Structural basis of EHEP-mediated offense against phlorotannin-induced defense from brown algae to protect akuBGL activity
- Faculty of Advanced Life Science, Hokkaido University, Japan
- Graduate School of Bioscience and Bioindustry, Tokushima University, Japan
eLife assessment
This important study presents convincing evidence on how the sea slug Aplysia kurodai optimizes its digestion of brown algae, in a classical predator-prey 'arms race' at the molecular level. The experimental protein structures and enzyme assays provide support for the claims of how A. kurodai avoids inhibition by algal compounds, and also hold promise for biotechnological applications.
https://doi.org/10.7554/eLife.88939.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Convincing: Appropriate and validated methodology in line with current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The defensive–offensive associations between algae and herbivores determine marine ecology. Brown algae utilize phlorotannin as their chemical defense against the predator Aplysia kurodai, which uses β-glucosidase (akuBGL) to digest the laminarin in algae into glucose. Moreover, A. kurodai employs Eisenia hydrolysis-enhancing protein (EHEP) as an offense to protect akuBGL activity from phlorotannin inhibition by precipitating phlorotannin. To underpin the molecular mechanism of this digestive–defensive–offensive system, we determined the structures of the apo and tannic acid (TNA, a phlorotannin analog) bound forms of EHEP, as well as the apo akuBGL. EHEP consisted of three peritrophin-A domains arranged in a triangular shape and bound TNA in the center without significant conformational changes. Structural comparison between EHEP and EHEP–TNA led us to find that EHEP can be resolubilized from phlorotannin precipitation at an alkaline pH, which reflects a requirement in the digestive tract. akuBGL contained two GH1 domains, only one of which conserved the active site. Combining docking analysis, we propose the mechanisms by which phlorotannin inhibits akuBGL by occupying the substrate-binding pocket, and EHEP protects akuBGL against this inhibition by binding with phlorotannin to free the akuBGL pocket.
Introduction
Over millions of years of evolution, predators have successfully coevolved with their prey to maintain an ecological balance (Becklin, 2008). In marine habitats, interactions between algae and marine herbivores dominate marine ecosystems. Most algae are consumed by marine herbivores (Jormalainen and Honkanen, 2008). They produce secondary metabolites as a chemical defense to protect themselves against predators. In brown algae Eisenia bicyclis, laminarin is a major storage carbohydrate, constituting 20–30% of algae dry weight. The sea hare Aplysia kurodai, a marine gastropod, preferentially feeds on E. bicyclis with its 110 and 210 kDa β-glucosidases (akuBGLs), hydrolyzing the laminarin and releasing large amounts of glucose. Interestingly, such a feeding strategy has attracted attention for producing glucose as a renewable biofuel source (Enquist-Newman et al., 2014). However, to protect themselves against predators, brown algae produce phlorotannin, a secondary metabolite, thereby reducing the digestion by A. kurodai by inhibiting the hydrolytic activity of akuBGLs. As the 110 kDa akuBGL is more sensitive to phlorotannin than the 210 kDa BGL (Tsuji et al., 2017), we focused on the 110 kDa akuBGL in this study (hereafter, akuBGL refers to 110 kDa akuBGL).
To counteract the antipredator adaptations of algae, herbivores use diverse approaches, such as detoxification, neutralization, defense suppression, and physiological adaptations (Erb and Reymond, 2019). A. kurodai inhibits the phlorotannin defense of brown algae through Eisenia hydrolysis-enhancing protein (EHEP), a protein from their digestive system that protects akuBGL activity from phlorotannin inhibition (Tsuji et al., 2017). Previous studies have shown that incubating E. bicyclis with akuBGL in the presence of EHEP results in increased glucose production because EHEP binds to phlorotannin and forms an insoluble complex (Tsuji et al., 2017).
The akuBGL–phlorotannin/laminarin–EHEP system exemplifies the digestion process of A. kurodai as well as the defense and antidefense strategies between E. bicyclis and A. kurodai. Although the defense/antidefense strategy has been established, the detailed molecular mechanism of this interplay remains unknown. Furthermore, phlorotannin inhibition hinders the potential application of brown algae as feedstocks for enzymatically producing biofuel from laminarin. Thus, understanding the underlying molecular mechanisms will be beneficial for the application of this system in the biofuel industry.
Despite the potential use of laminarin hydrolytic enzymes in the biofuel industry, only a few BGLs of glycoside hydrolases belonging to the GH3 and GH1 family are known to hydrolyze laminarin (e.g., Talaromyces amestolkiae BGL (Méndez-Líter et al., 2018), Ustilago esculenta BGL (Nakajima et al., 2012), and Vibrio campbellii BGL (Wang et al., 2015) from the GH3 family and Saccharophagus degradans 2-40T BGL (Kim et al., 2018) from the GH1 family). GH3 is a multidomain enzyme family characterized by N-terminal (β/α)8 (NTD) and C-terminal (β/α)6 (CTD) domains, with or without auxiliary domains (Mohsin et al., 2019); the nucleophile aspartate and the acid/base glutamate residues exist in the NTD and CTD, respectively. In contrast, the members of the GH1 family generally share a single (β/α)8-fold domain (hereafter referred to as the GH1 domain [GH1D]), and the two glutamic acid catalytic residues are located in the carboxyl termini of β-strands 4 and 7. Therefore, the two families may use different substrate recognition and catalytic mechanisms for laminarin. Intriguingly, although akuBGL possesses laminarin hydrolytic activity and belongs to the GH1 family, its molecular weight is considerably higher than that of other GH1 members. Sequence analysis has indicated that akuBGL consists of ≥2 GH1Ds. Because no structural information of BGL active on polysaccharides is available, the catalytic mechanism toward laminarin remains unclear.
There is limited information on EHEP, a novel cysteine-rich protein (8.2% of the amino acid content), because no structurally or functionally homologous protein exists in other organisms. EHEP was predicted to consist of three peritrophin-A domains (PADs) with a cysteine-spacing pattern of CX15CX5CX9CX12CX5–9C. The PADs consist of peritrophic matrix proteins, which have been proposed to play an important role in detoxifying ingested xenobiotics (Hegedus et al., 2009). For instance, Aedes aegypti intestinal mucin 1 (AeIMUC1) consists of a signal peptide followed by three PADs with an intervening mucin-like domain; its expression is induced by blood feeding. AeIMUC1-mediated blood detoxification during digestion is completed by binding to toxic heme molecules (Devenport et al., 2006). Despite the similar domain organization of EHEP and AeIMUC1, their functions and binding partners are different, implying their different characteristics. However, the characteristics of the EHEP–phlorotannin insoluble complex remain unknown; moreover, it remains unclear why and how EHEP protects akuBGL from phlorotannin inhibition.
In this study, we determined the structures of the apo and tannic acid (TNA, phlorotannin analog) bound forms of EHEP (EHEP, EHEP–TNA), as well as akuBGL, all isolated from A. kurodai. The structure of EHEP consists of three PADs arranged in a triangular shape, with TNA bound at the surface of the triangle center. A structural comparison of EHEP and EHEP–TNA revealed no significant changes in conformation upon TNA binding, implying that EHEP maintains its structure when precipitated with TNA. Then, we found the conditions to resolubilize EHEP–TNA precipitate for EHEP recycling. The obtained akuBGL structure suggests that only one GH1D (GH1D2) possesses laminarin hydrolytic activity; subsequently, ligand-docking experiments demonstrated that TNA/phlorotannin has a higher docking score than laminarin. Our results revealed the mechanisms by which EHEP protects akuBGL from phlorotannin inhibition and how phlorotannin inhibits the hydrolytic activity of akuBGL, providing structural support for the potential application of brown algae in biofuel production.
Results
Effects of TNA on akuBGL activity with or without EHEP
Phlorotannin, a type of tannin, is a chemical defense metabolite of brown algae. It is difficult to isolate a compound from phlorotannins because they are a group of polyphenolic compounds with different sizes and varying numbers of phloroglucinol units (Cassani et al., 2020), such as eckol, dieckol, and so on (Imbs and Zvyagintseva, 2018). Previous studies have reported that the phlorotannin-analog TNA has a comparable inhibitory effect on akuBGL to phlorotannin (Tsuji et al., 2017). Hence, we used TNA instead of phlorotannin to explore phlorotannin binding with EHEP and akuBGL. This activity assay system involves multiple equilibration processes: akuBGL ⇋ substrate, akuBGL ⇋ TNA, and EHEP ⇋ TNA. First, we confirmed TNA inhibition of akuBGL activity and clarified the protective effects of EHEP from TNA inhibition. The inhibition experiments showed that the galactoside hydrolytic activity of akuBGL decreased with increasing TNA concentration, indicating that TNA inhibits akuBGL activity in a dose-dependent manner (Figure 1A, Figure 1—figure supplement 1A). Approximately 70% akuBGL activity was inhibited at a TNA concentration of 40 μM. Moreover, protection ability analysis revealed that EHEP protects akuBGL activity from TNA inhibition in a dose-dependent manner, as indicated by the recovery of the inhibited akuBGL activity with increasing EHEP concentration (Figure 1B, Figure 1—figure supplement 1B). Approximately 80% of akuBGL activity was recovered at an EHEP concentration of 3.36 μM.
Figure 1 with 1 supplement see all
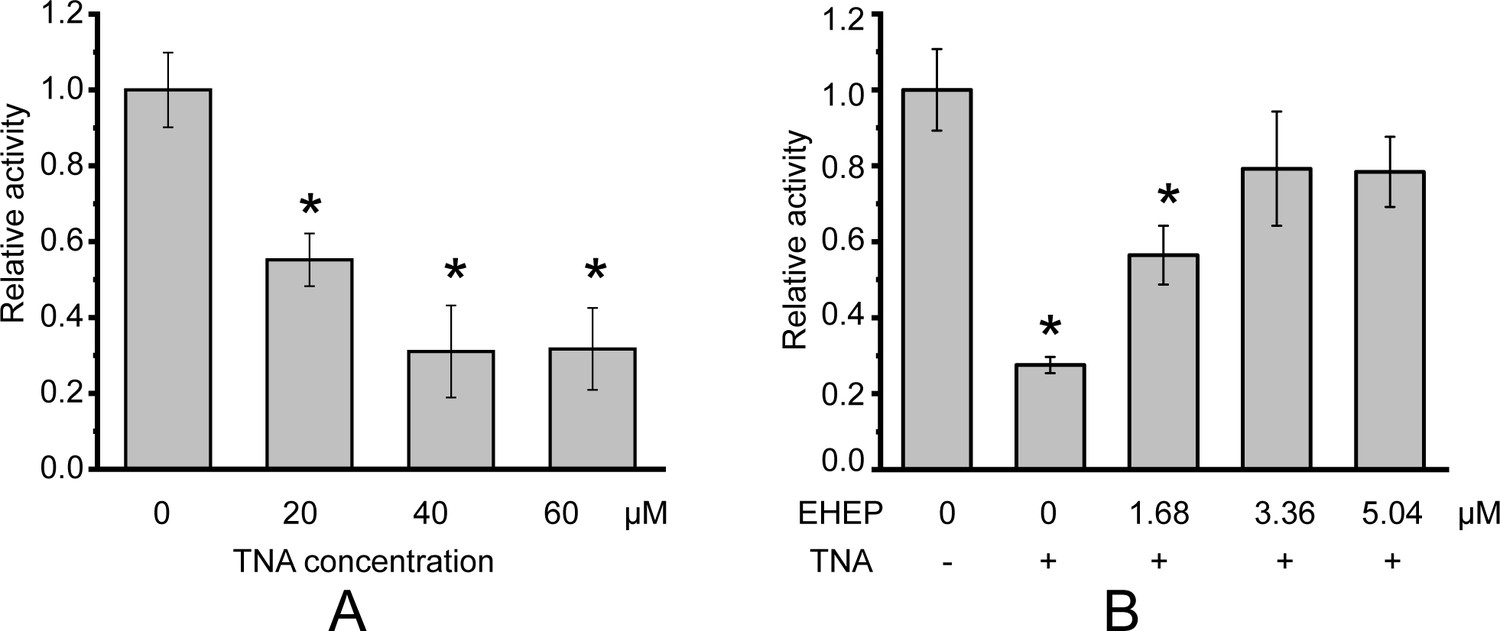
Galactoside hydrolytic activity of akuBGL toward 2.5 mM ortho-nitrophenyl-β-galactoside.
