The αC-β4 loop controls the allosteric cooperativity between nucleotide and substrate in the catalytic subunit of protein kinase A
- Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, United States
- Department of Chemistry and Supercomputing Institute, University of Minnesota, United States
- Department of Chemistry and Biochemistry, St. Catherine University, United States
- Department of Chemistry, University of Cambridge, United Kingdom
- Department of Pharmacology, University of California at San Diego, United States
- Department of Chemistry and Biochemistry, University of California at San Diego, United States
Figures
Figure 1
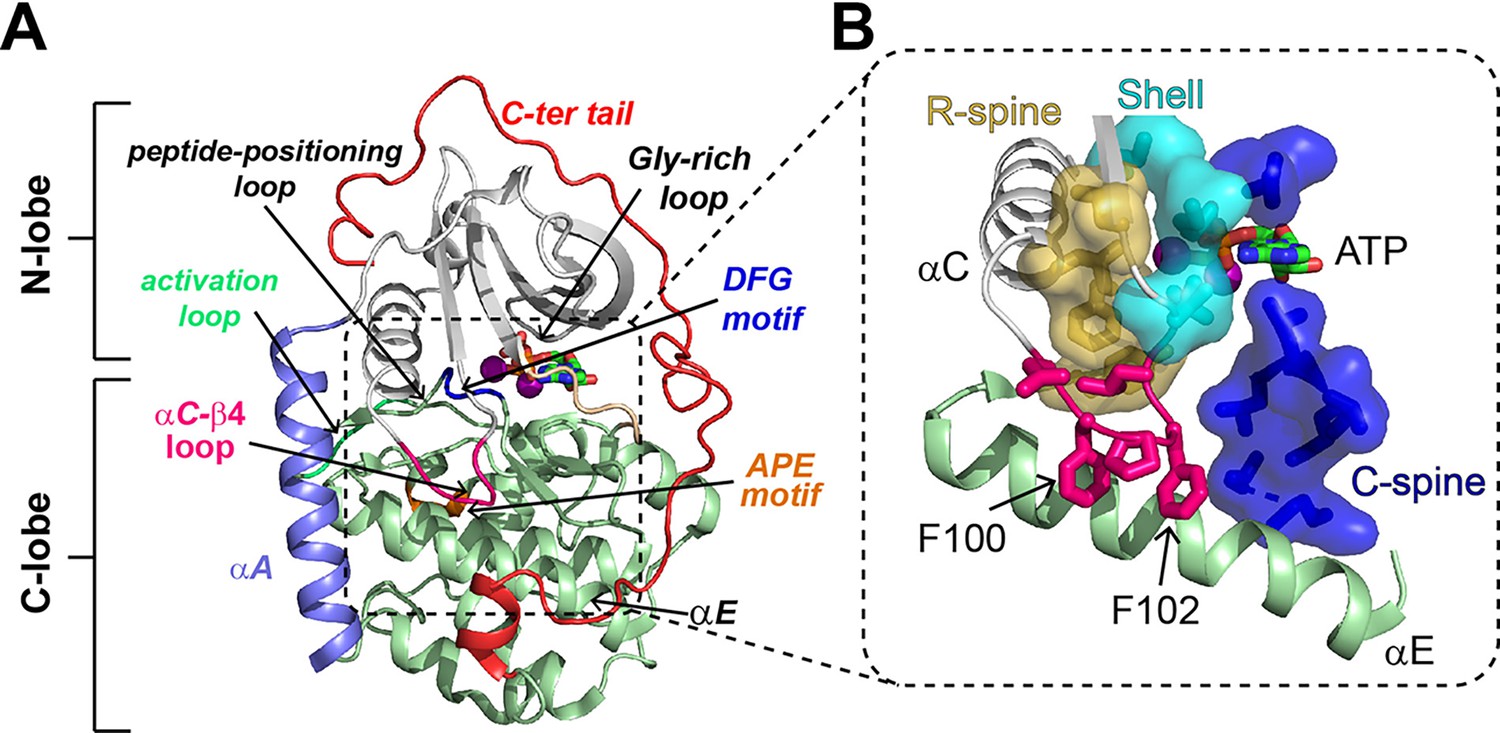
Structural and catalytic motifs of PKA-C.
(A) Backbone representation of the ternary complex of PKA-C bound to ATP and the PKI5-24 peptide (not depicted), PDB code 4WB5. Highlighted are key motifs including the αA, αC, and αE helices, C-terminal tail, activation loop, peptide-positioning loop, Gly-rich loop, and the DFG and APE motifs. (B) The hydrophobic core of PKA-C features the regulatory spine (R-spine, gold), catalytic spine (C-spine, blue), shell residues (cyan), and the αC-β4 loop (hot pink), which locks the αE helix and couples the two lobes of PKA-C.
Figure 2 with 4 supplements
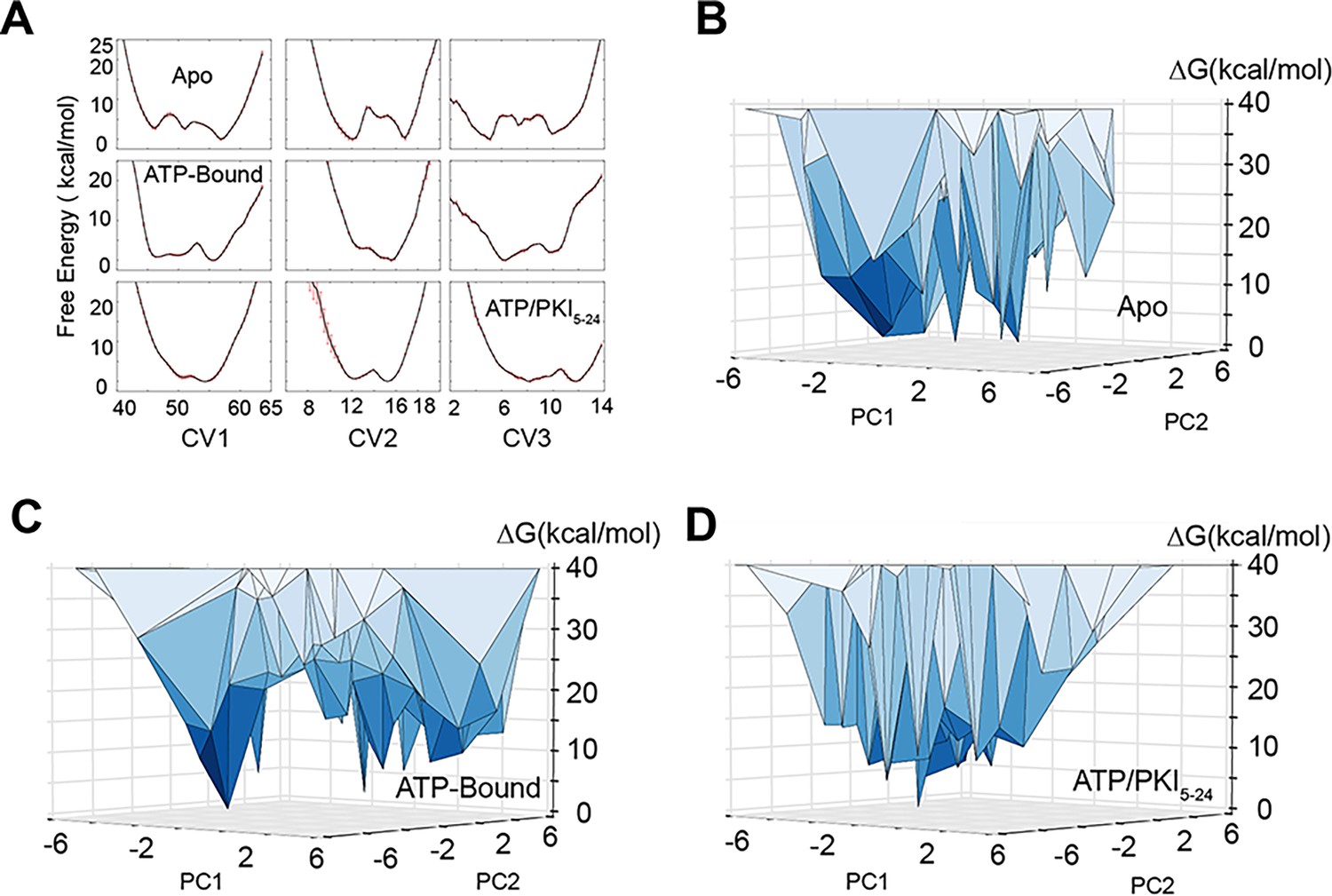
Free energy landscape of PKA-C obtained from replica-averaged metadynamics (RAM) simulations.
(A) Convergence of the bias deposition along the first three collective variables (CVs). The free energy (expressed in kcal/mol) of the different CVs was averaged over the last 100 ns of RAM simulations. The standard deviations are reported as red error bars. (B–D) Free energy landscape along the first two principal components (PC1 and PC2) of PKA-C in the apo, ATP-, and ATP/PKI-bound forms. PC1 and PC2 are projected from the first three CVs. The vertices represent conformational states. In the apo form, multiple states have comparable free energy with ΔG < 5 kcal/mol, whereas in the binary form, fewer states have ΔG < 5 kcal/mol. For the ternary form only a major ground state is populated.
-
Figure 2—source data 1
ΔG (kcal/mol) and relative population of the ground state and the first six excited states in different forms of PKA-C obtained from the replica-averaged metadynamics (RAM) simulations.
