Mitochondrial genomes of Pleistocene megafauna retrieved from recent sediment layers of two Siberian lakes
- Department of Biology, University of Konstanz, Germany
- Agroengineering Department/Department of Landscape Design and Sustainable Ecosystems, Agrarian and Technological Institute, RUDN University, Russian Federation
- Laboratory of Carbon Monitoring in Terrestrial Ecosystems, Institute of Physicochemical and Biological Problems of Soil Science of the Russian Academy of Sciences, Russian Federation
- Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research, Polar Terrestrial Environmental Systems, Germany
- Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, United States
- Howard Hughes Medical Institute, University of California, Santa Cruz, United States
- Embark Veterinary, Inc, United States
Figures
Figure 1
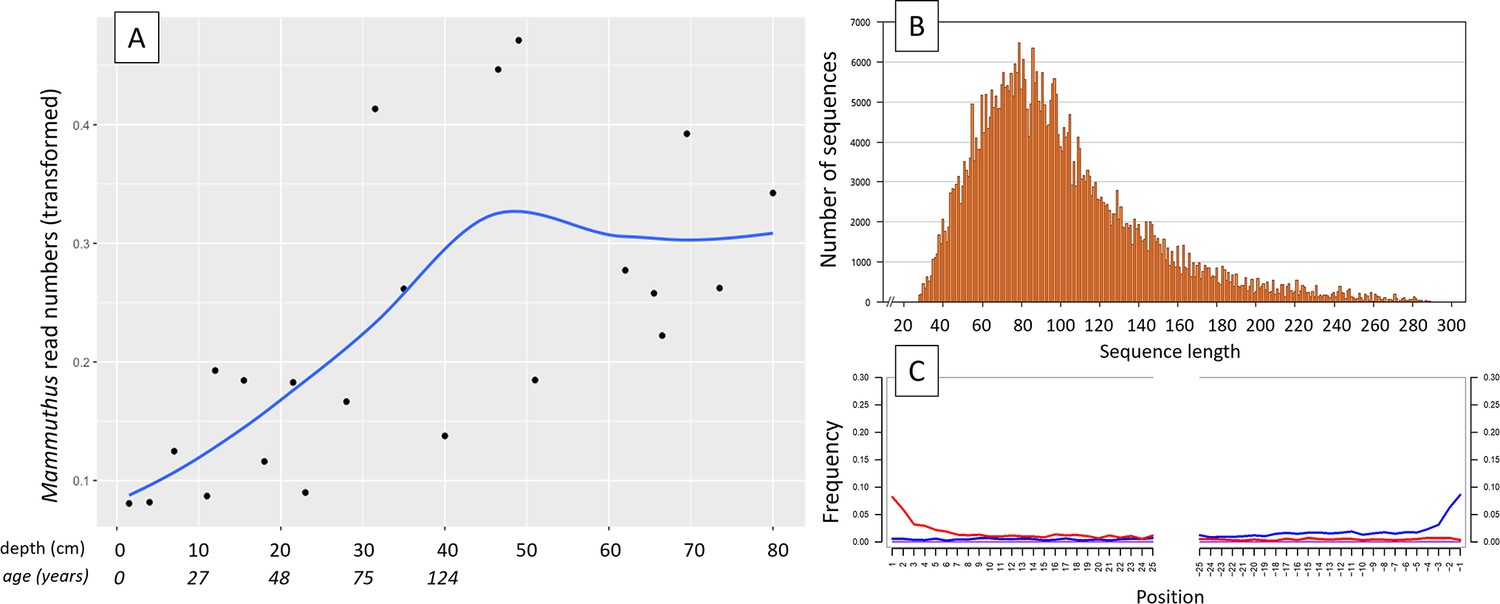
aDNA of Mammuthus in recent lake sediments.
(A) Read counts assigned to Mammuthus (square-root-transformed proportion of the respective number of raw reads per library) after hybridization capture enrichment of aeDNA of core LK-001 (shown are results of 22 libraries; one library was excluded as it did not produce any reads assigned to mammals); square-root transformation of percentage. Indicated are sample depths (in cm; 1.5–80 cm) and approximate ages as per 210Pb chronology (Appendix 1—table 7; to a maximum depth of 39.5 cm). The solid line indicates the general trend. Across the 22 libraries: (B) Fragment length distribution and (C) damage patterns (red indicates C-to-T transitions, blue G-to-A transitions. the Y-axis indicates the percentage of positions with a nucleotide change, the X-axis indicates the position along the fragment).