Activity (%) is shown as the fold increase relative to akuBGL without the addition of tannic acid (TNA) or Eisenia hydrolysis-enhancing protein (EHEP). (A) The hydrolytic activity of akuBGL (0.049 μM) with TNA at different concentrations. (B) The hydrolytic activity of akuBGL (0.049 μM) with 40 μM TNA and EHEP at different concentrations. The average and standard deviation of the relative activity were estimated from three independent replicates (N = 3). Asterisks in the top of error bars indicated significant (p<0.005) differences by t-test in comparison with 0 μM TNA (A) or 0 μM EHEP and no TNA (B).
Overall structure of EHEP
Considering the lack of known homologous proteins of EHEP, we determined the structure of EHEP using the native-SAD method at a resolution of 1.15 Å, with an Rwork and Rfree of 0.18 and 0.19, respectively (Table 1). The residues A21–V227 in purified EHEP (1–20 aa were cleaved during maturation) were built, whereas two C-terminal residues were disordered. The structure of EHEP consists of three PADs: PAD1 (N24–C79), PAD2 (I92–C146), and PAD3 (F164–C221), which are linked by two long loops, LL1 (Q80–N91) and LL2 (R147– G163), and arranged in a triangular shape (Figure 2A). These three PADs share a similar structure, with a root-mean-squared difference (RMSD) of 1.065 Å over 46 Cα atoms and only ~20.3% sequence identity (Figure 2B, C). The three PADs share a canonical CBM14 fold consisting of two β-sheets containing three N-terminal and two C-terminal antiparallel β-strands. Additionally, two small α-helices were appended to the N- and C-terminus in PAD1 and PAD3 but not in PAD2 (Figure 2B).
Table 1
X-ray data collection and structure-refinement statistics.
| EHEP1 | EHEP2 | EHEP–TNA | akuBGL | |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | PF BL17A | Spring 8 BL-41XU | PF BL17A | PF BL1A |
| Wavelength (Å) | 0.9800 | 1.0000 | 0.9800 | 1.0000 |
| Resolution range (Å) | 46.56–1.15 (1.20–1.15) | 47.02–1.4 (1.45–1.4) | 46.84–1.9 (1.97–1.9) | 49.65–2.7 (2.80–2.7) |
| Space group | P212121 | P212121 | P212121 | P62 |
| Unit-cell parameters a, b, c (Å) | 42.2, 65.3, 66.5 | 40.6, 65.6, 67.5 | 42.5, 65.4, 67.2 | 191.7, 191.7, 112.6 |
| Completeness (%) | 93.4 (75.8) | 99.30 (97.98) | 99.9 (99.7) | 99.9 (99.1) |
| Redundancy | 6.6 (6.1) | 6.4 (5.8) | 6.4 (6.4) | 10.7 (10.9) |
| Average I/σ(I) | 19.28 (1.97) | 14.34 (3.41) | 15.39 (1.83) | 10.69 (2.57) |
| Rmeas(%)* | 7.3 (90.5) | 8.6 73.7 (55.3) | 8.9 (89.4) | 19.4 (83.0) |
| CC1/2 (%) | 99.9 (70.4) | 99.8 (86.2) | 99.9 (73.7) | 99.5 (84.2) |
| Molecules/asymmetric unit | 1 | 1 | 1 | 2 |
| Refinement | ||||
| Rwork†/Rfree‡ (%) | 18.19/18.91 | 16.57/18.39 | 19.87/23.54 | 18.39/21.98 |
| No. of atoms | 1955 | 1861 | 1732 | 15,595 |
| No. of residues | 1600 | 1573 | 1580 | 15,256 |
| No. of water molecules | 343 | 273 | 87 | 96 |
| No. of ligands | 12 | 15 | 67 | 243 |
| RMSD from ideality | ||||
| Bond length (Å) | 0.005 | 0.006 | 0.008 | 0.004 |
| Bond angle (°) | 0.84 | 0.84 | 0.91 | 0.65 |
| Ramachandran plot (%) | ||||
| Favored | 99.02 | 98.54 | 98.52 | 96.16 |
| Allowed | 0.98 | 1.46 | 1.48 | 3.74 |
| Outliers | 0.00 | 0.00 | 0.00 | 0.11 |
| PDB accession code | 8IN3 | 8IN4 | 8IN6 | 8IN1 |
-
The highest resolution shell is shown in parentheses.
-
*
Rmeas = Σhkl{N(hkl)/[N(hkl) − 1]}1/2 Σi|Ii(hkl) − <I(hkl)> |/ΣhklΣi|Ii(hkl), where Ii(hkl) is the ith observation of the intensity of reflection hkl and 〈I(hkl)〉 is the mean over n observations.
-
†
Rwork = Σhkl||Fobs(hkl)| − |Fcalc(hkl)||/Σhkl|Fobs(hkl)|.
-
‡
Rfree was calculated with an approximate 5% fraction of randomly selected reflections evaluated from refinement.
Figure 2 with 1 supplement see all
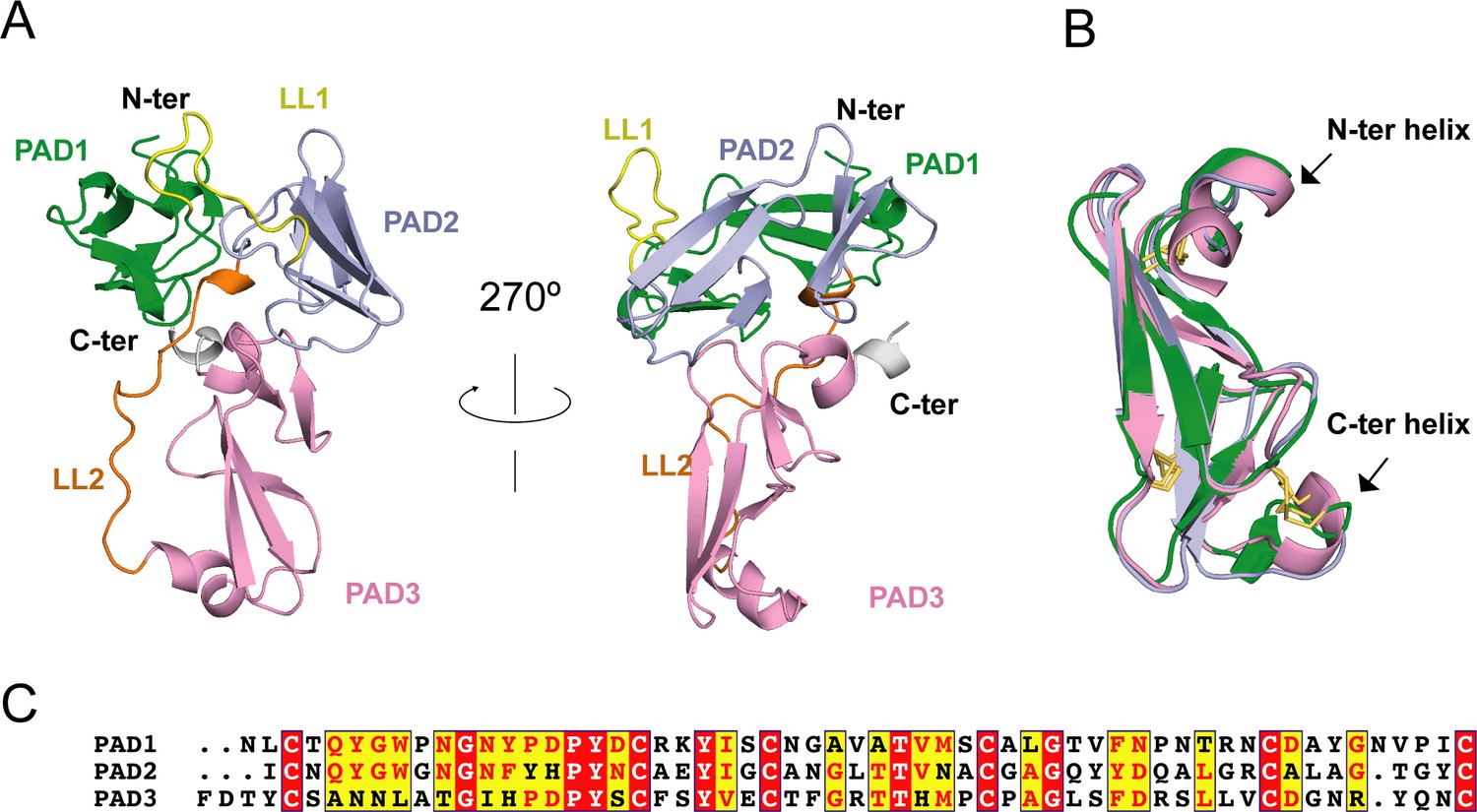
Eisenia hydrolysis-enhancing protein (EHEP) structure.
(A) Cartoon representation of EHEP. The three peritrophin-A domains (PADs) are colored green, light blue, and pink, respectively. Linker long loop1 (LL1) and loop2 (LL2) are colored yellow and blue. (B) Structural superposition of the three PAD domains of EHEP. The three domains are colored as in (A). The disulfide bonds are shown as yellow sticks. (C) Sequence alignment of three PAD domains. Alignment was performed by CLUSTALW and displayed with ESPript3.
