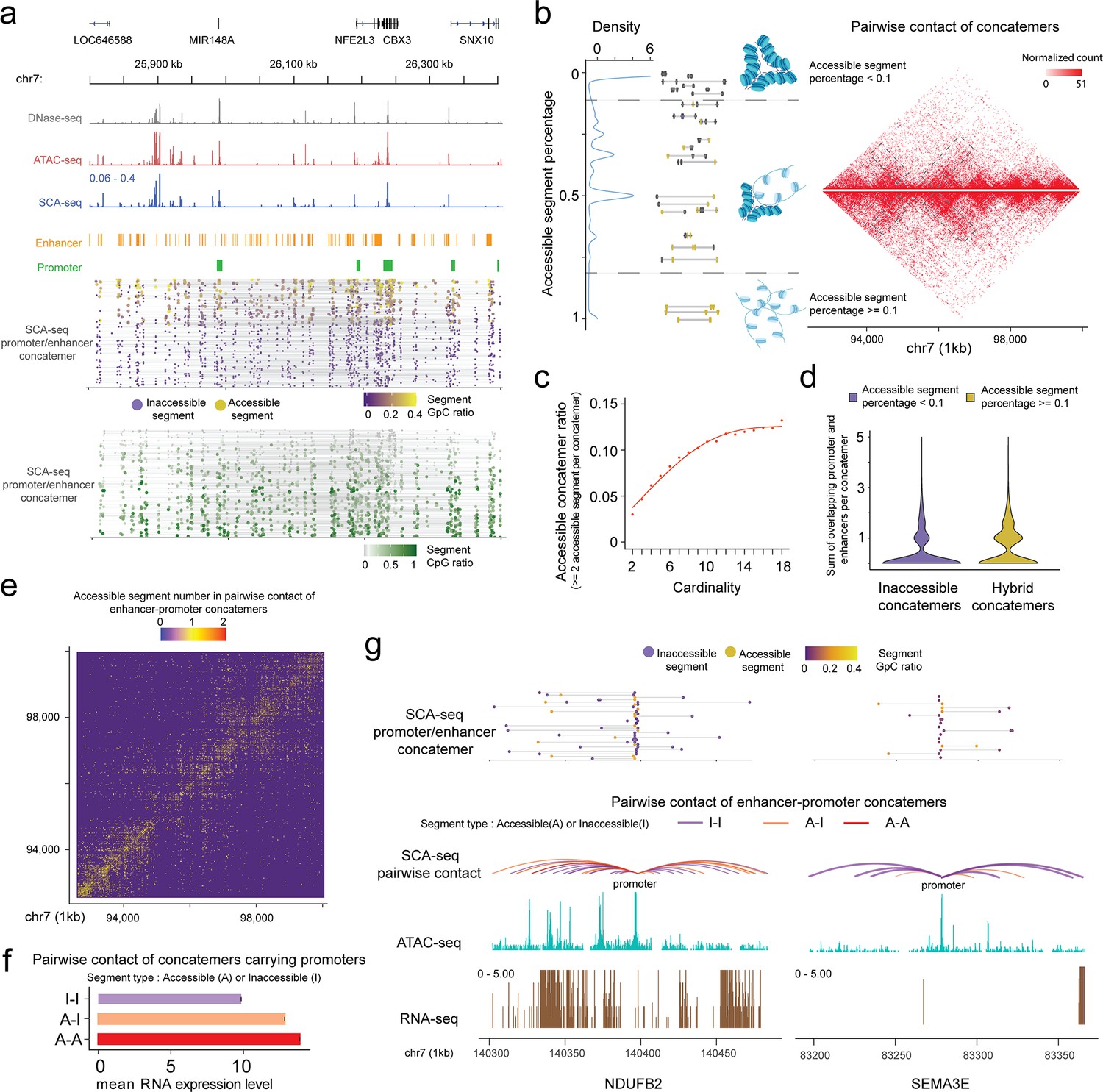
SCA-seq demonstrates spatial chromatin accessibility.