Figure 2
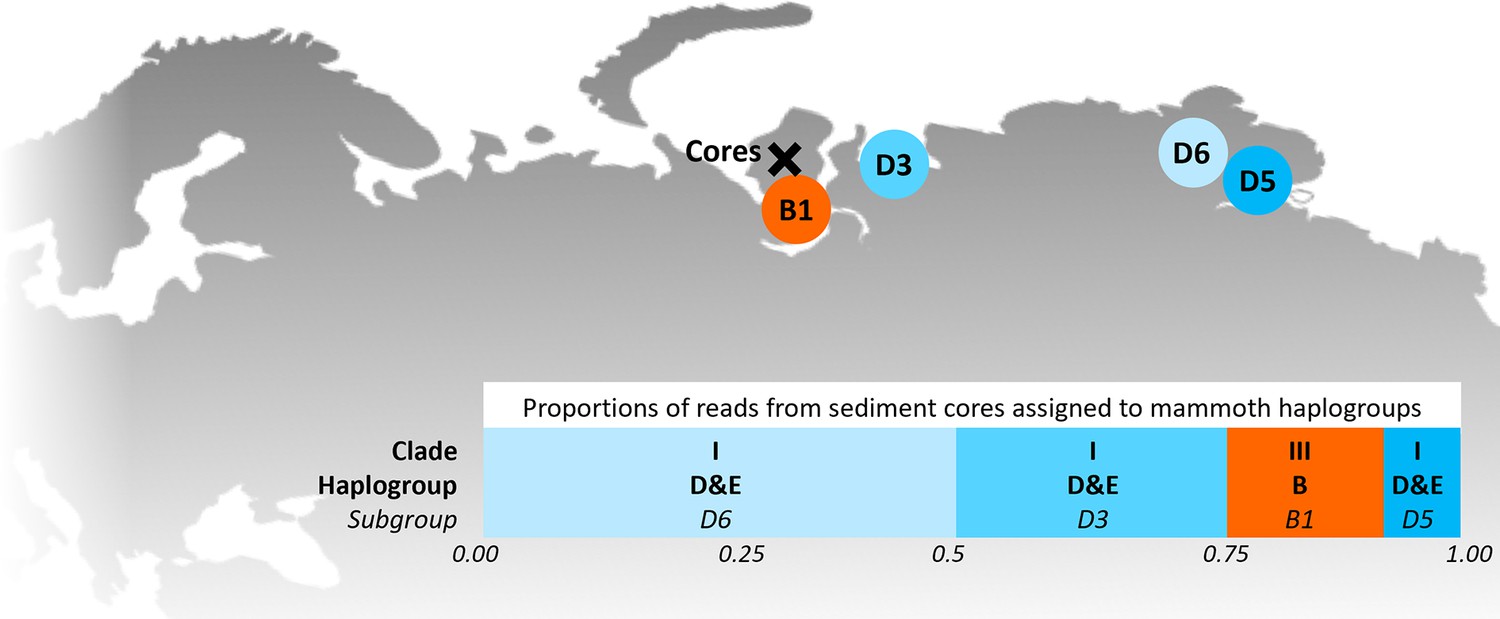
Locations of the sediment cores of the present study (Yamal peninsula, Siberia) and previously retrieved mammoth remains and their haplo(sub)groups (Appendix 1—table 6).
The bar chart indicates a maximum-likelihood estimate of the haplogroup proportions derived from the reads from the three sediment core libraries with the most mammoth reads.
Appendix 1—figure 1
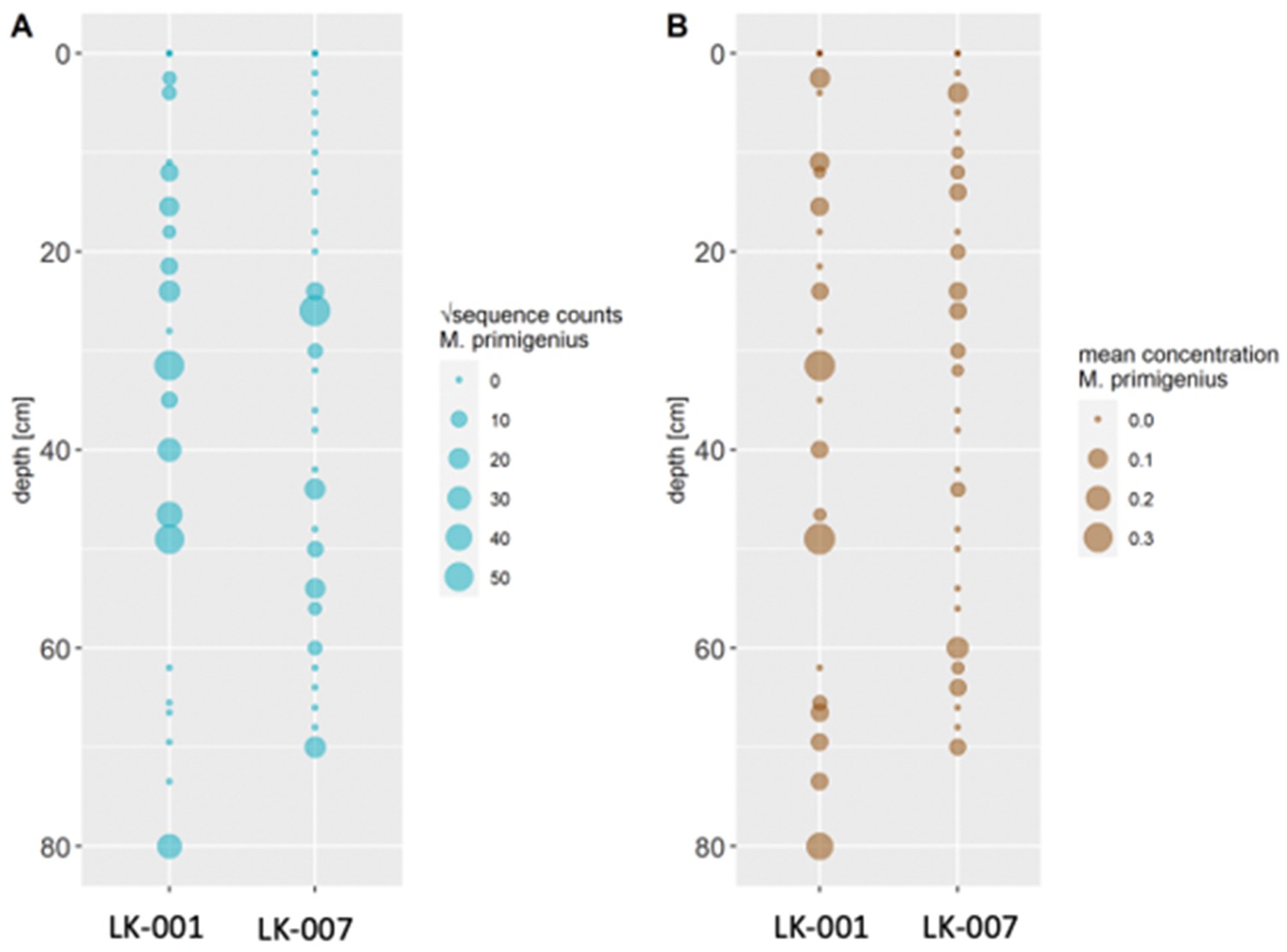
Comparison of transformed metabarcoding read counts (A) and ddPCR concentration estimations (B) of Mammuthus primigenius DNA in sediment cores LK-001 and LK-007 from the Yamal peninsula.