Although the Dali server (Holm and Rosenström, 2010) did not provide similar structures using the overall structure of EHEP as the search model, six structures showed similarities with a single PAD of EHEP. These structures were the members of the PAD family, including the chitin-binding domain of chitotriosidase (PDB ID 5HBF) (Fadel et al., 2016), avirulence protein 4 from Pseudocercospora fuligena [PfAvr4 (PDB ID 4Z4A)] (Kohler et al., 2016) and Cladosporium fulvum [CfAvr4 (PDBID 6BN0)] (Hurlburt et al., 2018), allergen Der p 23 (PDB ID 4ZCE) (Mueller et al., 2016), tachytitin (PDB ID 1DQC) (Suetake et al., 2000), and allergen Blot 12 (PDB ID 2MFK), with Z-scores of 4.7–8.4, RMSD values of 1.2–2.8 Å, and sequence identity of 19–37%. The highest sequence disparity was detected in PAD2, whereas the greatest structural differences were noted in PAD3. The CNo1X15CNo2X5CNo3X9CNo4X12CNo5X5–9CNo6 motif (superscripts and subscripts indicate the cysteine number and number of residues between adjacent cysteines, respectively) in each PAD of EHEP formed three disulfide bonds between the following pairs: CNo1–CNo3, CNo2–CNo6, and CNo4–CNo5 (Figure 2B). Such rich disulfide bonds may play a folding role in the structural formation of EHEP, with >70% of the backbone in a loop conformation. A similar motif with disulfide bonds was observed in tachycitin (Suetake et al., 2000), PfAvr4 (Kohler et al., 2016), CfAvr4 (Hurlburt et al., 2018), and the chitin-binding domain of chitinase (Fadel et al., 2016). Although these proteins share a highly conserved core structure, they have different biochemical characteristics. For example, the chitin-binding domain of human chitotriosidase, Avr4, and tachycitin possess chitin-binding activity, but the critical residues for chitin binding are not conserved (Fadel et al., 2016; Hurlburt et al., 2018; Madland et al., 2019), indicating that they employ different binding mechanisms. In contrast, EHEP and allergen Der p 23 do not possess chitin-binding activity (Tsuji et al., 2017; Mueller et al., 2016). Thus, the PAD family may participate in several biochemical functions.
Modification of EHEP
Consistent with the molecular weight results obtained using MALDI–TOF MS (Sun et al., 2020), the apo structure2 (1.4 Å resolution) clearly showed that the cleaved N-terminus of Ala21 underwent acetylation (Figure 2—figure supplement 1A), demonstrating that EHEP is acetylated in A. kurodai digestive fluid. N-terminal acetylation is a common modification in eukaryotic proteins. Such acetylation is associated with various biological functions, such as protein half-life regulation, protein secretion, protein–protein interaction, protein–lipid interaction (Silva and Martinho, 2015), metabolism, and apoptosis (Hollebeke et al., 2012). Furthermore, N-terminal acetylation may stabilize proteins (Lange and Overall, 2011). To explore whether acetylation affects the protective effects of EHEP on akuBGL, we used the E. coli expression system to obtain the unmodified recomEHEP (A21–K229). We measured the TNA-precipitating assay of recomEHEP. The results revealed that recomEHEP precipitated after incubation with TNA at a comparable level to that of natural EHEP (Figure 2—figure supplement 1B), indicating that acetylation is not indispensable for the phlorotannin-binding activity and stabilization of EHEP. Future studies are warranted to verify the exact role of N-terminal acetylation of EHEP in A. kurodai.
TNA binding to EHEP
To understand the mechanism by which TNA binds to EHEP, we determined the structure of EHEP complexed with TNA (EHEP–TNA) using the soaking method. In the obtained structure, both 2Fo–Fc and Fo–Fc maps showed the electron density of 1,2,3,4,6-pentagalloylglucose, a core part of TNA missing the five external gallic acids (Figure 3A, Figure 3—figure supplement 1). Previous studies have shown that acid catalytic hydrolysis of TNA requires a high temperature of 130°C (Jie Fu et al., 2015); even with a polystyrene-hollow sphere catalyst, a temperature of 80°C is required (Luo et al., 2018). Therefore, the five gallic acids could not be visualized in the EHEP–TNA structure, most likely due to the structural flexibility of TNA.
Figure 3 with 1 supplement see all
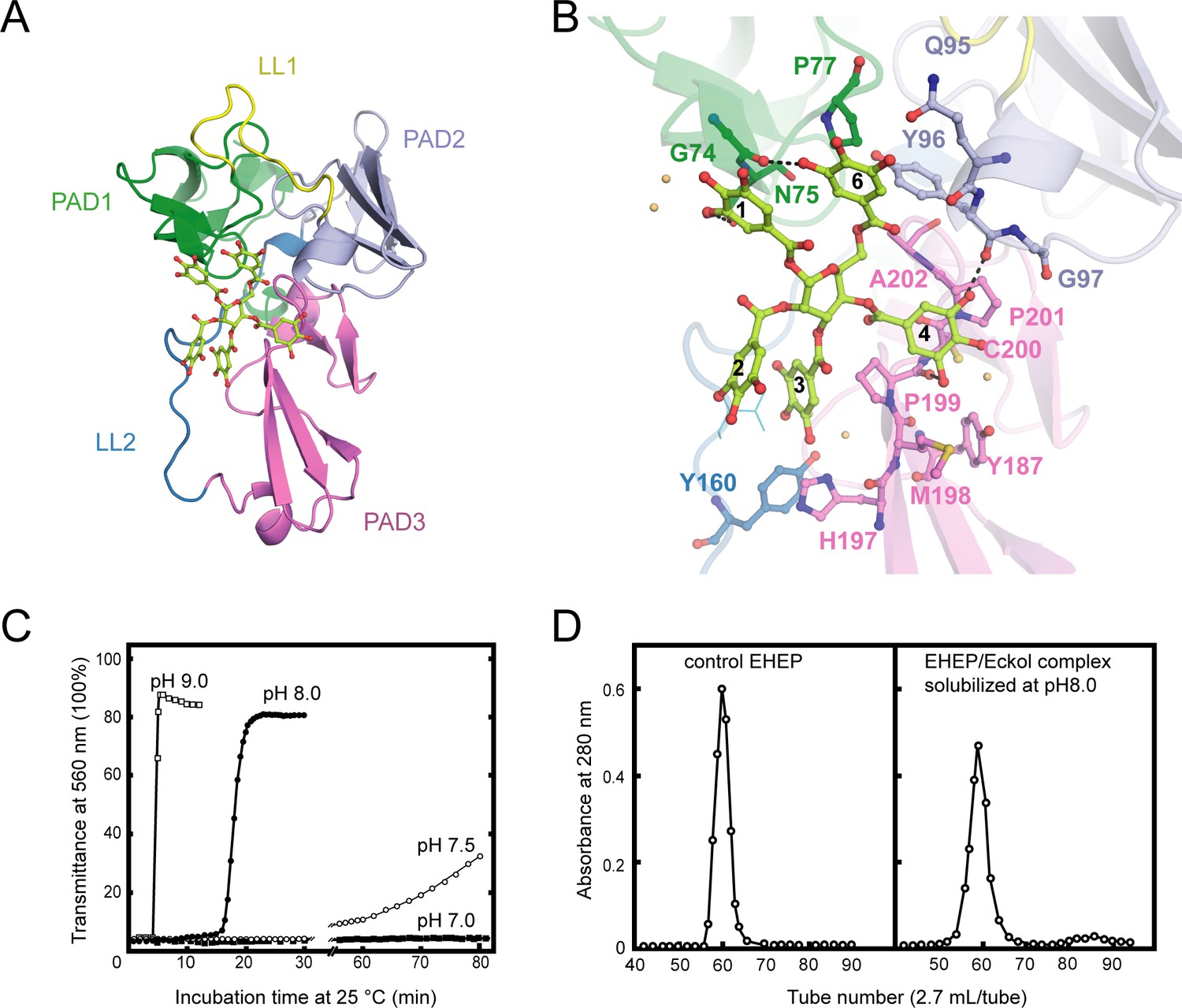
Structure of Eisenia hydrolysis-enhancing protein (EHEP)–tannic acid (TNA).
(A) The overall structure of EHEP–TNA. (A) The overall structure of EHEP–TNA. EHEP and TNA are shown by the cartoon and stick model, respectively. EHEP is colored as in Figure 1. The C and O atoms of TNA are colored lemon and red, respectively. (B) Interaction of TNA (ball-stick in the same color as (A)) with EHEP (cartoon in the same color as (A)) in EHEP–TNA structure. The residues of EHEP in contact are labeled and shown by a ball-stick with N, O, and S atoms in blue, red, and brown, respectively. The C and O atoms of TNA are colored the same as (A), lemon, and red, respectively. Dashed lines show hydrogen bonds. The water molecules stabilizing TNA were shown as light orange spheres. (C) Effect of pH on resolubilization of an EHEP–eckol precipitate. Buffers with pH 9.0, 8.0, 7.5, and 7.0 are presented as hollow square, solid circle, hollow circle, and solid square, respectively. (D) The EHEP–eckol precipitate was dissolved in 50 mM Tris–HCl (pH 8.0) and analyzed using a gel filtration column of Sephacryl S-100.
The apo EHEP and EHEP–TNA structures were highly similar, with an RMSD value of 0.283 Å for 207 Cα atoms (Figure 3—figure supplement 1B). However, the superposition of the two structures showed a decrease in the loop part of EHEP–TNA. TNA binding caused a slight increase in the α-helix and β-sheet contents of PAD2 and PAD3 (Figure 3—figure supplement 1B). In the EHEP–TNA structure, the residues C93–Y96 of PAD2 folded into an α-helix and each β-sheet of the first β-strand in PAD3 elongated by incorporating one residue in the first (G176) and second β-sheets (S186) and three residues in the third β-sheet (H197MP199).
The EHEP–TNA structure revealed that TNA binds to the center of the triangle formed by the three PADs, a positively charged surface (Figure 3A and Figure 3—figure supplement 1C). The binding pocket on the EHEP surface was formed by the C-terminal α-helix of PAD1, the N-terminal α-helix of PAD2, and the middle part (loop) of PAD3 assisted by two long linker loops (LL1 and LL2). TNA was primarily bound to EHEP via hydrogen bonds and hydrophobic interactions (Figure 3B). Gallic acid1, 4, and 6 interacted with EHEP via hydrogen bonds and additional hydrophobic contacts, whereas gallic acid2 and 3 only interacted hydrophobically with EHEP. The 3-hydroxyl groups of gallic acid1 and 6 individually formed a hydrogen bond with the main chain of G74 and the side chain of N75 in PAD1. The main chain of Y96 and P199 in PAD2 and PAD3 formed hydrogen bonds with the gallic acid4. Additionally, some hydrogen bonds were formed between TNA and water molecules. TNA binding was also stabilized by hydrophobic interactions between the benzene rings of gallic acid and EHEP. For instance, gallic acid4 and 6 are stacked with P201 and P77, respectively; moreover, gallic acid3 and 4 are stacked with P199.