- https://cdn.elifesciences.org/articles/91506/elife-91506-fig2-data1-v1.docx
Figure 2—figure supplement 1
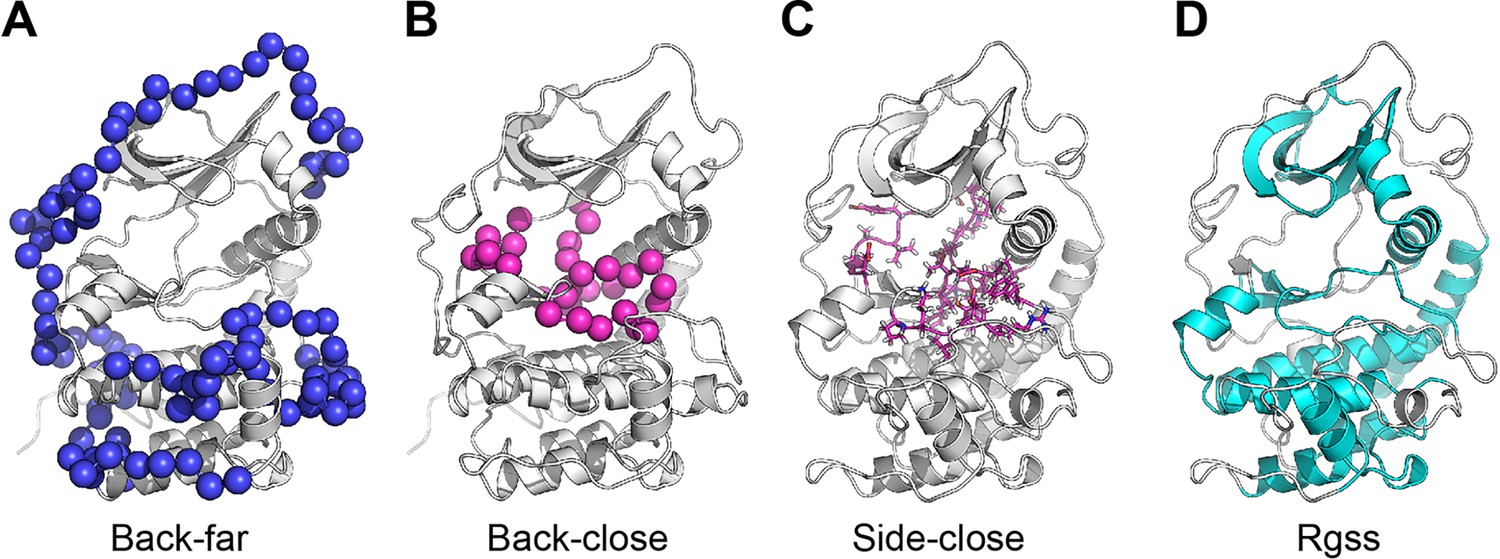
Illustration of the collective variables (CVs) used in the replica-averaged metadynamics (RAM) simulations.
(A) Backbone ψ angles of loops not in contact with ATP (Back-far). The Cα atoms are depicted as blue spheres. (B) Backbone ψ angles of loops in contact with ATP (Back-close). The Cα atoms are colored in magenta. (C) Side chains χ1 angles of loops in contact with ATP (Side-close). The side chains are represented in sticks colored in magenta. (D) The radius of gyration is calculated over the rigid part of the protein (Rgss), where the residues involved are colored in cyan.
Figure 2—figure supplement 2
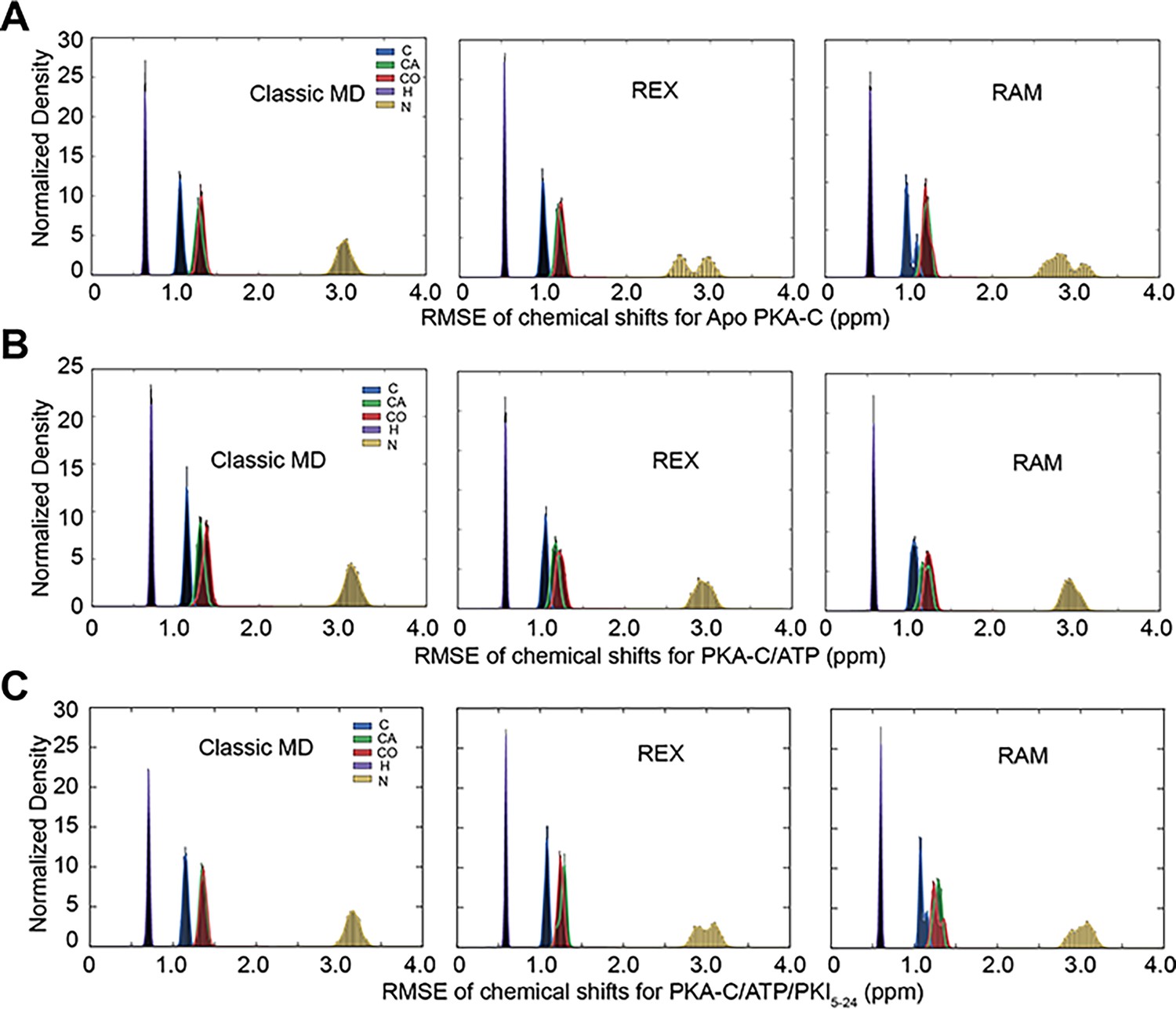
Distribution of the root-mean-square-error (RMSE) of the chemical shifts (CSs) for the different simulation schemes.
(A) RMSE of CS for apo PKA-C calculated from standard MD (left), replica exchange (REX; middle), and replica-averaged metadynamics (RAM) (right). (B) RMSE of CS for PKA-C/ATP calculated from standard MD (left), REX (middle), and RAM (right). (C) RMSE of CS for PKA-C/ATP/PKI5-24 from standard MD (left), REX (middle), and RAM (right). Color codes for different backbone atoms (C, Cα, CO, H, and N) are indicated in the left figures.