Tables
Table 1
Sediment cores retrieved from two lakes on the Yamal peninsula, Siberia.
| Lake | Coordinates | m above sea level | Area | Water depth | Core length |
|---|---|---|---|---|---|
| LK-001 | 70°16'45.6" N, 68°53'02.8" E | 28 | 38 ha | 17 m | 80 cm |
| LK-007 | 70°16'02.8" N, 68°59'35.7" E | 36 | 39 ha | 14 m | 75 cm |
Appendix 1—table 1
Sample depths and extraction protocols.
| (a) LK-001 | (b) LK-007 | ||
|---|---|---|---|
| Sample depth (cm) | Extraction protocol* | Sample depth (cm) | Extraction protocol* |
| 1.5 | PowerLyzer | 2.0 | PowerLyzer |
| 4.0 | 4.0 | PowerSoil Pro | |
| 7.0 | 6.0 | PowerLyzer | |
| 11.0 | 8.0 | PowerSoil Pro | |
| 12.0 | 10.0 | PowerLyzer | |
| 15,5 | 12.0 | PowerSoil Pro | |
| 18.0 | 14.0 | PowerSoil Pro | |
| 21.5 | 18.0 | PowerLyzer | |
| 23.0 | 20. | PowerSoil Pro | |
| 28.0 | 24.0 | PowerLyzer | |
| 31.5 | 26.0 | PowerSoil Pro | |
| 35.0 | 30.0 | PowerSoil Pro | |
| 40.0 | 32.0 | PowerSoil Pro | |
| 44.0 | 36.0 | PowerLyzer | |
| 46.5 | 38.0 | PowerSoil Pro | |
| 49.0 | 42.0 | PowerSoil Pro | |
| 51.0 | 44.0 | PowerLyzer | |
| 62.0 | 48.0 | PowerLyzer | |
| 65.5 | 50.0 | PowerLyzer | |
| 66.5 | 54.0 | PowerLyzer | |
| 69.5 | 56.0 | PowerLyzer | |
| 73.5 | 60.0 | PowerLyzer | |
| 80.0 | 62.0 | PowerSoil Pro | |
| 64.0 | PowerLyzer | ||
| 66.0 | PowerSoil Pro | ||
| 68.0 | PowerLyzer | ||
| 70.0 | PowerSoil Pro | ||
-
*
Kits from Qiagen (Hilden, Germany).
Appendix 1—table 2
PowerLyzer DNA extraction.
| Day 1: |
|
| Day 2: |
|
| PowerSoil Pro DNA extraction was performed as follows: | |
| Day 1: |
|
| Day 2: |
|
Appendix 1—table 3
Library preparation.
| End repair | per library | |
|---|---|---|
| Mix following components in a sterile low-binding PCR tube | ||
| NEBNext End Repair Buffer (10 X) | 5 µL | |
| NEBNext End Repair Enzyme Mix | 2.5 µL | |
| genomic DNA | 42.5 µL | |
| Incubate in a thermal cycler for 30 mins at 20 °C. Purify using QIAquick/MinElute PCR purification kit. Elution: add 32 µl buffer EB and incubate at 37 °C for 5 min before spinning down the DNA at 13,000 rpm for 1 min. | ||
| Adapter ligation | ||
| Mix following components in a sterile low-binding PCR tube | ||
| Quick Ligation Reaction Buffer (10 X) | 10 µL | |
| nuclease-free water | 4 µL | |
| P5/P7 adapter mix (50 µM stock) | 1 µL | |
| DNA as purified in step above | 30 µL | |
| Quick T4 DNA ligase | 4.8 µL | |
| *final adapter concentrations for ancient samples should be between 0.25–0.5 µM. **it is vital to add ligase after mixing DNA with adaptors. | ||
| Incubate for 15 min at 25 °C; purify using a QIAquick/MinElute PCR purification kit. Elution: 42 µL buffer EB and incubate at 37 ºC for 5 min before spinning down the DNA at 13,000 rpm for 1 min. | ||
| Fill-In Reaction | ||
| Add the following reagents into a low-binding PCR tube | ||
| ThermoPol Reaction Buffer | 5 µL | |
| dNTPs (10 mM) | 2 µL | |
| Bst DNA polymerase | 3 µL | |
| DNA as eluted above | 40 µL | |
| Incubate: 20 mins at 65 °C 20 mins at 80 °C No purification is needed after this step. | ||
Appendix 1—table 4
Indexing PCR.