(a) Demonstration of promoter–enhancer concatemers in the region from 25,800 to 26,400 kb on chromosome 7 on a single-molecule level. Gray, red, and blue tracks indicate chromatin accessibility signals from DNase-seq, ATAC-seq, and SCA-seq, respectively. The spatially inaccessible segments that were proximately ligated together to form one concatemer are shown as a single line in the bottom panel. Purple and green dots indicate chromatin accessibility (GpC) and CpG methylation ratio in one segment of a concatemer, respectively. Each row represents an SCA-seq concatemer. (b) Distribution of concatemers with specific accessible segment ratios. Each segment was considered accessible if at least one accessible region is identified; otherwise, it was considered inaccessible. Then the ratio of accessible segments was calculated as follows: Naccessible/(Naccessible+inaccessible). The colored line-dot plot shows GpC methylation of segments (dots) in each concatemer (each line). In total, 150,000 pairwise contacts were subsampled for the different types of concatemers in the genome region chr7:92,700,000–100,000,000 with a 25-kb resolution. Inaccessible concatemers tend to accumulate inside the TAD and hybrid concatemers have more TAD boundary interactions and distal interactions (gray box). (c) Dot plot with regression curve shows that accessible concatemers (accessible contacts >2 per concatemer) tend to correlate positively with concatemer cardinality (segment number in each concatemer). (d) Violin plot of the enhancer/promoter counts in each concatemer. Hybrid concatemers have significantly more enhancers/promoters than inaccessible concatemers (p < 2.6 × 10−16, Wilcoxon rank sum test). (e) Accessibility contact map of enhancer–promoter contacts. Inaccessible–inaccessible (I–I), inaccessible–accessible (I–A), and accessible–accessible (A–A) pairwise contacts are shown in blue, yellow, and red colors, respectively. The genome region 92,700,000–100,000,000 is shown at a 25-kb resolution as an example. (f) Bar plot showing the relationship of gene expression with different types of accessibility contacts. The y-axis indicates the A–I contact status. The x-axis is the mean RNA expression level the corresponding gene promoters (2 kb upstream of the gene). The accessible enhancer/promoter contacts significantly enhanced gene expression (p < 2.6 × 10−16, Student’s t-test). (g) An example of two genes with similar accessibility on the promoter (2.1% accessibility, promoter-centered plot) but different number of A–A contacts. The curve line track is the chromatin interaction detected by SCA-seq. The cyan and brown tracks are ATAC-seq and RNA-seq by counts on the genome. The top line-dot plot shows the higher-order interaction and accessibility at a single concatemer resolution. The NDUFB2 gene, with a greater number of accessible contacts, had a notably stronger expression level (528 RPKM) than SEMA3E (0 RPKM), which had few accessible contacts.
Figure 4—figure supplement 1
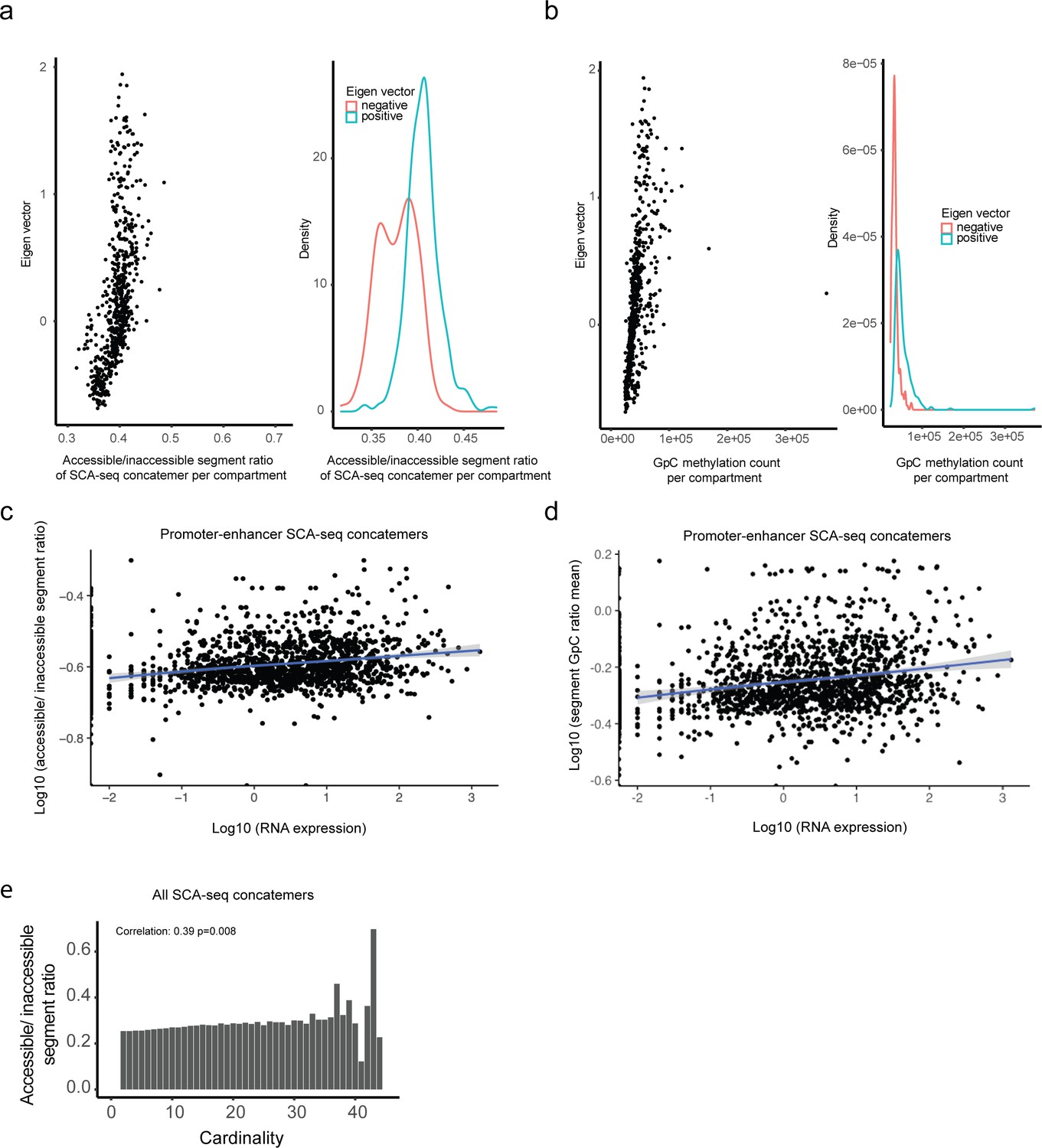
Correlation between compartmental eigenvalue, RNA expression, and spatial accessibility.