Figure 2—figure supplement 3
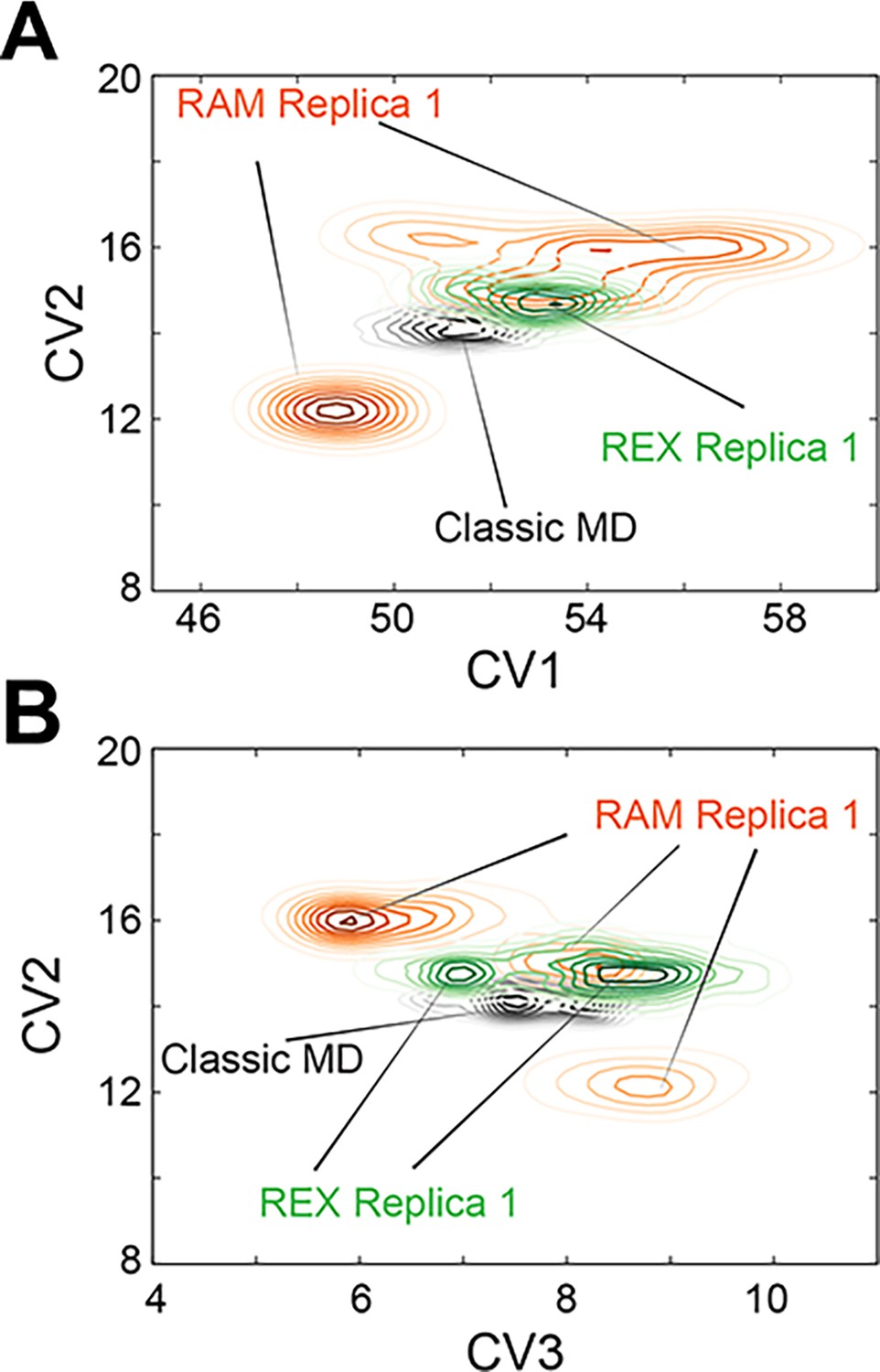
Replica-averaged metadynamics (RAM) simulations explore a larger conformational space than standard MD and replica exchange (REX) simulations.
(A) Comparison of conformational space (CV1 vs. CV2) sampled by RAM Replica 1, standard MD, and REX Replica 1 for PKA-C. (B) Comparison of conformational space (CV3 vs. CV2) sampled by RAM Replica 1, standard MD, and REX Replica 1 for apo PKA-C.
Figure 2—figure supplement 4
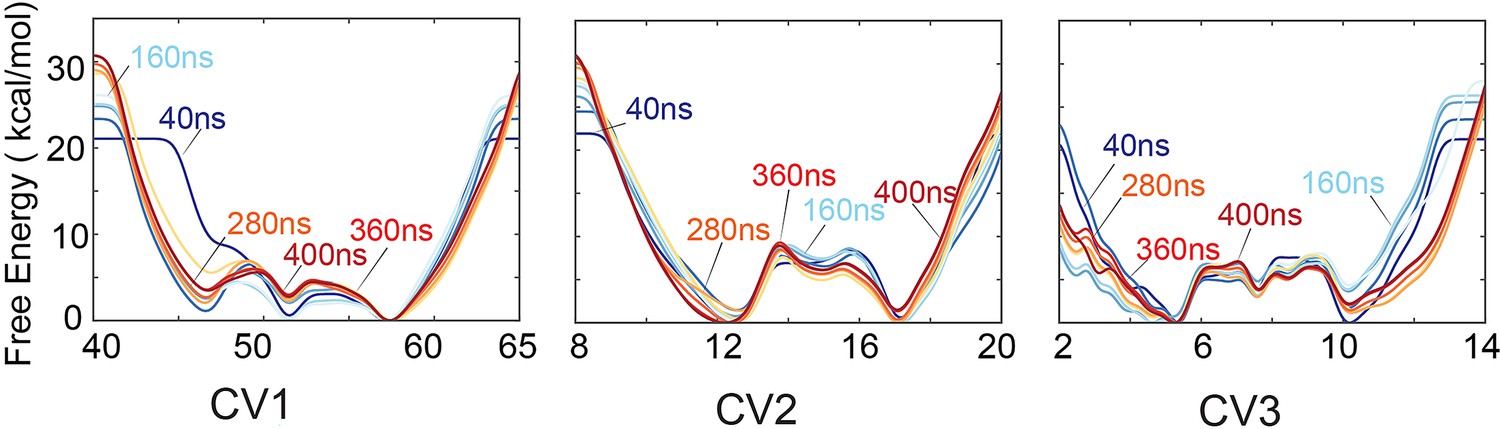
Accumulative deposition of history-dependent biases along the first three collective variables (CVs) for the replica-averaged metadynamics (RAM) simulations of the apo PKA-C.
The accumulative biases converged after ~300 ns in the three CVs.
Figure 3 with 3 supplements
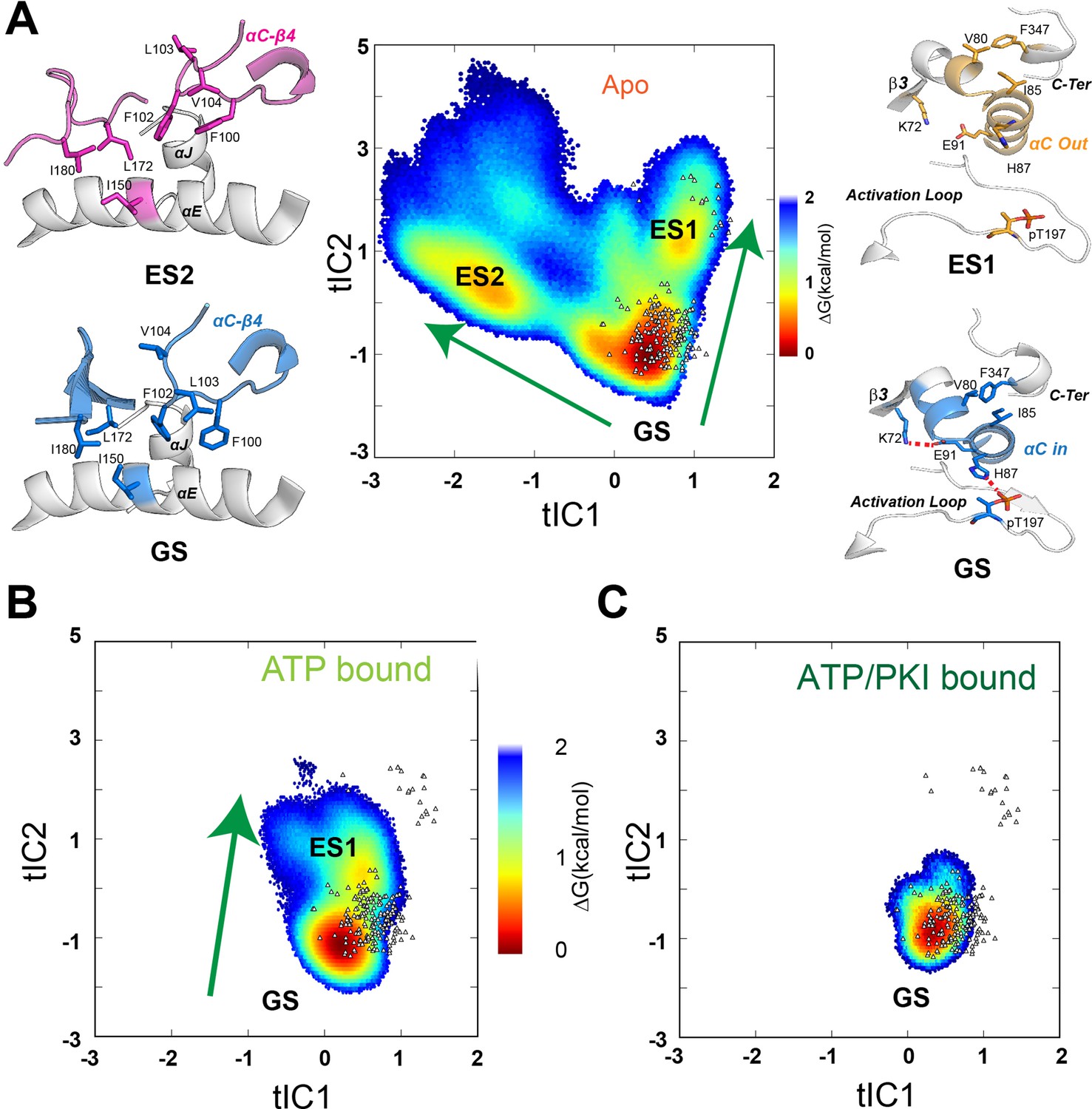
Free energy surfaces and dynamic transitions determined by a Markov State Model (MSM) for apo, ATP-, and ATP/PKI-bound PKA-C.
(A) Free energy landscape projected along the first two time-lagged independent components (tICs) of apo PKA-C, featuring three basins, ground state (GS), ES1, and ES2. The transition from GS to ES1 (arrow) highlights the changes around the αB-αC loop, with the disruption of the K72–E91 salt bridge and the PIF pocket (V80–I85–F347) hydrophobic interactions. The GS to ES2 transition (arrow) displays the rearrangement of the hydrophobic packing around the αC-β4 loop. (B, C) Free energy surfaces projected along the first two tICs for the ATP- and ATP/PKI-bound PKA-C, respectively. Known crystal structures for the three forms are indicated by small white triangles.