| Reagent | µL | |
|---|---|---|
| H2O | 3.45 | |
| Platinum Hifi Taq Buffer 10 X (Thermo Fisher Scientific) | 2.50 | |
| dNTPs (25 mM) | 0.25 | |
| bovine serum albumin (New England Biolabs) | 1.00 | |
| MgSO4 (50 mM) | 1.00 | |
| Platinum HiFi (5 U/µL; Thermo Fisher Scientific) | 0.20 | |
| total | 8.40 | |
| Index P5 (10 µM) | 0.80 | |
| Index P7 (10 µM) | 0.80 | |
| template DNA | 15.00 | |
| Thermocycling | ||
| °C | t | |
| 94 | 1 min | |
| 94 | 15 s | 8 cycles |
| 60 | 20 s | |
| 68 | 60 s | |
| 68 | 3 min | |
| 20 | store | |
Appendix 1—table 5
Mitogenome templates of 17 mammal species for design of RNA baits.
| Order | Species | NCBI accession | # of baits |
|---|---|---|---|
| Artiodactyla | Bison bison | NC_012346.1 | 446 |
| Bos primigenius | NC_020746.1 | 456 | |
| Saiga tatarica | NC_013996.1 | 538 | |
| Ovis canadensis | NC_015889.1 | 522 | |
| Ovibos moschatus | NC_020631.1 | 542 | |
| Cervus elaphus | NC_007704.2 | 523 | |
| Rangifer tarandus | NC_007703.1 | 526 | |
| Alces alces | NC_020677.1 | 520 | |
| Camelus ferus | NC_009629.2 | 583 | |
| Perissodactyla | Equus przewalskii | NC_024030.1 | 575 |
| Coelodonta antiquitatis | NC_012681.1 | 571 | |
| Lagomorpha | Lepus arcticus | NC_044769.1 | 586 |
| Ochotona collaris | NC_003033.1 | 591 | |
| Proboscidea | Mammuthus primigenius | NC_007596.2 | 588 |
| Eulipotyphla | Sorex tundrensis | NC_025327.1 | 584 |
| Rodentia | Castor canadensis | NC_033912.1 | 584 |
| Dicrostonyx torquatus | NC_034646.1 | 575 | |
| 9,310 | |||
Appendix 1—table 6
Cladonia rangiferina sequences for RNA bait design (shown are the NCBI accessions).
| ITS-1 |
|---|
| MN756840.1; DQ394367.1; JQ695919.1; MK179592.1; KP031549.1; KP001202.1; AF458306.1; KT792792.1; MK811970.1; KT792788.1; MK508944.1; GU169225.1; KP001197.1; KP001201.1; MK812260.1; MK811708.1; KY119381.1; MK508952.1; KT792789.1; EU266113.1; KY266884.1; KP001192.1; KP001190.1; JQ695918.1; KT792790.1; JQ695920.1; KP001191.1; AF458307.1; KP001200.1; MK508943.1; MK812460.1; MK508937.1; KT792791.1 resulting in 23 baits |
| ITS-2 |
| KT792789.1; KP001190.1; AF458307.1; JQ695919.1; MK179592.1; DQ394367.1; KP001194.1; KP001193.1; KY266884.1; KP001199.1; KP001192.1; MK300750.1; MN756487.1; MK508937.1; KP001200.1; MK812460.1; MK508943.1; KP001191.1; KP031549.1; KP001202.1; AF458306.1; MK811970.1; KY119381.1; KP001198.1; MK812260.1; MK811708.1; GU169225.1; JQ695918.1; KP001201.1 resulting in 6 baits |
Appendix 1—table 7
Reference mitogenomes used for mapping.