(a) Plot of compartmental eigenvalues against the average concatemer accessible/inaccessible ratio in the compartments. The average concatemer accessible/inaccessible ratio was correlated with the compartmental eigenvalue (Pearson’s correlation; p < 2.6 × 10−16). The average concatemer accessible/inaccessible ratio was significantly higher in the A compartments (positive eigenvalue) than in the B compartments (negative eigenvalue) (p < 2.6 × 10−16, Student’s t-test). (b) High correlation of compartmental eigenvalues with labeled GpC methylations. The labeled GpC methylations were highly correlated with the compartmental eigenvalue (Pearson’s correlation; p < 2.6 × 10−16). The labeled GpC methylations were significantly more abundant in the A compartments (positive eigenvalue) than in the B compartments (negative eigenvalue) (p < 2.6 × 10−16, Student’s t-test). (c) For enhancer/promoter accessibility contacts in Figure 4d, we summarized the mean contactable accessibility on the promoters/enhancers (inaccessible/inaccessible 0, inaccessible/accessible 1, accessible/accessible 2). The mean contactable accessibilities on promoters highly correlated with the expression level of the corresponding gene (Pearson’s correlation, p < 2.6 × 10−16). (d) Similarly, we summarized the mean GpC methylation ratio on the concatemers with promoter/enhancer regions. Mean concatemer GpC methylation ratios on promoters/enhancers significantly correlated with corresponding gene expression levels (Pearson’s correlation, p < 2.6 × 10−16). (e) The accessible/inaccessible ratios of concatemers were positively correlated with the cardinality (Pearson’s correlation, p = 0.0008). As shown in Figure 3—figure supplement 2b, c, we found that high-order concatemers indicate long-distance interactions.
Figure 5

CpG methylation and different types of spatial contacts of CpG islands.
(a) Schematic illustration of orphan CGI (oCGI) spatial contacts with CTCF, promoter, and enhancer. oCGIs are CpG islands that are far away (at least 1 kb) from enhancers or promoters. The enhancer and promoter annotations were downloaded from ENCODE. The eCGIs and pCGIs are defined as CpG islands near or overlapping with the enhancers and promoters. oCGIs comprised 65% of all CGIs that did not directly contact enhancers or promoters. The pie chart shows the abundance of different types of CGIs. (b) Number of overlaps between three types of CGIs containing concatemers and histone marks/CTCF site. About 83% of the reads contained oCGI with low CpG methylation level, mainly located with active histone markers that shows high level of chromatin accessibility, such as H3K27ac and H3K4me3, as well as the CTCF-binding motif. H3K27me3 and H3K36me3, which are inactive histone makers, were on the opposite DNA methylation state. (c) The upset plots indicate types of interaction between oCGI and other elements in one concatemer. On average, one concatemer has five DNA segments. These segments with oCGIs included CTCF-binding motifs, enhancers, promoters, and other proteins. The y-axis indicates the number of concatemers. (d) Stacked bar plot shows the abundance of each type of elements interacting with concatemers harboring oCGI or random-sampled concatemers (control). The number of interacting elements was normalized by the total number for itself. oCGIs preferentially interacted with CTCF-binding motifs and promoters. (e) Density plot demonstrating the distribution of the number of elements on concatemers. Thirty-nine percent of concatemers have more than one enhancer and CTCF-binding motif with oCGIs. Promoters were in contact only with oCGIs.
Additional files
-
Supplementary file 1
Data table for datasets used in this paper.
- https://cdn.elifesciences.org/articles/87868/elife-87868-supp1-v1.xlsx
-
Supplementary file 2
Metrics of pore-C and SCA-seq data in this paper.
- https://cdn.elifesciences.org/articles/87868/elife-87868-supp2-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/87868/elife-87868-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Spatial chromatin accessibility sequencing resolves high-order spatial interactions of epigenomic markers
eLife 12:RP87868.
https://doi.org/10.7554/eLife.87868.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}