The EHEP–TNA structure clearly showed that TNA binds to EHEP without covalent bonds, and the binding does not induce significant structural changes; thus, we attempted to recover EHEP from EHEP–TNA precipitates by adjusting the pH. As hypothesized, resolubilization of the EHEP–phlorotannin precipitate is pH dependent (Figure 3C). The EHEP–TNA precipitate did not resolubilize at pH 7.0; however, after incubating for >1 hr at pH 7.5, the precipitate started resolubilizing. Most of the precipitate rapidly resolubilized at an alkaline pH (≥8.0) after incubation for 15 min. Furthermore, the resolubilized EHEP had the same elution profile as that of the natural EHEP (Figure 3D) in SEC, suggesting that resolubilized EHEP maintained the native structure and its phlorotannin-precipitate activity (Figure 3—figure supplement 1D).
Two domains of akuBGL
To reveal the structural basis of akuBGL recognition of laminarin and its inhibition by TNA, we attempted to determine its structure. We soaked crystals in TNA as well as various substrate solutions but finally obtained the optimal resolution using crystal soaking in TNA. There was no electron density of TNA or something similar in the 2Fo–Fc and Fo–Fc map of the obtained structure; thus, we considered this structure as the apo form of akuBGL.
Two akuBGL molecules were observed in an asymmetric unit (MolA and MolB). These molecules lacked the N-terminal 25 residues (M1–D25), as confirmed by N-terminal sequencing analysis of purified natural akuBGL. This N-terminal fragment was predicted to be a signal peptide using the web server SignalP-5.0. The residues L26–P978 were constructed in MolA and MolB with glycosylation, whereas the remaining C-terminal residues (A979–M994) could not be visualized as they were disordered. The electron density map of Fo–Fc revealed N-glycosylation at three residues, that is, N113, N212, and N645 (Figure 4—figure supplement 1A, B). N-glycosylation of GH enzymes prevents proteolysis and increases thermal stability (Amore et al., 2017; Han et al., 2020b). Additionally, a study on β-glucosidase Aspergillus terreus BGL demonstrated that N-glycosylation of N224 affected the folding stability, even when it is located close to a catalytic residue (Wei et al., 2013). In akuBGL, all N-glycosylation sites were present on the surface, far from the catalytic site. Therefore, we speculate that akuBGL glycosylation does not affect its activity. Except for the difference in visualized glycans resulting from glycosylation, MolA and MolB were similar, with an RMSD value of 0.182 for 899 Cα atoms; therefore, we used MolA for further descriptions and calculations.
The structure of akuBGL consisted of two independent GH1 domains, GH1D1 (L26–T494) and GH1D2 (D513–P978), linked by a long loop (D495–Y512) (Figure 4A). There was little interaction between GH1D1 and GH1D2, only in a buried surface area comprising 2% of the total surface (708.9 Å2) (Figure 4—figure supplement 1C). GH1D1 and GH1D2 have a sequence identity of 40.47% and exhibit high structural similarity with an RMSD value of 0.59 Å for 371 Cα atoms (Figure 4—figure supplement 2A, upper panel).
Figure 4 with 3 supplements see all
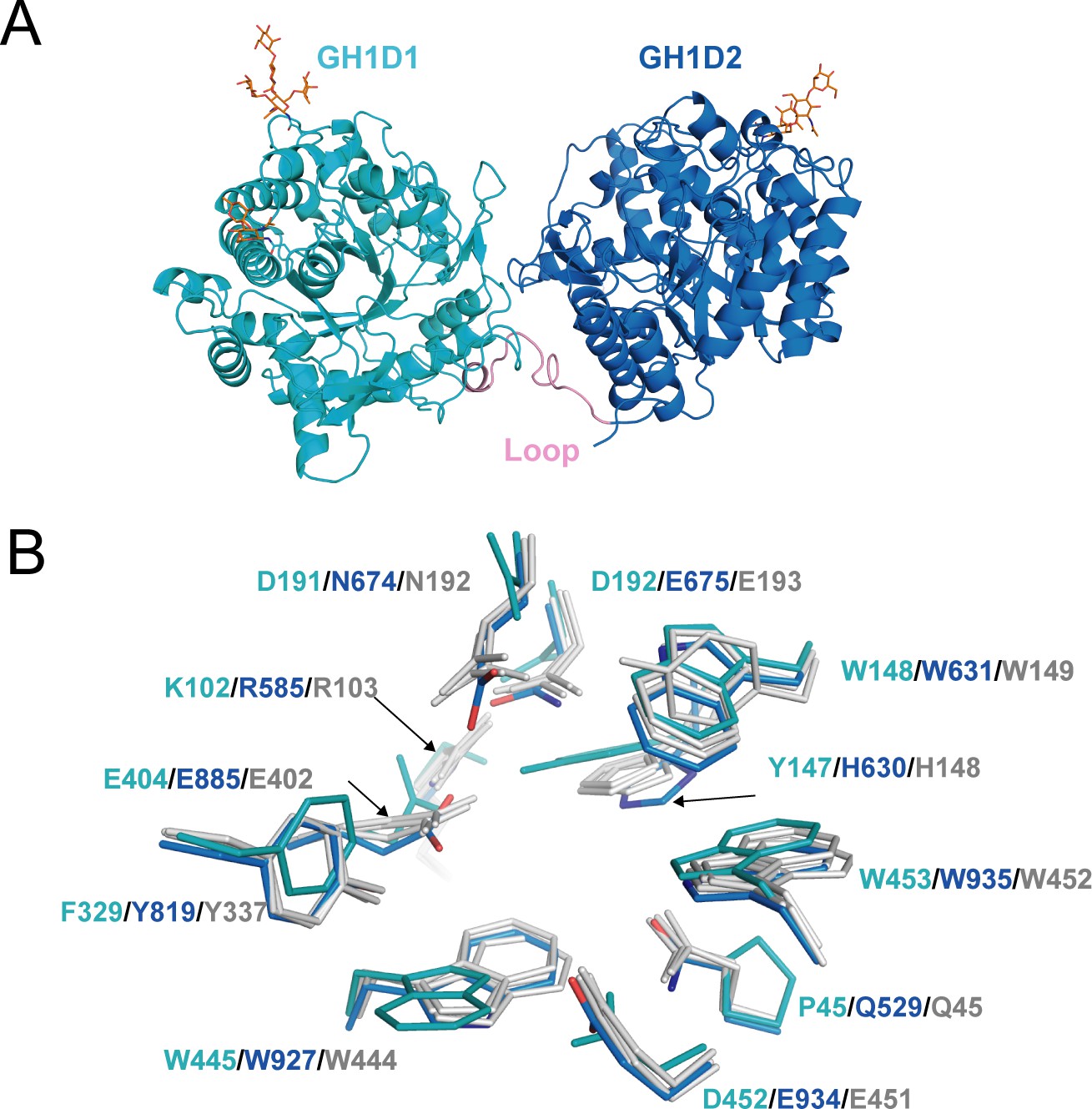
Structure of akuBGL.
(A) Overall structure. The GH1D1 (light blue) and GH1D2 (cyan) domains are linked by a long loop (linker loop) colored in pink. The N-linked glycans were shown in the orange stick. (B) Residues superposition of the glycone-binding site (GBS) and catalysis-related residue (CR) site of the domains GH1D1 (cyan), GH1D2 (light blue) with β-glucosidase structures from termite Neotermes koshunensis (NkBGL, gray PDB ID 3VIH) (Jeng et al., 2012), β-glucosidase from rice (OsBGL, gray, PDB ID 2RGL) (Chuenchor et al., 2008), and β-glucosidase from Bacillus circulans sp. Alkalophilus (gray, PDB ID 1QOX) (Hakulinen et al., 2000). Only the residue numbers of GH1D1 (cyan), GH1D2 (light blue), and NkBGL (gray) are shown for clarity.
Glucosidases of the GH1 family utilize a retaining mechanism with two glutamic acids to catalyze glucoside hydrolysis. In general, the distance between the two catalytic oxygen atoms of the side chain of two glutamic acids is approximately 5 Å (Hayashi et al., 2007). Sequence and structure alignment of GH1D1 and GH1D2 of akuBGL with other members of the GH1 family revealed that the second glutamate is conserved (E404), but the first glutamate is replaced by D192 in GH1D1. The oxygen atoms of the side chains between D192 and E404 of GHD1 were 8.4 Å apart. In contrast, GH1D2 conserved two glutamic acids (E675 and E885) at the carboxyl termini of β-strands 4 and 7; the distance between the oxygen atoms of the E675 and E885 side chains was 5.1 Å (Figure 4—figure supplement 2A bottom panel), similar to that of Neotermes koshunensis BGL (NkBGL, PDB ID 3VIH) (Jeng et al., 2012), Nannochloropsis oceanica BGL (NoBGL, PDB ID 5YJ7) (Dong et al., 2021), and Spodoptera frugiperda BGL (PDB ID 5CG0) (3.9–4.9 Å) (Tamaki et al., 2016). Furthermore, regarding the two other conserved essential regions for β-glucosidase activity, namely, the glycone-binding site (GBS) and catalysis-related residues (CR), GH1D1 conserved neither GBS nor CR, whereas GH1D2 conserved both (Figure 4B). Altogether, we suggest that GH1D1 does not possess catalytic activity. We expressed and purified the recombinant GH1D1, which did not show any hydrolytic activity toward O-PNG (Figure 4—figure supplement 1D, E), although we could not rule out the effect of N-glycosylation.