Figure 3—figure supplement 1
R spine and shell residues selected for two time-lagged independent components (tICA) and Markov State Model (MSM) analysis.
(A) Atom motions of key residues that define tIC1 of apo PKA-C colored according to the superposition deviations. Backbone atoms of Val104 show the largest change in tIC1. (B) Atom motions of key residues that define tIC2 of apo PKA-C colored according to the superposition deviations. Backbone atoms of Phe185 and Val104 show the largest change in tIC2.
Figure 3—figure supplement 2
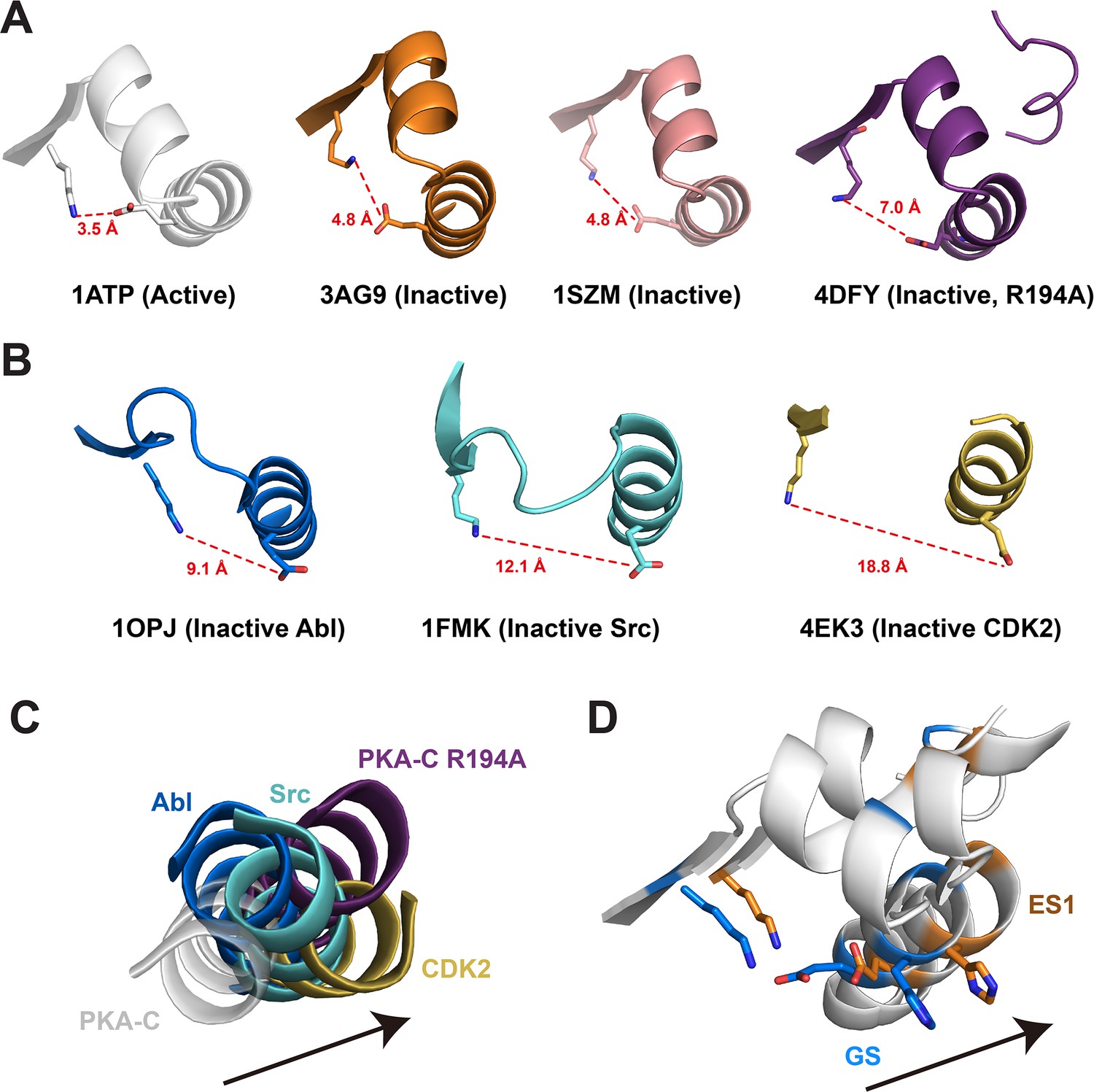
Structural features of αC-out transition (ES1) in various inactive kinases.
(A) Crystal structures of PKA-C in active and inactive states highlight the αB-αC loop alterations. Crystal structures of PKA-C in the active (1ATP) and inactive conformations (3AG9, 1SZM, 4DFY), highlighting the electrostatic interactions between K72 in β3 and E91 in the αC. (B) Disruption of the K72–E91 salt bridge in the inactive structures of Abl (1OPJ), Src (1FMK), and CDK2 (4EK3). (C) The αC helix orientation of active PKA-C compared to the orientation in inactive kinases. (D) Structural transition from the ground state (GS) to ES1 state characterized by the outward movement of the αC helix (i.e., kinase inactivation).
Figure 3—figure supplement 3
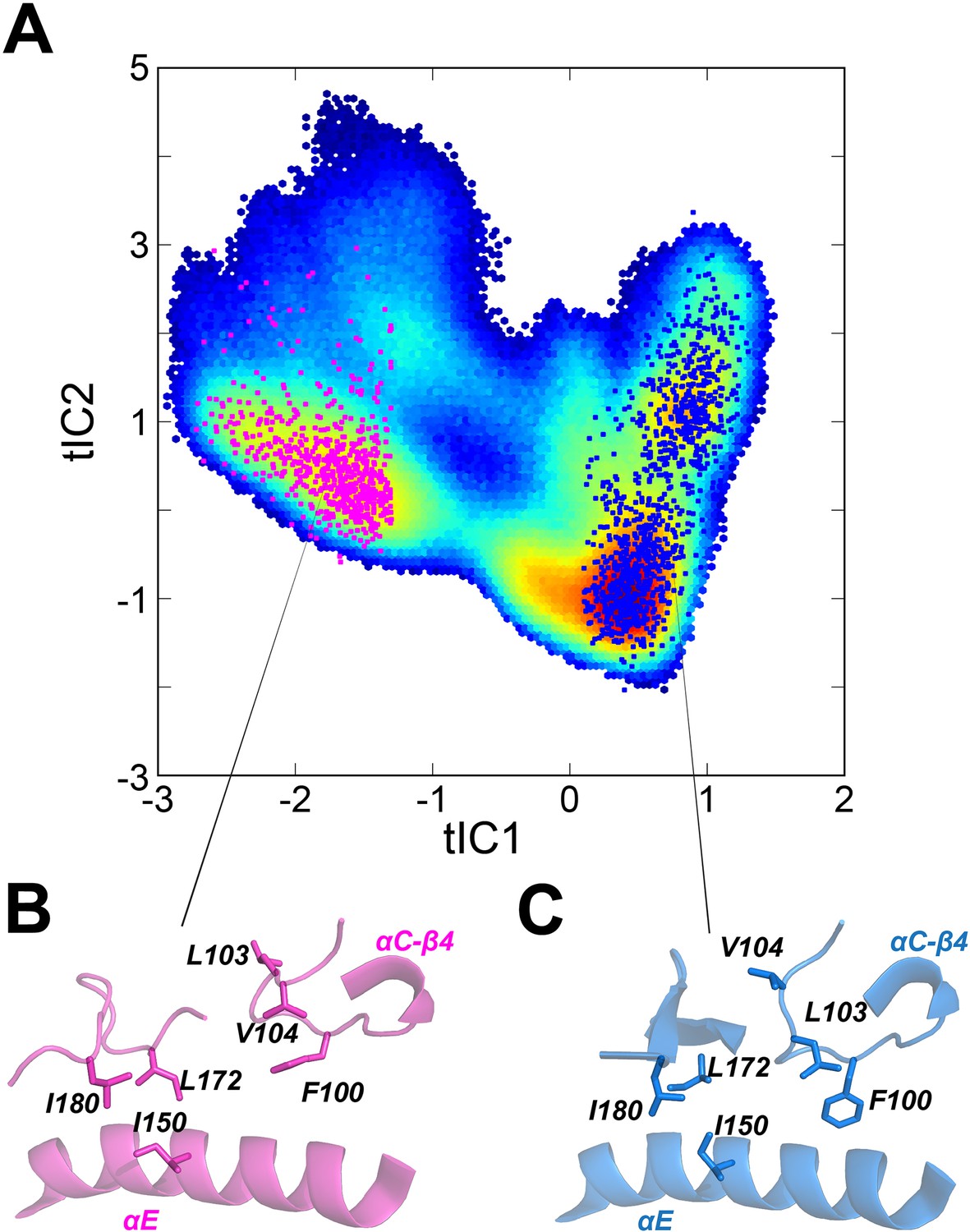
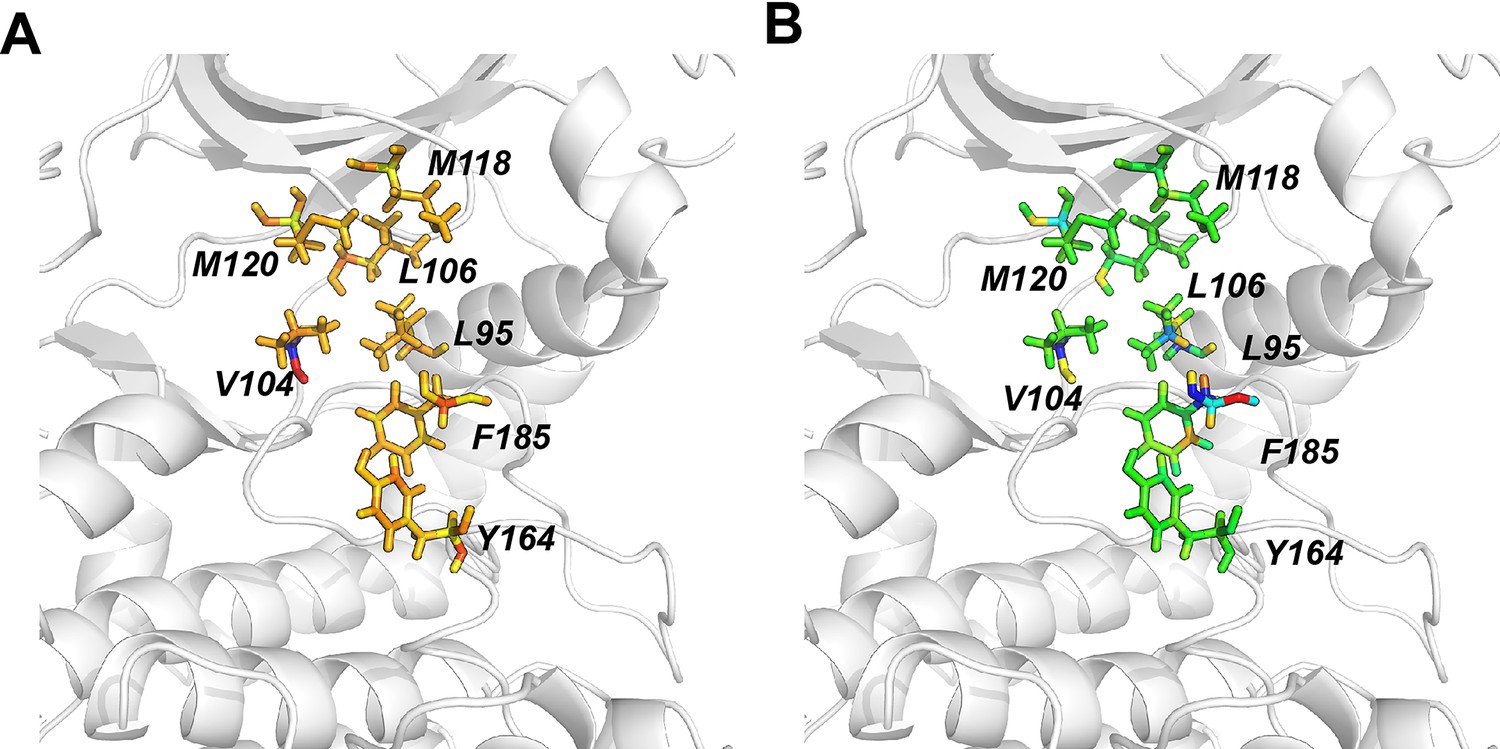
Distinct hydrophobic packing for residues around the αC-β4 loop in the ground state (GS) and ES states of apo PKA-C.