| NC_020679.1 | Antilocapra americana | NC_018783.1 | Equus ovodovi |
| NC_012346.1 | Bison bison | HM118851.1 | Equus hemionus |
| NC_020746.1 | Saiga tatarica | MK982180.1 | Equus asinus |
| NC_013996.1 | Bos primigenius | NC_012681.1 | Coelodonta antiquitatis |
| NC_015889.1 | Ovis canadensis | NC_007596.2 | Mammuthus primigenius |
| NC_020630.1 | Oreamnos americanus | FR691686.1 | Castor fiber |
| NC_020631.1 | Ovibos moschatus | NC_033912.1 | Castor canadensis |
| NC_027233.1 | Bison priscus | NC_034313.1 | Dicrostonyx groenlandicus |
| NC_009629.2 | Camelus ferus | NC_034646.1 | Dicrostonyx torquatus |
| KR822422.1 | Camelops cf. hesternus | JN181159.1 | Peromyscus leucopus |
| NC_013836.1 | Cervus elaphus xanthopygus | NC_006853.1 | Bos taurus |
| NC_007704.2 | Cervus elaphus | KM093871.1 | Capra hircus |
| NC_013840.1 | Cervus elaphus yarkandensis | NC_015241.1 | Microtus fortis fortis |
| KP405229.1 | Alces alces cameloides | KP200876.1 | Vulpes lagopus |
| NC_020677.1 | Alces alces | HM236180.1 | Ovis aries |
| NC_020729.1 | Odocoileus hemionus | KT448275.1 | Canis latrans |
| NC_015247.1 | Odocoileus virginianus | JN632610.1 | Capreolus capreolus |
| NC_007703.1 | Rangifer tarandus | KJ681493.1 | Capreolus pygargus |
| KY987554.1 | Platygonus compressus | JN632629.1 | Dama dama |
| NC_002008.4 | Canis lupus familiaris | KM982549.1 | Lynx lynx |
| NC_009686.1 | Canis lupus lupus | KP202265.1 | Panthera pardus |
| NC_013445.1 | Cuon alpinus | NC_026460.1 | Rhinolophus macrotis |
| NC_026529.1 | Vulpes lagopus | Y07726.1 | Ceratotherium simum |
| NC_028302.1 | Panthera leo | NC_005089.1 | Mus musculus |
| NC_022842.1 | Panthera onca | AM711900.1 | Meles meles |
| NC_010642.1 | Panthera tigris | KM091450.1 | Mustela erminea |
| NC_014456.1 | Lynx rufus | NC_005358.1 | Ochotona princeps |
| NC_020642.1 | Martes americana | NC_012095.1 | Sus scrofa domesticus |
| NC_020641.1 | Neovison vison | DQ480489.1 | Canis lupus familiaris |
| NC_024942.1 | Mustela nigripes | NC_020670.1 | Crocuta crocuta |
| NC_020664.1 | Martes pennanti | NC_011116.1 | Arctodus simus |
| NC_020639.1 | Mustela nivalis | NC_027963.1 | Sorex araneus |
| NC_009685.1 | Gulo gulo | NC_025327.1 | Sorex tundrensis |
| NC_011112.1 | Ursus spelaeus | KJ397607.1 | Lepus arcticus |
| NC_003426.1 | Ursus americanus | NC_001640.1 | Equus caballus |
| NC_003427.1 | Ursus arctos | NC_024030.1 | Equus przewalskii |
| NC_003428.1 | Ursus maritimus |
Appendix 1—table 8
Command lines and software used for initial mapping, processing, and taxonomic assignment.
| adapter trimming, filtering, and merging: leeHom Renaud et al., 2014: | src/leeHom -t 120 –ancientdna –auto -fq1 file_R1.fastq.gz -fq2 file_R2.fastq.gz -fqo leehom_out |
| mapping: BWA Li and Durbin, 2009 against 75 mammal mitogenomes Appendix 1—table 3: | bwa index reference_mitogenomes.fasta bwa aln reference_mitogenomes.fasta leehom_out.fq.gz -l 16,000 n 0.01 -O 2 -o 2 t 8>bwa_out.sai bwa samse reference_mitogenomes.fasta bwa_out.sai leehom_out.fq.gz>bwa_out.sam samtools view -q ≥ 30 S -b bwa_out.sam>bwa_out.bam |
| remove duplicates: samtools Li and Durbin, 2009 | samtools collate -o bwa_out_col.bam bwa_out.bam samtools fixmate -m bwa_out_col.bam bwa_out_fixmate.bam samtools sort -o bwa_out_pos.bam bwa_out_fixmate.bam samtools markdup bwa_out_pos.bam bwa_out_mark.bam samtools fastq bwa_out_mark.bam>bwa_out.fastq |
| fastq to fasta | sed -n ‘1~4 s/^@/>/p;2~4 p’ bwa_out.fastq>bwa_out.fasta |
| #alignment: blastn Altschul et al., 1990 blastn -db ncbi_nt -query bwa_out.fasta -evalue 0.01 -out blastn_out.fasta | |
| aDNA damage: mapDamage Ginolhac et al., 2011 | bwa index reference.fasta bwa aln reference.fasta sample.fasta >sample.sai bwa samse referecne.fasta sample.sai sample.fasta >sample.sam samtools view -q 25 S -b sample.sam >sample.bam mapDamage -i sample.bam -r reference.fasta mapDamage -d results_ sample/ -y 0.1 --plot-only mapDamage -i sample.bam -r reference.fasta –rescale mapDamage -d results_sample / --forward --stats-only -v -r reference.fasta |
Appendix 1—table 9
Reference mammoth mitogenomes used for panel.
| Clade | Haplogroup | Subgroup | Accession numbers | Proportion of reads assigned to haplogroup |
|---|---|---|---|---|
| II | A | EU153451, EU153450 | - | |
| III | B | B0 | KX027526, KX027531 | - |
| B1 | KX027526 | 0.1615 | ||
| B2 | KX027531 | - | ||
| I | C | KX027498, KX027565, KX027567, JF912200, KX027499, KX027502 | - | |
| I | D&E | D0 | DQ316067, EU153454, EU153447, EU153456, EU153449, EU153455, EU153446 | - |
| D1 | DQ316067 | - | ||
| D2 | EU153454 | - | ||
| D3 | EU153447 | 0.2790 | ||
| D4 | EU153456 | - | ||
| D5 | EU153449 | 0.0765 | ||
| D6 | EU153455 | 0.4829 | ||
| D7 | EU153446 | - | ||
| I | F | KX027503, KX027512, NC_015529, KX027511, KX027548, KX027547, KX027556, KX027559, KX027550 | - | |
| Krestovka mammoth | K | PRJEB42269 (European Nucleotide Archive) | - | |
| Conventional PCR | ||||
| 94 °C | 2 min | |||
| 94 °C | 30 s | 55 cycles | ||
| 54 °C | 30 s | |||
| 68 °C | 20 s | |||
| 68 °C | 1 min | |||
Appendix 1—table 10
Conventional PCR.