Structural comparison of GH1D2 with other BGLs, including NkBGL (PDB ID 3VIH) (Jeng et al., 2012), rice (Oryza sativa L.) BGL (OsBGL, PDB ID 2RGL) (Chuenchor et al., 2008), and microalgae NoBGL (PDB ID 5YJ7) (Dong et al., 2021), revealed the characteristics of each active pocket (Figure 4—figure supplement 2B). OsBGL and NoBGL have deep, narrow, and straight pockets, whereas GH1D2 and NkBGL have broad and crooked pockets. Such active pocket shapes reflect the substrate preferences of OsBGL and NoBGL; they hydrolyze laminaribiose with no detectable activity toward laminaritetraose (Dong et al., 2021; Opassiri et al., 2003). Furthermore, the difference in the features of large active pockets between NkBGL and GH1D2, wherein GH1D2 often possesses an auxiliary site with several aromatic residues bound to the carbohydrate via CH–π interactions (Hudson et al., 2015), may explain their substrate specificity. NkBGL efficiently hydrolyzes laminaribiose and cellobiose but has weak hydrolytic activity toward laminarin (Ni et al., 2007). In contrast, the GH1D2 of akuBGL has similar activity levels toward cellobiose and laminarin (Tsuji et al., 2013). Therefore, the GH1D2 of akuBGL may recognize larger substrates than that of other BGLs. Laminarin typically has a curved conformation; accordingly, narrow- and straight-shaped pockets are incompatible for binding. Furthermore, we docked GH1D2 with laminaritetraose, wherein the four glucose units formed extensive contacts with GH1D2. Hydrogen bonds are involved in the catalytic residues E675. In addition, several aromatic residues, such as W631, F677, W681, F689, Y819, Y846, W857, and W935, formed CH–π stacking interactions (Figure 4—figure supplement 3). Some interacting residues belonged to GBS and CR sites, such as E675, W631, Y819, and W935. Additionally, the docking structure revealed that the +3 and +4 glucose of laminaritetraose are located at the auxiliary binding site and that atom O1 of the +4 glucose is positioned outside the pocket (Figure 4—figure supplement 3), implying that the auxiliary binding site with several aromatic residues (F677 and W681) of GH1D2 facilitates laminarin binding.
Inhibitor binding of akuBGL
As we could not obtain the complex structure of akuBGL with TNA, we performed docking calculations of akuBGL GH1D2 with TNA to explore the inhibition mechanism. The docking model of akuBGL–TNA showed that seven gallic acid rings of TNA formed an extensive hydrogen bond network with akuBGL in the binding pocket (Figure 5). The hydroxyl groups of TNA formed hydrogen bonds with the residues N552, E675, D735, K739, K759, Q840, T844, D852, and K859 of GH1D2. Moreover, benzene rings showed hydrophobic interactions with several hydrophobic residues. In particular, stable π–π stacking was observed between TNA and residues F547, W631, F689, Y846, W857, and W935. Among these residues, the conserved E675 was the catalytic residue, and W631, W935, and E934 contributed to GBS and CR sites.
Figure 5 with 1 supplement see all
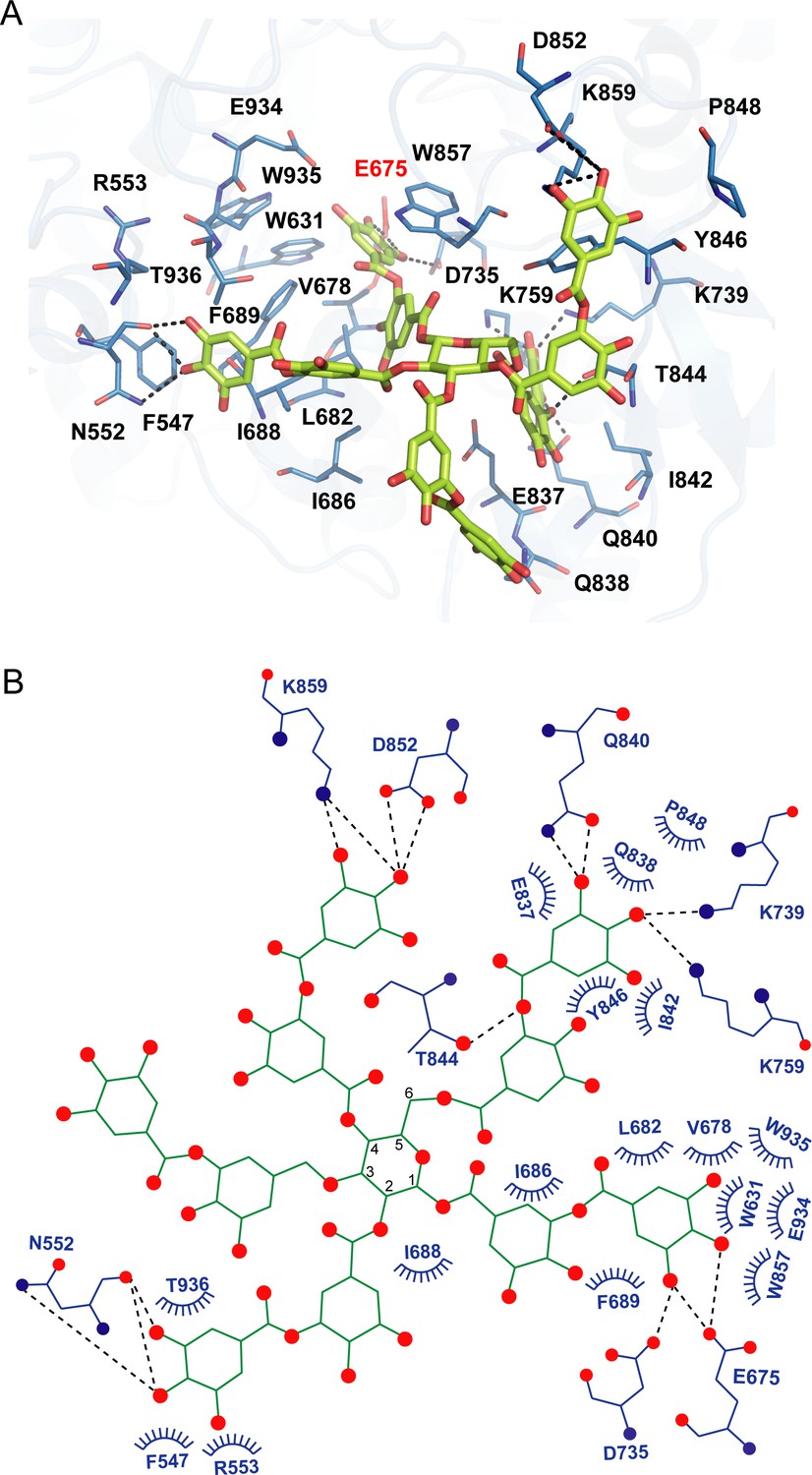
Docking model of akuBGL with tannic acid (TNA).
(A) Detailed interaction between akuBGL and TNA in the docking model. TNA is shown in the green stick model. The hydrogen bonds are shown as dashed lines. (B) A 2D diagram of the interaction between akuBGL and TNA shown in (A). The hydrogen bonds are shown as dashed lines, and the hydrophobic contacts as circular arcs.
In addition, we performed a docking calculation of GH1D2 with the characteristic inhibitors eckol and phloroglucinol (Jung et al., 2010). The binding mechanisms of eckol and phloroglucinol were similar to those of TNA but with different contact residues (Figure 5—figure supplement 1). For eckol, the six hydroxyl groups formed hydrogen bonds with residues E675, D735, E737, K759, E885, and E934. Additionally, residues W631, F677, F689, Y819, W857, W935, F943, and W927 formed π–π stacking interactions with eckol. For phloroglucinol, the three hydroxyl groups formed hydrogen bonds with E675, E885, and E934, whereas residues W631, F689, Y819, W857, W935, and W927 formed π–π stacking interactions with the benzene ring.
The docking scores of the inhibitors TNA, eckol, and phloroglucinol were −8.8, −7.3, and −5.7 kcal/mol, respectively, whereas the substrate laminaritetraose had a docking score of −6.6 kcal/mol. The docking scores corroborated well with the inhibition activity toward akuBGL, that TNA had a more robust inhibition activity than phloroglucinol (Tsuji et al., 2017), indicating that the docking results are reasonable. In summary, the three inhibitors interacted with akuBGL through similar binding mechanisms to occupy the substrate-binding site, suggesting a reversible competitive inhibition mechanism.
Discussion
In marine habitats, the ecological interactions between brown algae and herbivores dominate marine ecosystems (Amsler and Fairhead, 2005). The akuBGL–phlorotannin/laminarin–EHEP system represents the feeding defense–offense associations between A. kurodai and brown algae. We focused on this system to understand the molecular mechanism at the atomic level. In contrast to most GH1 BGLs containing one catalytic GH1 domain, akuBGL consists of a noncatalytic GH1D1 and a catalytic GH1D2. The noncatalytic GH1D1 may act as a chaperone for GH1D2, as we successfully overexpressed GH1D1 but failed to do the same for GH1D2. Such multi-GH1D assembly and a similar function have been suggested in β-glucosidase CjCEL1A of Corbicula japonica (Sakamoto et al., 2009) and glycosidase LpMDGH1 of the shipworm Lyrodus pedicellatus (Sabbadin et al., 2018). CjCEL1A has two tandem GH1Ds with a sequence identity of 43.41% (Sakamoto et al., 2009). Two catalytic glutamic acids and the residues related to substrate binding are conserved in the second GH1D, whereas the first GH1 domain lacks these conserved residues and may play a role in folding the catalytic domain. LpMDGH1 consists of six GH1Ds, among which GH1D2, 4, 5, and 6 contain the conserved residues for activity, whereas others do not contain these residues and might be involved in protein folding or substrate interactions (Sabbadin et al., 2018). Future assays of GH1D2 and its inactive mutants are the complement to validate the molecular mechanism of akuBGL.
BGLs have different substrate preferences in the degree of polymerization and type of glycosidic bond. In general, BGLs prefer to react with mono-oligo sugars over polysaccharides. For instance, OsBGL, NoBGL, and NkBGL hydrolyze disaccharides (cellobiose and laminaribiose) but display no or weak activity toward polysaccharides (cellulose and laminarin) (Dong et al., 2021; Opassiri et al., 2003; Ni et al., 2007). The structure of GH1D2 explained the substrate preference for the polysaccharide laminarin. GH1D2 contains an additional auxiliary site composed of aromatic residues (Figure 4—figure supplement 2B) in the substrate entrance pocket, which putatively enables it to accommodate a long substrate, contributing to akuBGL activity toward laminarin, as supported by docking calculations (Figure 4—figure supplement 3). In addition, docking analysis of akuBGL GH1D2 with inhibitors (TNA, phlorotannin, eckol, and phloroglucinol) revealed that these inhibitors bind to the substrate-binding site via hydrogen bonds and hydrophobic interactions similar to laminarin. Such binding mechanisms suggest the presence of competitive inhibition to occupy the binding site, consistent with previous research (Tsuji et al., 2017). Future kinetic experiments are required to quantitatively validate the competitive inhibition of phlorotannin against akuBGL.