(A) Projections of randomly selected conformations for the GS (blue) and ES (magenta) onto the conformational landscape of apo PKA-C. Snapshots with tIC1 <1.2 were clustered to separate the ES and GS, whereas those confomers with tIC1 >0.2 were clustered as GS. (B, C) Close up of the hydrophobic packing in the ES and GS (C) states, highlighting Leu103, Val104, Ile150, Leu172, and Ile180 that show slow exchange in the Carr–Purcell–Meiboom–Gill (CPMG) experiments.
Figure 4
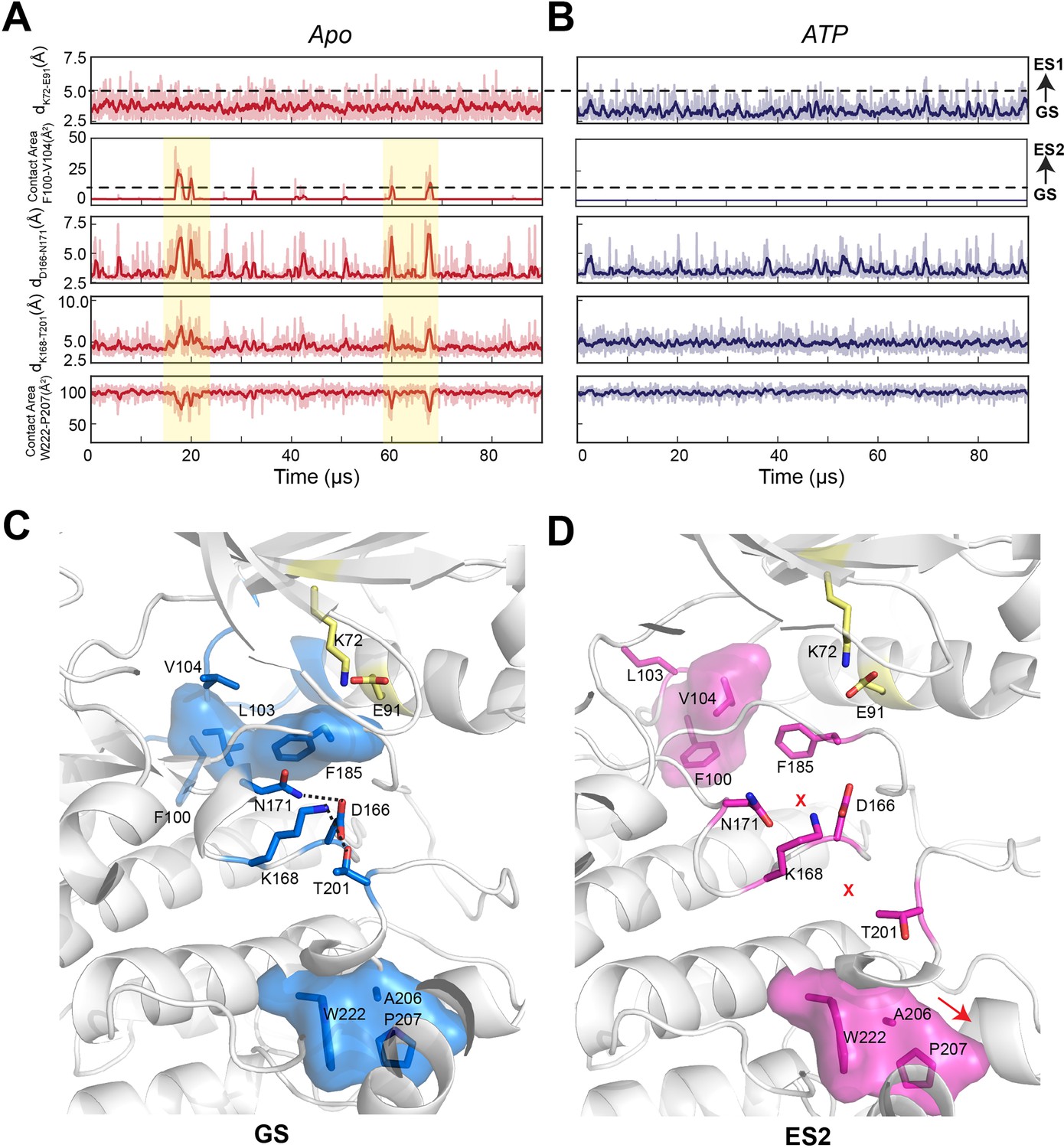
Conformational transitions of apo and ATP-bound PKA-C from ground state (GS) to ES1 and ES2 states as shown by the kinetic Monte Carlo trajectories.
(A, B) Time course of the structural transitions from GS to ES1 and GS to ES2 for apo and ATP-bound PKA-C, respectively. The GS to ES1 transition is characterized by the disruption of the K72–E91 salt bridge and occurs frequently for both PKA-C forms. In contrast, the structural transition from GS to ES2 occurs only for the apo PKA-C, and it features the interactions between F100 and V104 that cause allosteric changes between D166–N171, K168–T201, and W222–A206–P207. The dark color traces indicate the moving averages calculated every 10 frames. (C) Structural snapshot of the GS conformation showing that the key catalytic motifs are poised for phosphoryl transfer. (D) Structural snapshot of the ES2 conformation with a disrupted configuration of key catalytic motifs typical of inactive kinase.