| Reagent | µL | |
|---|---|---|
| H2O | 14.85 | |
| Platinum Hifi Taq Buffer 10 X (Thermo Fisher Scientific) | 2.5 | |
| bovine serum albumin (New England Biolabs) | 0.2 | |
| MgSO4 (50 mM) | 1.00 | |
| dNTPs (25 mM) | 0.25 | |
| Platinum HiFi (5 U/µL; Thermo Fisher Scientific) | 0.20 | |
| Blocking primer R (1 µM) | 0.5 | |
| Blocking primer F (1 µM) | 0.5 | |
| total | 20.00 | |
| template DNA | 3.00 | |
| Thermocycling | ||
| °C | t | |
| 94 | 5 min | |
| 94 | 30 s | 40 cycles |
| 50 | 30 s | |
| 68 | 30 s | |
| 68 | 10 min | |
| RT | store | |
Appendix 1—table 11
Conventional PCR.
| Load data | obi import --quality-sanger file_R1.fastq reads1 obi import --quality-sanger file_R2.fastq reads2 |
| Import tags | obi import --ngsfilter-input taglist.txt ngsfile |
| Align paired-end reads | obi alignpairedend -R reads2 reads1 aligned_reads |
| Grep entries whose mode are alignment | obi grep -a mode:alignment aligned_reads good_sequences |
| Assign alignments to individual PCRs | obi ngsfilter -t ngsfile -u unidentified_sequences good_sequences identified_sequences |
| Filter out sequences | obi grep -p "sequence[’score']>50" identified_sequences identified_sequences_filtered obi grep -p "sequence[’score_norm']>0.9" identified_sequences_filtered identified_sequences_filtered_adj |
| Dereplicate Sequences | obi uniq -m sample identified_sequences_filtered_adj dereplicated_sequences_filtered |
| Keep only useful tags | obi annotate -k COUNT -k MERGED_sample dereplicated_sequences_filtered cleaned_metadata_sequences |
| Discard sequences that are shorter than 60 bp (based on primer pair) | obi grep -p "len(sequence) ≥ 60 and sequence['COUNT'] ≥ 10" cleaned_metadata_sequences denoised_sequences |
| Clean the sequences from PCR/sequencing errors | obi clean -s MERGED_sample -r 0.05 H denoised_sequences cleaned_sequences |
| Load database | cp STD_MAM_1.dat.gz ~/edna_LauraB/master/mammalia/database/ |
| Import it into DMS | obi import /data/scc/edna/LauraBa/master/mammalia/database/STD_MAM_1.dat.gz database_mam obi import --embl EMBL embl_refs |
| Download the taxonomy | wget https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz |
| Import the taxonomy in the DMS | obi import --taxdump /data/scc/edna/LauraBa/master/mammalia/taxdump.tar.gz taxonomy/my_tax |
| Cleaning the database with in silico PCR | obi ecopcr -e 3 l 50 L 150 F CGAGAAGACCCTATGGAGCT -R CCGAGGTCRCCCCAACC --taxonomy taxonomy/my_tax embl_refs mam_refs |
| Filter sequences | obi grep --require-rank=species --require-rank=genus --require-rank=family --taxonomy taxonomy/my_tax mam_refs mam_refs_clean |
| Dereplicate identical sequences | obi uniq --taxonomy taxonomy/my_tax mam_refs_clean mam_refs_uniq |
| Add taxid at the family level | obi grep --require-rank=family --taxonomy taxonomy/my_tax mam_refs_uniq |
| Build the reference database | obi build_ref_db -t 0.97 --taxonomy taxonomy/my_tax mam_refs_uniq_clean mam_db_97 |
| Assign each sequence to a taxon | obi ecotag -m 0.97 --taxonomy taxonomy/my_tax -R mam_db_97 cleaned_sequences assigned_sequences |
| Align the sequences | obi align -t 0.95 assigned_sequences aligned_assigned_sequences |
| Export tables for downstream data analysis | obi grep -A SCIENTIFIC_NAME assigned_sequences assigned_for_metabR |