EHEP, expressed in the midgut of A. kurodai, was identified as an antidefense protein, protecting the hydrolysis activity of akuBGL from phlorotannin inhibition (Tsuji et al., 2017). Such an ecological balance also exists between plants and their predatory mammals and insects. Similar to brown algae, plants use the toxic secondary metabolite tannins as their defense mechanism against predators, constituting 5%–10% of the dry weight of leaves. In vertebrate herbivores, tannins reduce protein digestion (Barbehenn and Peter Constabel, 2011). In phytophagous insects, tannins may be oxidized in the alkaline pH of the insect midgut and cause damage to cells (Marsh et al., 2020). The evolution of plant–herbivore survival competition has led to the development of remarkably unique adaptation strategies. Mammals feeding on plants that contain tannin may overcome this defense by producing tannin-binding proteins, proline-rich proteins, and histatins (Shimada, 2006; De Smet and Contreras, 2005). Proline constitutes at least 20% of the total amino acid content in proline-rich proteins; for some species, the proportion of proline reaches 40%. Histidine constitutes 25% of the total amino acid content in histatins. Both proline-rich proteins and histatins are unfolded proteins with random coils in solution. In caterpillars, the oxidation damage of tannin is reduced by the low oxygen level. Some insects use the peritrophic membrane to transport tannins into the hemolymph, where they are excreted (War et al., 2020). Additionally, the peritrophic envelop protects insects from tannins by forming an impermeable barrier to tannins (Barbehenn and Peter Constabel, 2011). A. kurodai uses a similar strategy to mammals by secreting the tannin-binding protein EHEP. Although EHEP has a completely different amino acid composition with proline-rich proteins and histatins, EHEP also binds to phlorotannin. Therefore, EHEP may be a specific counteradaptation that allows A. kurodai to feed on brown algae, as there are no homologous proteins in other organisms.
The three PADs of EHEP are arranged in a triangular shape, forming a large cavity on the surface at the triangle center to provide a ligand-binding site. EHEP has a positively charged surface at a pH of <6.0, whereas the surface becomes negatively charged at a pH of >7.0 (Figure 3—figure supplement 1C). Meanwhile, TNA has a pKa of 4.9–8 (Lin et al., 2009; Ge et al., 2019; Yi et al., 2011), showing minor negative charges at an acidic pH and the highest negative charge at a pH of >7.0 (Dultz et al., 2021). Therefore, TNA binds to EHEP at a pH of <6.0 (pH of crystallization = 4.5) but shows charge repulsion with EHEP at a pH of >8.0. Altogether, TNA is protonated and behaves as a hydrogen bond donor when the pH is below its pKa, whereas when the pH is above its pKa, TNA is deprotonated and the hydrogen bonding cannot be maintained. As losing hydrogen bonds and increasing repulsive forces at a pH >8.0, the precipitated EHEP–TNA could dissolve in the buffer of pH >8.0. This pH-induced reversible interaction also occurred in other proteins, such as BSA, pepsin, and cytochrome C (Han et al., 2020a). The phlorotannin members share a similar structure with TNA; thus, we speculate that the EHEP–phlorotannin complex also exhibits a pH-induced reversible interaction. In vivo, the pH of the digestive fluid of A. kurodai is approximately 5.5 (Tsuji et al., 2017), which favors the binding of EHEP to phlorotannin. In the alkaline hindgut (Lemke et al., 2003), the EHEP–phlorotannin disassociates (Figure 6), and the phlorotannin is subsequently excreted from the anus.
Figure 6
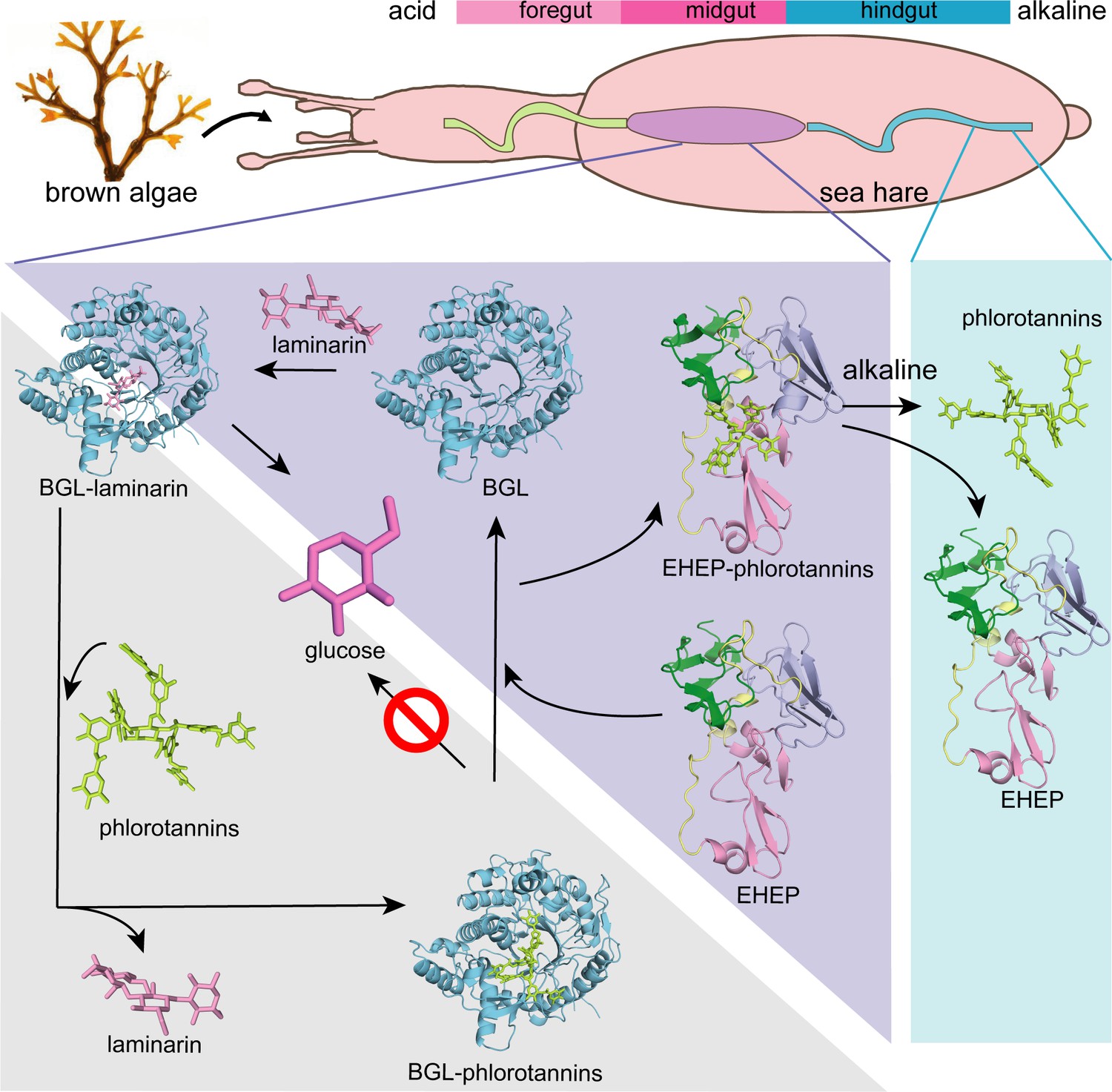
Proposed molecular mechanisms.
Proposed molecular mechanisms of tannic acid (TNA) inhibition of akuBGL activity and Eisenia hydrolysis-enhancing protein’s (EHEP) protective effects of akuBGL in akuBGL–phlorotannin/laminarin–EHEP system (light purple triangle). The digestive tract of A.kurodai consists of foregut (blue), midgut (purple), and hindgut (blue) (top). The bar chart above depicts the pH of the digestive tract, with pink denoting acid and blue denoting alkalinity.
Based on the EHEP–TNA structure and docking models of akuBGL–inhibitor/substrate, we proposed a mechanism of phlorotannin inhibition on akuBGL activity and EHEP protection from phlorotannin inhibition (Figure 6). Because laminarin lacks the benzene rings essentially to form CH–π stacking interactions with EHEP, the EHEP can be considered not bind with laminarin. In the absence of EHEP, phlorotannin occupies the substrate-binding site of akuBGL, inhibiting the substrate from entering the active site and resulting in no glucose production. In the presence of EHEP, it competitively binds to phlorotannin, freeing the akuBGL pocket. Then, the substrate can enter the active pocket of akuBGL, and glucose can be produced normally. The digestive fluid of A. kurodai contains EHEP at a high concentration (>4.4 µM) (Tsuji et al., 2017), which is slightly higher than the concentration of EHEP (3.36 µM) that protects akuBGL activity (Figure 1B). The high concentration of EHEP allows A. kurodai to feed on phlorotannin-rich brown algae. The balance between phlorotannin inhibition and protection is controlled by the concentrations of phlorotannin and EHEP in vivo. The kinetic analysis of the akuBGL–phlorotannin/laminarin–EHEP system will provide detailed reaction parameters.
The akuBGL–phlorotannin/laminarin–EHEP system represents the digestive–defensive–offensive associations between algae and herbivores. Our study presented the molecular mechanism of this system at the atomic level, providing a molecular explanation for how the sea hare A. kurodai utilizes EHEP to protect akuBGL activity from phlorotannin inhibition. Furthermore, such a feeding strategy has attracted attention for producing glucose as a renewable biofuel source, so our studies provide a molecular basis for the biofuel industry applications of brown algae.
Materials and methods
EHEP and akuBGL preparation
Request a detailed protocolNatural EHEP (22.5 kDa) and akuBGL (110 kDa) were purified from A. kurodai digestive fluid as previously described (Tsuji et al., 2017). For crystallization, we added one step for purification of EHEP by size-exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE, America) equilibrated in 20 mM MES [2-(4-morpholino) ethanesulfonic acid]–NaOH buffer (pH 6.5). Collected EHEP was then concentrated to 15–25 mg/ml using Vivaspin-4 10K columns (Sartorius, Göttingen, Germany). For the akuBGL, we exchanged the buffer from 20 mM Tris–HCl pH 7.0–20 mM Bis-Tris pH 6.0 with Amicon-Ultra (molecular weight cut-off of 50 kDa) and concentrated to 11 mg/ml.
To verify whether the chemical modification indicated by the previous study (Sun et al., 2020) affects the function of EHEP, we prepared recombinant EHEP (recomEHEP) without the N-terminal signal peptide (1–20 aa) and the chemical modification (Sun et al., 2020). EHEP cDNA was obtained via reverse transcription-polymerase chain reaction using the total RNA of A. kurodai as the template. The reamplified fragment was digested and ligated to a plasmid derived from pET28a (Novagen, Darmstadt, Germany). The primers used were shown in Supplementary file 1. The resulting plasmid encoding recomEHEP with an N-terminal hexahistidine-tag was transformed into E. coli B834(DE3) pARE2 cells. The cells were cultured in lysogeny broth (LB) medium with the antibiotics kanamycin (25 mg/l) and chloramphenicol (34 mg/l) until the optical density at 600 nm (OD600) reached 0.6. Subsequently, overexpression was induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranoside for 20 hr at 20 ℃. The cells were harvested by centrifugation at 4000 × g, resuspended in a buffer containing 50 mM Tris–HCl pH 7.4, 300 mM NaCl, DNase, and lysozyme, and disrupted by sonication. The insoluble part was removed by centrifugation at 40,000 × g for 30 min at 4°C. We loaded the supernatant onto a 5-ml HisTrap HP column (GE, America). The recomEHEP was eluted using increasing concentrations of imidazole (0–500 mM). The purified proteins were dialyzed against a solution containing 50 mM Tris–HCl pH 7.4 and 50 mM NaCl and subsequently loaded onto a HiTrap Q HP column (GE, America) and eluted by a linear gradient of a solution containing 50 mM Tris–HCl and 1 M NaCl. Fractions containing recomEHEP were concentrated and then purified using a gel filtration column (HiLoad 16/60 Superdex 75 pg) (GE, America) equilibrated with 20 mM sodium acetate pH 6.0 and 100 mM NaCl. We collected the fractions containing recomEHEP and concentrated them to 2.1 mg/ml using Amicon (Merck, America).