Figure 5 with 1 supplement
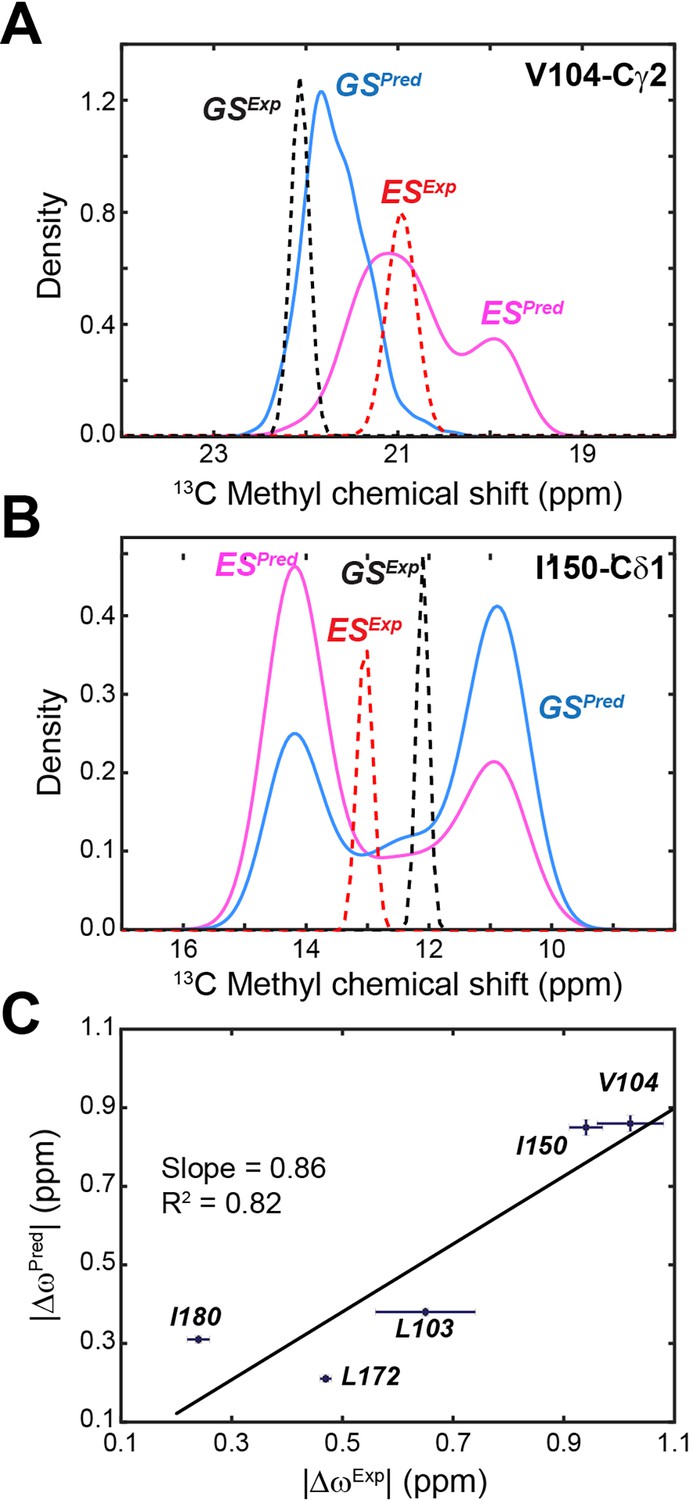
Comparison of experimental versus calculated 13C chemical shifts (CSs) of methyl groups of PKA-C.
(A) Experimental and calculated CS for Val104-Cγ1 of apo PKA-C. The ground state (GS) is in blue and the ES in magenta. (B) Corresponding CS profiles for Ile150-Cδ1. The experimental CS is shown in dotted lines for GS (black) and ES (red). (C) Correlation between predicted |ΔωPred| and experimental |ΔωExp| CS differences for methyl groups near the αC-β4 loop. The fitted linear correlation has a slope of 0.86 and R2 of 0.82. The experimental errors were estimated from the signal-to-noise ratios of the CEST spectra.
Figure 5—figure supplement 1
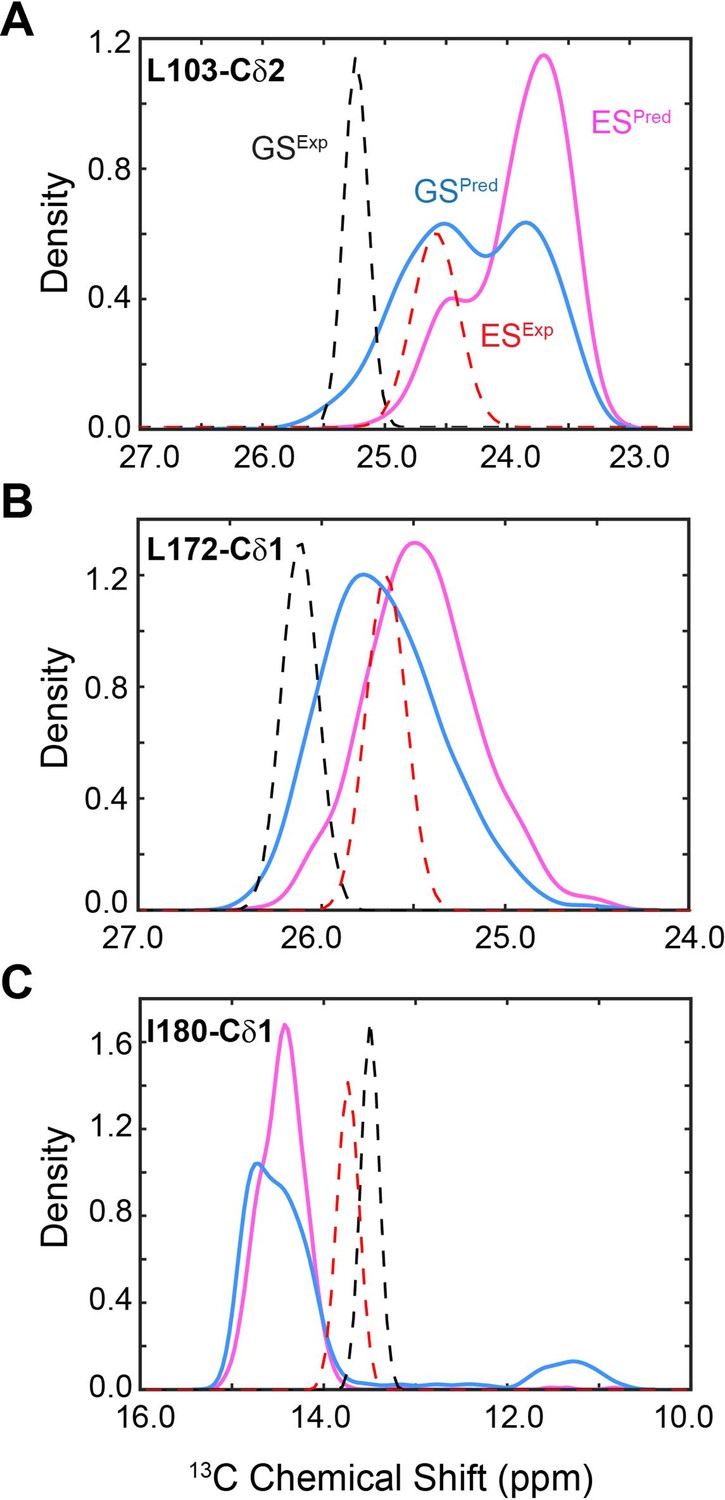
Distribution of predicted and experimental 13C chemical shift (CS) of selected methyl groups.
ES (magenta) and ground state (GS; blue) of apo PKA-C for Leu103-Cδ2 (A), Leu172-Cδ1 (B), and Ile180-Cδ1 (C). The experimental CSs are shown as dotted lines for GS (black) and ES (red).
Figure 6 with 1 supplement
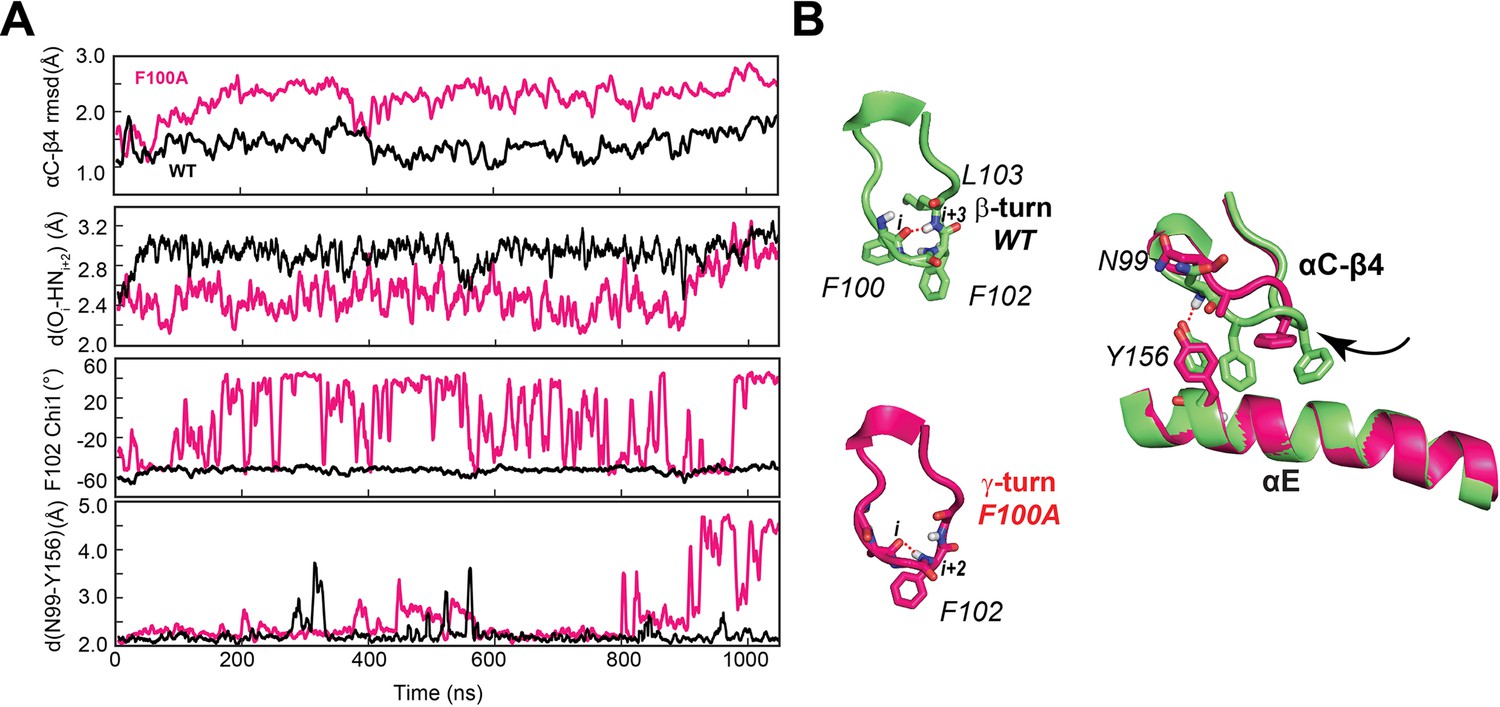
F100A mutation increases the dynamics of the αC-β4 loop, perturbing the local hydrophobic packing and its anchoring to the αE helix.