| Output two tables required by metabaR | obi annotate -k MERGED_sample assigned_for_metabR assigned_for_metabR_reads_tableobi export --tab-output --output-na-string 0 assigned_for_metabR_reads_table >mam_reads_01.txtobi annotate --taxonomy taxonomy/my_tax \ --with-taxon-at-rank superkingdom \ --with-taxon-at-rank kingdom \ --with-taxon-at-rank phylum \ --with-taxon-at-rank subphylum \ --with-taxon-at-rank class \ --with-taxon-at-rank subclass \ --with-taxon-at-rank order \ --with-taxon-at-rank suborder \ --with-taxon-at-rank infraorder \ --with-taxon-at-rank superfamily \ --with-taxon-at-rank family \ --with-taxon-at-rank genus \ --with-taxon-at-rank species \ --with-taxon-at-rank subspecies \ assigned_for_metabR assigned_for_metabR_taxInfo obi annotate \ -k BEST_IDENTITY -k TAXID -k SCIENTIFIC_NAME -k COUNT -k seq_length \ -k superkingdom_name \ -k kingdom_name \ -k phylum_name \ -k subphylum_name \ -k class_name \ -k subclass_name \ -k order_name \ -k suborder_name \ -k infraorder_name \ -k superfamily_name \ -k family_name \ -k genus_name \ -k species_name \ assigned_for_metabR_taxInfo assigned_for_metabR_motus obi export --tab-output assigned_for_metabR_motus >mam_motus_01.txt |
| Further processing of the data sets was done using RStudio. | |
| Editing files for metabar | reads<- dt_reads %>% dplyr::select(-c("DEFINITION", "NUC_SEQ"))%>% as.data.frame() %>% janitor::row_to_names(row_number = 897, remove_rows_above = FALSE, remove_row = TRUE) %>% mutate_if(is.integer,as.numeric) |
| Assign name to first column | reads <- cbind(rownames(reads),reads) rownames(reads) <- NULL colnames(reads) <- c(names(reads)) colnames(reads)(1) <- "pcr_id" |
| Edit the names of the column | reads$pcr_id = strsplit(reads$pcr_id,"[.]") reads$pcr_id = sapply(reads$pcr_id, function(x) x[length(x)]) rownames(reads)<- reads$pcr_id |
| Organizing the MOTUs table | motus<- dplyr::select(dt_motus, 'ID', 'NUC_SEQ', 'COUNT','BEST_IDENTITY', 'TAXID', 'SCIENTIFIC_NAME', ’superkingdom_name', ’species_name', 'class_name', 'order_name', 'family_name', 'genus_name', 'kingdom_name', 'phylum_name', ’subphylum_name', ’subclass_name', ’suborder_name') names(motus)[names(motus) == 'NUC_SEQ'] <- ’sequence' |
Appendix 1—table 12
Conventional PCR.
| Reagent | µL | |
|---|---|---|
| ddPCR Supermix for probes | 11 | |
| H2O (DEPC) | 6.8 | |
| 20 x Target-Primers/Probe (FAM) | 1.1 | |
| 20 x Target-Primers/Probe (HEX) | 1.1 | |
| total | 20 | |
| template DNA | 2.0 | |
| Thermocycling | ||
| °C | t | |
| 95 | 10 min | |
| 94 | 30 sec | 40 cycles |
| 50 | 30 sec | |
| 60 | 30 sec | |
Appendix 1—table 13
210Pb chronology of Yamal lake sediment core LK-001.
| Depth | Chronology | Sedimentation Rate | ||||
|---|---|---|---|---|---|---|
| Date | Age | |||||
| cm | g cm–2 | AD | y | ± | g cm–2 y–1 | cm y–1 |
| 0.00 | 0.0 | 2019 | 0 | 0 | ||
| 0.25 | 0.2 | 2018 | 1 | 1 | 0.34 | 0.37 |
| 1.25 | 1.1 | 2016 | 3 | 2 | 0.34 | 0.37 |
| 2.25 | 1.9 | 2013 | 6 | 2 | 0.34 | 0.37 |
| 3.25 | 2.8 | 2010 | 9 | 2 | 0.34 | 0.37 |
| 4.25 | 3.8 | 2008 | 11 | 2 | 0.34 | 0.37 |
| 5.25 | 4.7 | 2005 | 14 | 2 | 0.34 | 0.37 |
| 6.25 | 5.7 | 2002 | 17 | 2 | 0.34 | 0.37 |
| 7.25 | 6.7 | 1999 | 20 | 3 | 0.36 | 0.37 |
| 8.25 | 7.6 | 1996 | 23 | 3 | 0.39 | 0.41 |
| 10.25 | 9.7 | 1992 | 27 | 3 | 0.49 | 0.47 |
| 12.25 | 11.8 | 1988 | 31 | 4 | 0.53 | 0.49 |
| 14.25 | 14.0 | 1984 | 35 | 4 | 0.55 | 0.51 |
| 14.75 | 14.5 | 1983 | 36 | 4 | 0.55 | 0.51 |
| 15.50 | 15.4 | 1981 | 38 | 4 | 0.55 | 0.51 |
| 16.50 | 16.5 | 1980 | 39 | 4 | 0.55 | 0.51 |
| 17.50 | 17.6 | 1978 | 41 | 4 | 0.55 | 0.51 |
| 18.50 | 18.6 | 1976 | 43 | 4 | 0.55 | 0.51 |
| 20.50 | 20.7 | 1971 | 48 | 5 | 0.55 | 0.51 |
| 22.50 | 22.7 | 1968 | 51 | 6 | 0.55 | 0.51 |
| 23.50 | 23.7 | 1966 | 53 | 6 | 0.55 | 0.51 |
| 24.50 | 24.8 | 1964 | 55 | 6 | 0.55 | 0.51 |
| 25.50 | 25.9 | 1962 | 57 | 7 | 0.44 | 0.42 |
| 26.50 | 27.0 | 1961 | 58 | 7 | 0.37 | 0.36 |
| 27.50 | 28.0 | 1957 | 62 | 8 | 0.30 | 0.29 |
| 28.50 | 29.0 | 1952 | 67 | 9 | 0.28 | 0.22 |
| 29.50 | 30.2 | 1949 | 70 | 10 | 0.23 | 0.19 |
| 31.50 | 32.6 | 1938 | 81 | 10 | 0.23 | 0.19 |
| 33.50 | 34.7 | 1933 | 86 | 10 | 0.23 | 0.19 |
| 35.50 | 36.9 | 1927 | 92 | 11 | 0.23 | 0.19 |
| 37.50 | 39.1 | 1913 | 106 | 13 | 0.23 | 0.19 |
| 39.50 | 41.4 | 1895 | 124 | 17 | 0.23 | 0.19 |
Appendix 1—table 14
Radiocarbon dating results.