DNA encoding akuBGL without a signal peptide was codon optimized (GENEWIZ, China) for overexpression in E. coli. The cDNA encoding the GH1D1 domain was subcloned into a modified pET-32a vector (Invitrogen, America) with an N-terminal TrxA tag and a 6×His tag, followed by a TEV protease recognition site. The primers used were shown in Supplementary file 1. The plasmid pET-32a-GH1D1 was electorally transformed into E. coli origami2 cells. We grew the cells in LB medium supplemented with 100 µg/ml of ampicillin at 37°C until the OD600 reached 0.6. Then, the cultures were cooled and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside at 20°C for 16 hr. After resuspending the harvested cells in a buffer containing 50 mM Tris–HCl (pH 7.4), 250 mM NaCl, and 5% glycerol, we disrupted the cells by sonication. The cell lysate was centrifugated at 40,000 × g for 30 min at 4°C. The supernatant was filtered by 0.45 μm membrane and then loaded onto a HisTrap HP column (GE, America). After washing with lysis buffer supplemented with 20 mM imidazole, the recomGH1D1 was eluted linearly from the column with lysis buffer supplemented with 500 mM imidazole. The fractions containing recomGH1D1 were added with TEV protease at a ratio of 1:10 and dialyzed against a buffer containing 50 mM Tris (pH 7.4), 50 mM NaCl, and 2 mM DTT at 4°C overnight. Then, we purified the recomGH1D1 by a HisTrap HP column again (GE, America) and collected the flowthrough. Finally, we purified the recomGH1D1 by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 pg column (GE, America) equilibrated with a buffer containing 20 mM Bis-Tris (pH 6.0). The recomGH1D1 was concentrated to 1.5 mg/ml by centrifugation using Amicon-Ultra (molecular weight cut-off of 10 kDa).
N-terminal sequencing of akuBGL
Request a detailed protocolWe performed an N-terminal sequencing of purified akuBGL using the Edman degradation method. akuBGL was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), followed by electrophoretic transfer onto a PVDF (polyvinylidene fluoride) membrane (GE, America). The membrane was subsequently stained with Ponceau S solution. The band corresponding to akuBGL was excised and analyzed using a PPSQ-53A Protein sequencer (Shimadzu, Japan) at the Instrumental Analysis Service of Hokkaido University.
Effects of TNA on akuBGL activity with or without EHEP
Request a detailed protocolDue to the yellow color of TNA, which affects absorbance at 420 nm of the reaction product o-nitrophenol of akuBGL, we used high-performance liquid chromatography (HPLC) to measure the akuBGL activity in the reaction system containing TNA. Ortho-nitrophenyl-β-galactoside (ONPG) was used as a substrate to measure akuBGL activity. The reaction system (100 μl) included 2.5 mM ONPG, 49 nM akuBGL, and different TNA concentrations (0, 20, 40, and 60 μM) in a reaction buffer (50 mM CH3COONa pH 5.5, 100 mM NaCl, and 10 mM CaCl2). After incubation for 10 min at 37°C, 100 μl of methanol was added to each sample to terminate the reaction. Then, the mixture was centrifuged for 10 min at 15,000 × g at 4°C and the supernatant was used for analyzing akuBGL activity via HPLC. To measure the protective effect of EHEP on akuBGL, we added different amounts of EHEP (1.68, 3.36, and 5.04 μM) to the reaction system (2.5 mM ONPG, 49 nM akuBGL, 40 μM TNA, 50 mM CH3COONa pH 5.5, 100 mM NaCl, and 10 mM CaCl2).
RecomGH1D1 activity assay
Request a detailed protocolWe measured simply the activity of the recomGH1D1 using a spectrophotometer because the reaction product, o-nitrophenol, is yellow. The reaction system (100 μl) included 2.5 mM ONPG, 49 nM akuBGL or recomGH1D1 in a reaction buffer (50 mM CH3COONa pH 5.5, 100 mM NaCl, and 10 mM CaCl2). After incubation for 10 min at 37°C, the reaction was terminated by adding 100 μl of 500 mM Na2CO3. The absorbance at 420 nm was measured using a SpectraMax spectrophotometer (Molecular Devices, Japan).
Binding assay for recomEHEP with TNA
Request a detailed protocolWe measured the binding activity of recomEHEP using precipitation analysis in the same method as natural EHEP, as previously described (Tsuji et al., 2017). Briefly, recomEHEP or EHEP was incubated with TNA at 25℃ for 90 min and centrifuged for 10 min at 12,000 × g at 4℃. Then, we washed the precipitates twice and resuspended them in an SDS–PAGE loading buffer for binding analysis.
Resolubilization of the EHEP–eckol precipitate
Request a detailed protocolA mixture of 2 mg of EHEP and 0.4 mg of eckol was incubated at 37°C for 1 hr, followed by centrifugation at 12,000 × g for 10 min, and the supernatant was removed. The sediment was dissolved in a 50 mM Tris–HCl buffer at different pH (7.0–9.0), and the absorbance at 560 nm was measured over time. After checking the elution peak by SDS–PAGE, the resolubilized EHEP was analyzed by a Sephacryl S-100 HR column (2.0 × 110 cm). Moreover, the eckol-/dieckol-binding activity of resolubilized EHEP was assessed, as mentioned above in this section.
Crystallization and data collection
Request a detailed protocolThe crystallization, data collection, and initial phase determination of EHEP were described previously (Sun et al., 2020). As EHEP precipitates when bound to TNA, we could not cocrystallize EHEP with TNA. Therefore, we used the soaking method to obtain the EHEP–TNA complex. Owing to the poor reproducibility of EHEP crystallization, we used a co-cage-1 nucleant (Yao and Li, 2020), a metal–organic framework (Matsuzaki et al., 2014) to prepare EHEP crystals for forming the complex with TNA. Finally, we obtained high-quality EHEP crystals under the reservoir solution containing 1.0 M sodium acetate and 0.1 M imidazole (pH 6.5) with co-cage-1 nucleant (Yao and Li, 2020). Subsequently, we soaked the EHEP crystals in a reservoir solution containing 10 mM TNA at 37℃ for 2 days; then, they were maintained at 20℃ for 2 weeks. Next, we soaked the EHEP crystals in a reservoir solution containing 10 mM phloroglucinol. For data collection, the crystal was soaked in a cryoprotectant solution containing 20% (vol/vol) glycerol along with the reservoir solution. Diffraction data were collected under a cold nitrogen gas stream at 100 K using Photon Factory BL-17 (Tsukuba, Japan) or Spring 8 BL-41XU (Hyogo, Japan).
For akuBGL crystallization, the initial crystallization screening was performed using the sitting-drop vapor-diffusion method with Screen Classics and Classics II crystallization kits (QIAGEN, Hilden, Germany) and PACT kits (Molecular Dimensions, Anatrace, Inc) at 20℃. Crystallization drops were set up by mixing 0.5 μl of the protein solution with an equal volume of the reservoir solution. The initial crystals were obtained under condition no. 41 (0.1 M sodium acetate pH 4.5 and 25% polyethylene glycol [PEG] 3350) of Classics II, no. 13 (0.1 M MIB buffer [25 mM sodium malonate dibasic monohydrate, 37.5 mM imidazole, and 37.5 mM boric acid] with pH 4.0 and 25% PEG 1500), and no. 37 (0.1 M MMT buffer [20 mM DL-malic acid, 40 mM MES monohydrate, and 40 mM Tris] with pH 4.0 and 25% PEG 1500) of PACT. After optimization by varying the buffer pH and precipitant concentration and adding co-cage-1 nucleant (Yao and Li, 2020), the optimal crystals were obtained using 0.1 M sodium acetate pH 4.5 and 20% PEG 3350 as a reservoir solution at a protein concentration of 5.4 mg/ml with a co-cage-1 nucleant (Yao and Li, 2020). Diffraction data were collected under a cold nitrogen gas stream at 100 K using Photon Factory BL-1A (Tsukuba, Japan) after cryoprotection by adding glycerol to a 20% final concentration into the reservoir solution. The optimal resolution of diffraction data was obtained by soaking a crystal with 5 mM TNA in the reservoir buffer at 37℃ for 4 hr.
All datasets were indexed, integrated, scaled, and merged using XDS/XSCALE program (Kabsch, 2010). Statistical data collection and process are summarized in Table 1.
Structure determination and refinement
Request a detailed protocolFor EHEP structure determination, after initial phasing via the native-SAD method (Sun et al., 2020; Yu et al., 2020), the model was obtained and refined with auto-building using Phenix AutoBuild of the PHENIX software suite (Adams et al., 2010). The obtained native-SAD structure was used as a model for rigid body refinement using phenix.refine (Afonine et al., 2012) of the PHENIX software suite with the native data at a high resolution of 1.15 Å. The structure of EHEP was automatically rebuilt using Phenix AutoBuild of the PHENIX software suite again (Adams et al., 2010). Several rounds of refinement were performed using phenix.refine of the PHENIX software suite (Adams et al., 2010), alternating with manual fitting and rebuilding using COOT program (Emsley and Cowtan, 2004). The final refinement statistics and geometry are shown in Table 1.
The structure of the EHEP–TNA complex was determined by the molecular replacement (MR) method using the EHEP structure as a search model with Phaser of the PHENIX software suite (McCoy et al., 2007). The electron density block of TNA was clearly shown in both 2Fo–Fc and Fo–Fc maps. Subsequently, the TNA structure was manually constructed, followed by several rounds of refinement using phenix.refine (Adams et al., 2010), with manual fitting and rebuilding using COOT (Emsley and Cowtan, 2004). We also determined the structure of phloroglucinol-soaked crystals at a resolution of 1.4 Å by the MR method using the refined EHEP structure as a search model with phenix.phaser. However, no electron density block of phloroglucinol was obtained. Therefore, we referred to this structure as the apo form (apo structure2). The final refinement statistics and geometry are shown in Table 1.