(A) Time series of the αC-β4 loop dynamics, H-bond occurrence for the β- and γ-turns, F102 χ1 angle, and N99 and Y156 for WT (black) and F100A (red) in the ATP-bound state. (B) Representative structural snapshots showing the β-turn conformation for PKA-CWT (green) and γ-turn for PKA-CF100A (magenta).
Figure 6—figure supplement 1
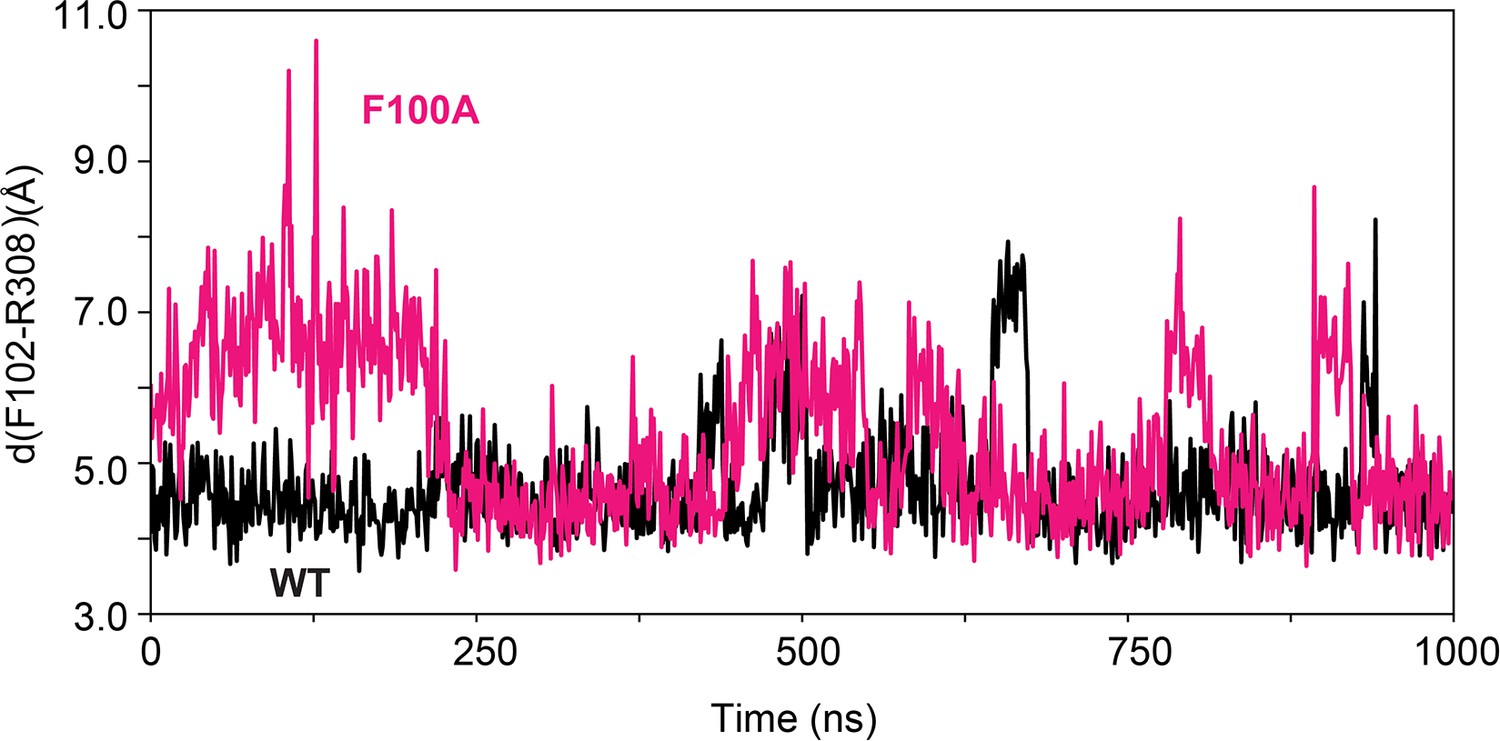
Time series of the distance between F102 and R308 for PKA-CWT (black) and the PKA-CF100A mutant (magenta).
The π-cation interactions between the aromatic ring of F102 and the guanidine group of R308 are more persistent in the WT enzyme than in the F100A mutant.
Figure 7
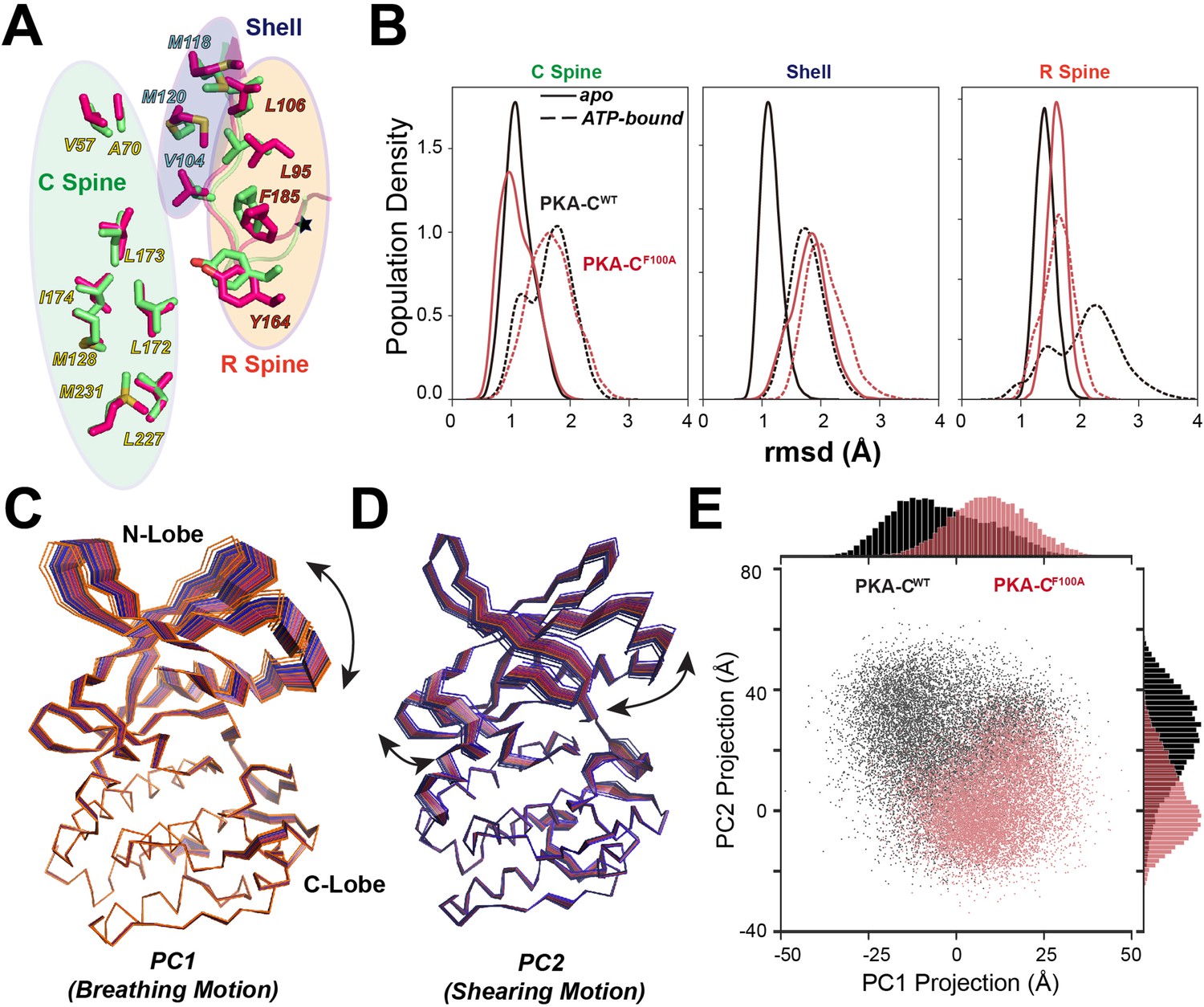
Structural responses to ATP binding of PKA-CF100A mutant.