| Sample label | F14C | ± (abs) | Age (y) | ± (y) | Weight (µg C) | Comment |
|---|---|---|---|---|---|---|
| LK-001_36.5 cm | 0.9595 | 0.0095 | 332 | 79 | 35 | |
| LK-001_51 cm | 0.3396 | 0.0056 | 8677 | 132 | 148 | Off; lateral input |
| LK-001_74 cm | 0.8248 | 0.0234 | 1547 | 228 | 13 |
Appendix 1—table 15
Numbers of sequences assigned to mammals (core LK-001).
| depth | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sum | 1.5 | 4.0 | 7.0 | 11.0 | 12.0 | 15.5 | 18.0 | 21.5 | 23.0 | 28.0 | 31.5 | 35.0 | 40.0 | 46.5 | 49.0 | 51.0 | 62.0 | 65.5 | 66.5 | 69.5 | 73.5 | 80.0 | |
| Mammuthus primigenius | 19,640 | 191 | 162 | 524 | 290 | 1205 | 605 | 194 | 666 | 277 | 800 | 3697 | 305 | 1188 | 1374 | 1923 | 631 | 674 | 519 | 1110 | 1083 | 814 | 1408 |
| Rangifer tarandus | 18,055 | 253 | 63 | - | 34 | 328 | 688 | 644 | 1816 | 545 | 902 | 2186 | 1240 | 1571 | 1574 | 86 | 284 | 2714 | 56 | 345 | 991 | 114 | 1,621 |
| Dicrostonyx torquatus | 16,870 | 211 | 112 | 34 | 111 | 1211 | 614 | 271 | 1035 | 522 | 1049 | 850 | 1298 | 314 | 761 | 193 | 318 | 656 | 200 | 2763 | 1011 | 1408 | 1928 |
| Lepus | 6371 | 148 | 134 | 105 | 23 | 584 | 245 | 40 | 369 | 336 | 630 | 382 | 231 | 404 | 799 | 285 | 141 | 116 | 126 | 352 | 330 | 293 | 298 |
| Coelodonta antiquitatis | 2737 | 33 | - | - | 28 | 119 | 153 | 347 | 55 | - | 97 | 128 | 467 | 298 | 141 | 27 | 103 | 152 | - | 250 | 144 | 4 | 191 |
| Homo sapiens | 387 | - | - | - | - | - | - | 38 | 217 | - | - | 23 | - | 25 | - | 69 | - | 15 | - | - | - | - | - |
| Ovibos moschatus moschatus | 145 | - | 69 | - | - | - | - | - | - | - | 38 | - | - | - | 38 | - | - | - | - | - | - | - | - |
| Castor fiber | 135 | - | - | - | - | 18 | - | - | - | - | 15 | - | - | - | 27 | 51 | - | 6 | 1 | - | - | - | 17 |
| Bos | 105 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 105 | - | - | - | - | - | - | - |
| Cervinae | 92 | - | - | - | - | - | - | - | 33 | - | - | - | - | 31 | 28 | - | - | - | - | - | - | - | - |
| Saiga tatarica | 91 | - | - | - | - | - | - | - | - | - | 57 | - | 34 | - | - | - | - | - | - | - | - | - | - |
| Sus scrofa cristatus | 85 | - | - | - | - | - | - | - | 34 | - | - | - | - | - | - | 24 | - | - | - | - | 27 | - | - |
| Ochotona | 74 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 14 | - | - | - | 60 |
| Ovis aries musimon | 70 | - | - | - | - | 70 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Bison | 54 | - | - | - | - | 24 | - | 30 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Crocuta crocuta | 53 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 53 | - | - | - |
| Chrotopterus auritus | 43 | - | - | - | - | - | 43 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Sorex tundrensis | 32 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 32 | - | - | - | - | - | - |
| Equus | 32 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 7 | - | - | - | 25 |
| Peromyscus maniculatus bairdii | 29 | - | - | - | - | - | - | - | - | - | - | - | - | - | 29 | - | - | - | - | - | - | - | - |
| Murinae | 28 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 28 | - |
| Capra | 21 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 21 | - | - | - | - | - | - |
| Myopus schisticolor | 18 | - | - | - | - | - | - | - | 18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Lemmus trimucronatus | 16 | 16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Mitochondrial genomes of Pleistocene megafauna retrieved from recent sediment layers of two Siberian lakes
eLife 12:RP89992.
https://doi.org/10.7554/eLife.89992.3




{kind=link}
{kind=link}
{kind=link}