We determined the structure of akuBGL by the MR method using Phaser of the PHENIX software suite (McCoy et al., 2007). We used one GH domain (86–505 aa) of β-klotho (PDB entry: 5VAN) (Lee et al., 2018) as the search model. The GH domain of β-klotho shares 30% sequence identity with akuBGL. Four GH domains of two molecules were found in an asymmetric unit and rebuilt with Phenix_autobuild of Phenix software suite (Adams et al., 2010). Finally, refinement of akuBGL structure was performed as described for EHEP.
Docking studies of akuBGL with phlorotanins and laminarins
Request a detailed protocolWe used the Schrodinger Maestro program to perform docking studies (Sastry et al., 2013). First, we superimposed the structure of the OsBGL mutant complexed with cellotetraose (PDB ID 4QLK; Pengthaisong and Ketudat Cairns, 2014) onto that of akuBGL GH1D2 to define the ligand position in the ligand-binding cavity. Then, we modified the structure of the akuBGL GH1D2 using the wizard module to remove water molecules and add hydrogen atoms for docking. The 2D structures of the inhibitory ligands, including TNA, phloroglucinol, and eckol, were downloaded from PubChem (Wang et al., 2009) and further converted to 3D structures using the LigPrep module of the Schrodinger Maestro program. The structure of the substrate laminaritetraose was extracted from the Zobellia galactanivorans β-glucanase–laminaritetraose complex structure (PDB ID: 4BOW; Labourel et al., 2014). Then, a receptor grid was constructed in the center of the ligand-binding cavity. We performed docking using the Glide standard precision mode without any constraints. The optimal binding pose was determined using the lowest Glide score, and the docked structures were analyzed using PyMol.
Data availability
The atomic coordinates were deposited in the PDB with the accession codes as follows: EHEP with 1.15 Å resolution (8IN3), EHEP with 1.4 Å resolution (8IN4), EHEP complexed with tannic acid (8IN6), akuBGL (8IN1).
-
RCSB Protein Data BankID 8IN3. Eisenia hydrolysis-enhancing protein from Aplysia kurodai.
-
RCSB Protein Data BankID 8IN4. Eisenia hydrolysis-enhancing protein from Aplysia kurodai.
-
RCSB Protein Data BankID 8IN6. Eisenia hydrolysis-enhancing protein from Aplysia kurodai with tannic acid.
-
RCSB Protein Data BankID 8IN1. beta-glucosidase protein from Aplysia kurodai.
References
-
PHENIX: a comprehensive Python-based system for macromolecular structure solutionActa Crystallographica. Section D, Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
Towards automated crystallographic structure refinement with phenix.refineActa Crystallographica. Section D, Biological Crystallography 68:352–367.https://doi.org/10.1107/S0907444912001308
-
Defensive and sensory chemical Ecology of Brown algaeAdvances in Botanical Research 43:1–91.https://doi.org/10.1016/S0065-2296(05)43001-3
-
Tannins in plant-herbivore interactionsPhytochemistry 72:1551–1565.https://doi.org/10.1016/j.phytochem.2011.01.040
-
A coevolutionary arms race: understanding plant-herbivore interactionsThe American Biology Teacher 70:288–292.https://doi.org/10.1662/0002-7685(2008)70[288:ACARUP]2.0.CO;2
-
Structural insights into rice BGlu1 beta-glucosidase oligosaccharide hydrolysis and transglycosylationJournal of Molecular Biology 377:1200–1215.https://doi.org/10.1016/j.jmb.2008.01.076
-
Human antimicrobial peptides: defensins, cathelicidins and histatinsBiotechnology Letters 27:1337–1347.https://doi.org/10.1007/s10529-005-0936-5
-
Structural insight into a GH1 β-glucosidase from the oleaginous microalga, Nannochloropsis oceanicaInternational Journal of Biological Macromolecules 170:196–206.https://doi.org/10.1016/j.ijbiomac.2020.12.128
-
Effects of solution chemistry on conformation of self-aggregated tannic acid revealed by laser light scatteringThe Science of the Total Environment 754:142119.https://doi.org/10.1016/j.scitotenv.2020.142119
-
Coot: model-building tools for molecular graphicsActa Crystallographica. Section D, Biological Crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Molecular interactions between plants and insect herbivoresAnnual Review of Plant Biology 70:527–557.https://doi.org/10.1146/annurev-arplant-050718-095910
-
Polyphenol-mediated assembly of proteins for engineering functional materialsAngewandte Chemie 59:15618–15625.https://doi.org/10.1002/anie.202002089
-
Klotho-related protein is a novel cytosolic neutral beta-glycosylceramidaseThe Journal of Biological Chemistry 282:30889–30900.https://doi.org/10.1074/jbc.M700832200
-
New insights into peritrophic matrix synthesis, architecture, and functionAnnual Review of Entomology 54:285–302.https://doi.org/10.1146/annurev.ento.54.110807.090559
-
N-terminal acetylation and other functions of Nα-acetyltransferasesBiological Chemistry 393:291–298.https://doi.org/10.1515/hsz-2011-0228
-
Dali server: conservation mapping in 3DNucleic Acids Research 38:W545–W549.https://doi.org/10.1093/nar/gkq366
-
Carbohydrate-aromatic interactions in proteinsJournal of the American Chemical Society 137:15152–15160.https://doi.org/10.1021/jacs.5b08424
-
Phlorotannins are polyphenolic metabolites of brown algaeRussian Journal of Marine Biology 44:263–273.https://doi.org/10.1134/S106307401804003X
-
High-resolution structures of Neotermes koshunensis β-glucosidase mutants provide insights into the catalytic mechanism and the synthesis of glucoconjugatesActa Crystallographica. Section D, Biological Crystallography 68:829–838.https://doi.org/10.1107/S0907444912013224
-
A greener process for gallic acid production from tannic acid hydrolysis with hydrochloric acidAsian Journal of Chemistry 27:3328–3332.https://doi.org/10.14233/ajchem.2015.18689
-
BookMacroalgal chemical defenses and their roles in structuring temperate marine communitiesIn: Amsler CD, editors. Algal Chemical Ecology. Springer. pp. 57–89.https://doi.org/10.1007/978-3-540-74181-7
-
Molecular docking studies of phlorotannins from Eisenia bicyclis with BACE1 inhibitory activityBioorganic & Medicinal Chemistry Letters 20:3211–3215.https://doi.org/10.1016/j.bmcl.2010.04.093
-
XDSActa Crystallographica. Section D, Biological Crystallography 66:125–132.https://doi.org/10.1107/S0907444909047337
-
The β-glucanase ZgLamA from Zobellia galactanivorans evolved a bent active site adapted for efficient degradation of algal laminarinThe Journal of Biological Chemistry 289:2027–2042.https://doi.org/10.1074/jbc.M113.538843
-
TopFIND, a knowledgebase linking protein termini with functionNature Methods 8:703–704.https://doi.org/10.1038/nmeth.1669
-
Physicochemical conditions and microbial activities in the highly alkaline gut of the humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae)Applied and Environmental Microbiology 69:6650–6658.https://doi.org/10.1128/AEM.69.11.6650-6658.2003
-
Networked-cage microcrystals for evaluation of host-guest interactionsJournal of the American Chemical Society 136:17899–17901.https://doi.org/10.1021/ja5109535
-
Phaser crystallographic softwareJournal of Applied Crystallography 40:658–674.https://doi.org/10.1107/S0021889807021206
-
Crystal Structure of a GH3 β-Glucosidase from the Thermophilic Fungus Chaetomium thermophilumInternational Journal of Molecular Sciences 20:5962.https://doi.org/10.3390/ijms20235962
-
Serological, genomic and structural analyses of the major mite allergen Der p 23Clinical and Experimental Allergy 46:365–376.https://doi.org/10.1111/cea.12680
-
Identification, cloning, and characterization of β-glucosidase from Ustilago esculentaApplied Microbiology and Biotechnology 93:1989–1998.https://doi.org/10.1007/s00253-011-3538-2
-
Uncovering the molecular mechanisms of lignocellulose digestion in shipwormsBiotechnology for Biofuels 11:59.https://doi.org/10.1186/s13068-018-1058-3
-
Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichmentsJournal of Computer-Aided Molecular Design 27:221–234.https://doi.org/10.1007/s10822-013-9644-8
-
Salivary proteins as a defense against dietary tanninsJournal of Chemical Ecology 32:1149–1163.https://doi.org/10.1007/s10886-006-9077-0
-
Developmental roles of protein N-terminal acetylationProteomics 15:2402–2409.https://doi.org/10.1002/pmic.201400631
-
Chitin-binding proteins in invertebrates and plants comprise a common chitin-binding structural motifThe Journal of Biological Chemistry 275:17929–17932.https://doi.org/10.1074/jbc.C000184200
-
Crystallographic analysis of Eisenia hydrolysis-enhancing protein using a long wavelength for native-SAD phasingActa Crystallographica. Section F, Structural Biology Communications 76:20–24.https://doi.org/10.1107/S2053230X19016716
-
PubChem: a public information system for analyzing bioactivities of small moleculesNucleic Acids Research 37:W623–W633.https://doi.org/10.1093/nar/gkp456
-
A novel Vibrio beta-glucosidase (LamN) that hydrolyzes the algal storage polysaccharide laminarinFEMS Microbiology Ecology 91:fiv087.https://doi.org/10.1093/femsec/fiv087
-
Co-Evolution of Secondary Metabolites795–822, Plant defense and insect adaptation with reference to secondary metabolites, Co-Evolution of Secondary Metabolites, Springer, 10.1007/978-3-319-96397-6_60.
-
N-glycosylation affects the proper folding, enzymatic characteristics and production of a fungal ß-glucosidaseBiotechnology and Bioengineering 110:3075–3084.https://doi.org/10.1002/bit.24990
-
A solution-free crystal-mounting platform for native SADActa Crystallographica. Section D, Structural Biology 76:938–945.https://doi.org/10.1107/S2059798320011584
Article and author information
Author details
Funding
Japan Society for the Promotion of Science (21H01754)
- Min Yao
Japan Agency for Medical Research and Development (JP18am0101071)
- Min Yao
Japan Agency for Medical Research and Development (JP19am0101083)
- Min Yao
The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported in part by Grant-in-Aid for Scientific Research (B) (Grant Number 21H01754 to MY) and Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from Japan Agency for Medical Research and Development (AMED) under Grant Number JP18am0101071 and JP19am0101083. We are grateful to the Photon Factor and SPring-8 (No. 2017B2545, 2017A2551, and 2018B2538) for beam time and the beamline staff for their assistance with data collection.
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.88939. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Sun, Ye et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,021
- views
-
- 66
- downloads
-
- 1
- citation
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for Version of Record https://doi.org/10.7554/eLife.88939.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structural basis of EHEP-mediated offense against phlorotannin-induced defense from brown algae to protect akuBGL activity
eLife 12:RP88939.
https://doi.org/10.7554/eLife.88939.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}