(A) Superposition of the hydrophobic cores (C spine, R spine, and shell residues) for PKA-CWT (lime) and PKA-CF100A (hot pink), highlighting the structural perturbations of the R spine and shell residues. (B) Structural perturbation upon ATP binding for the hydrophobic core residues of PKA-CWT and PKA-CF100A shown as changes in the population densities versus rmsd. (C, D) First (PC1) and second (PC2) principal components describing breathing and shearing motions of the two lobes. (E) 2D projections and distributions of PC1 and PC2 for PKA-CWT and PKA-CF100A.
Figure 8
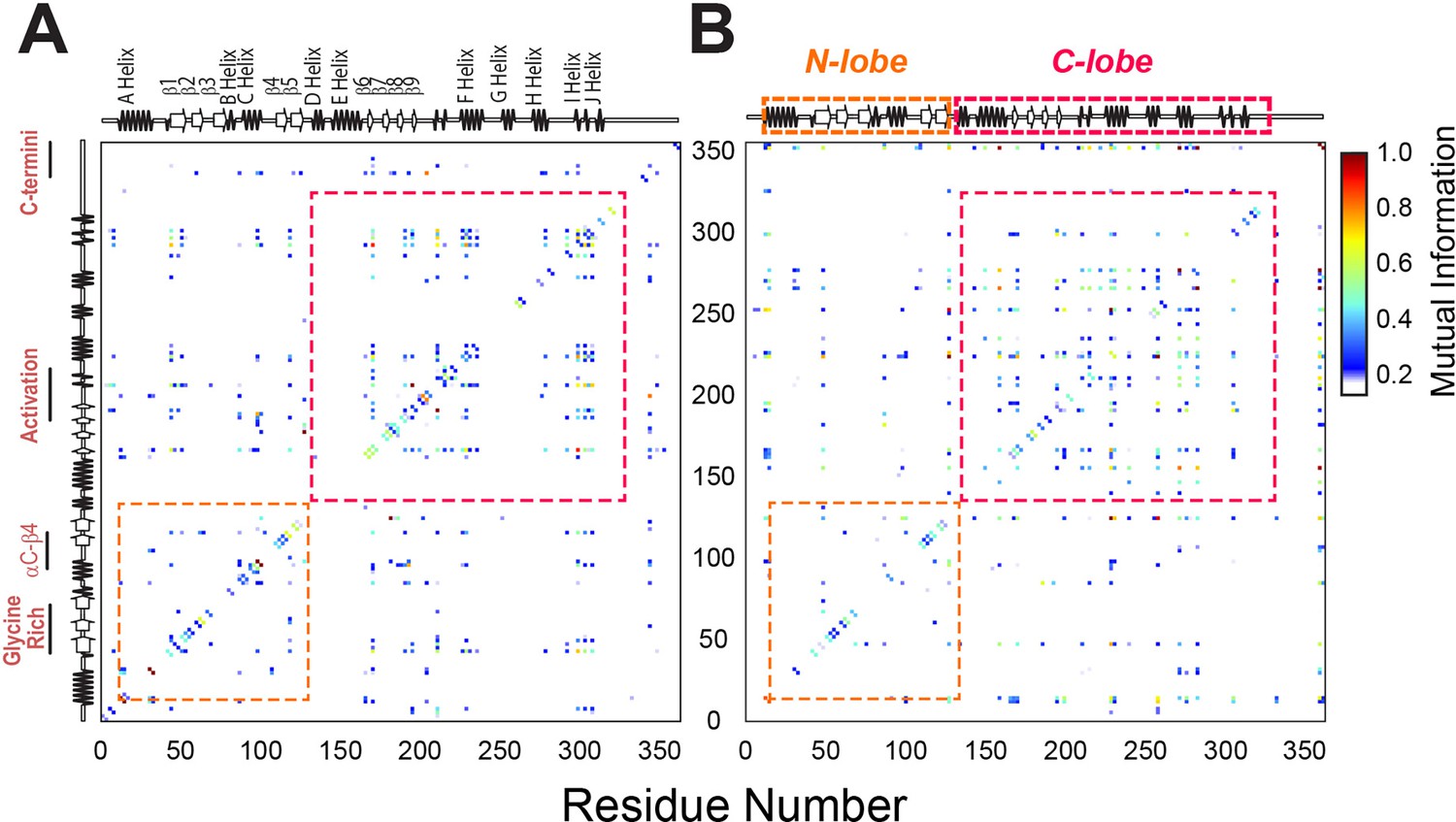
Mutual information analysis of backbone and side chain rotamers of ATP-bound PKA-CWT and PKA-CF100A.
(A) Mutual information matrix for PKA-CWT showing well-organized clusters of interactions within each lobe and distinct inter-lobe communication typical of an active kinase. (B) Mutual information matrix for PKA-CF100A revealing an overall reorganization of the allosteric network caused by the disruption of the hydrophobic core. Effects of the F100A mutation on the catalytic efficiency and binding thermodynamics of PKA-C.
Figure 9 with 2 supplements
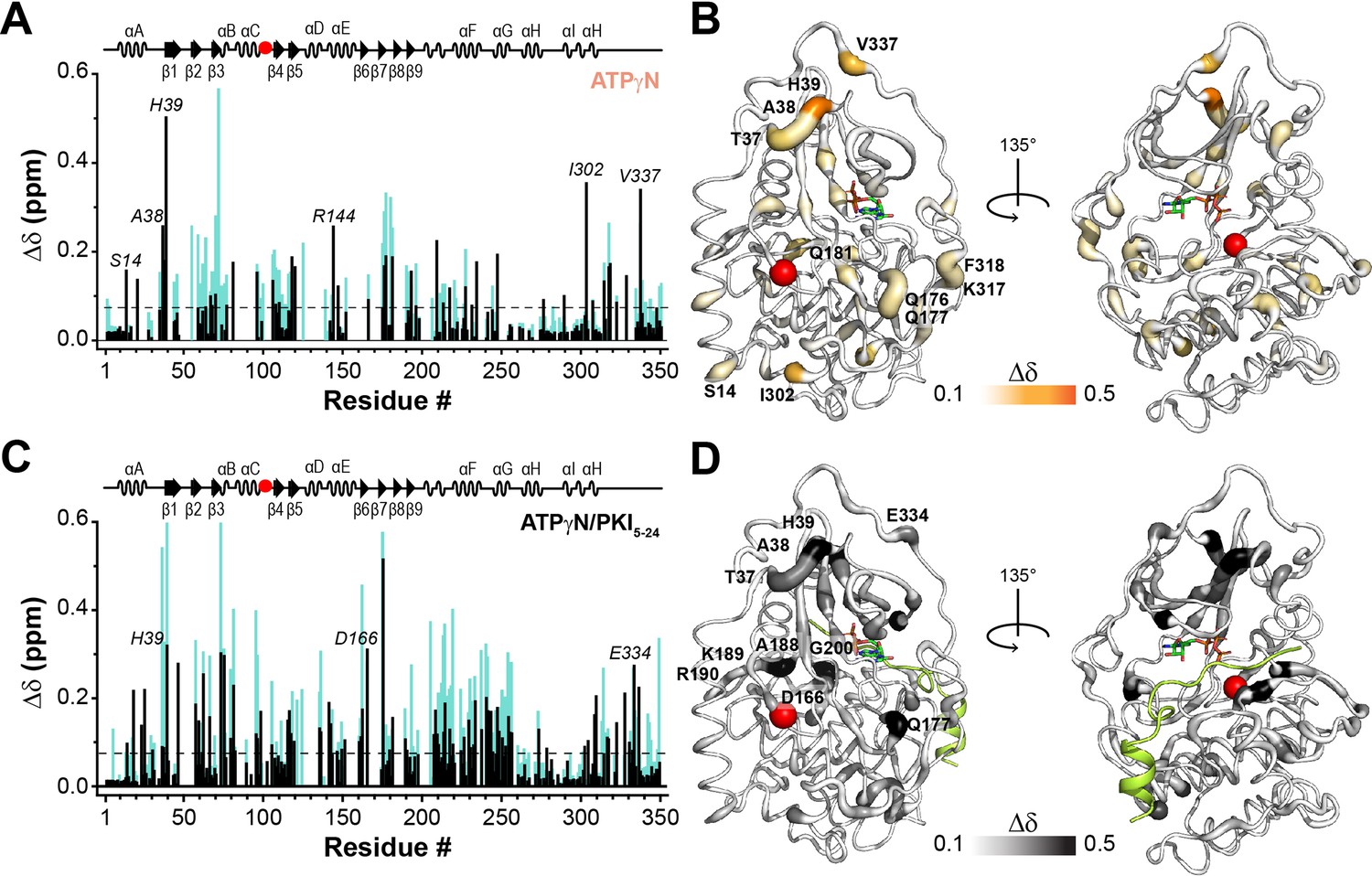
NMR map of the structural response of PKA-CF100A to nucleotide and protein kinase inhibitor (PKI) binding.
(A) Comparison of the chemical shift perturbation (CSP) of the amide resonances for PKA-CF100A (black) and PKA-CWT (cyan) upon ATPγN binding. The dashed line indicates one standard deviation from the average CSP. (B) CSPs of PKA-CF100A/ATPγN amide resonances mapped onto the crystal structure (PDB: 4WB5). (C) Comparison of the CSPs of the amide resonances for PKA-CF100A and PKA-CWT upon binding ATPγN and PKI5-24 (black). (D) CSPs for the F100A/ATPγN/PKI complex mapped onto the crystal structure (PDB: 4WB5).
Figure 9—figure supplement 1
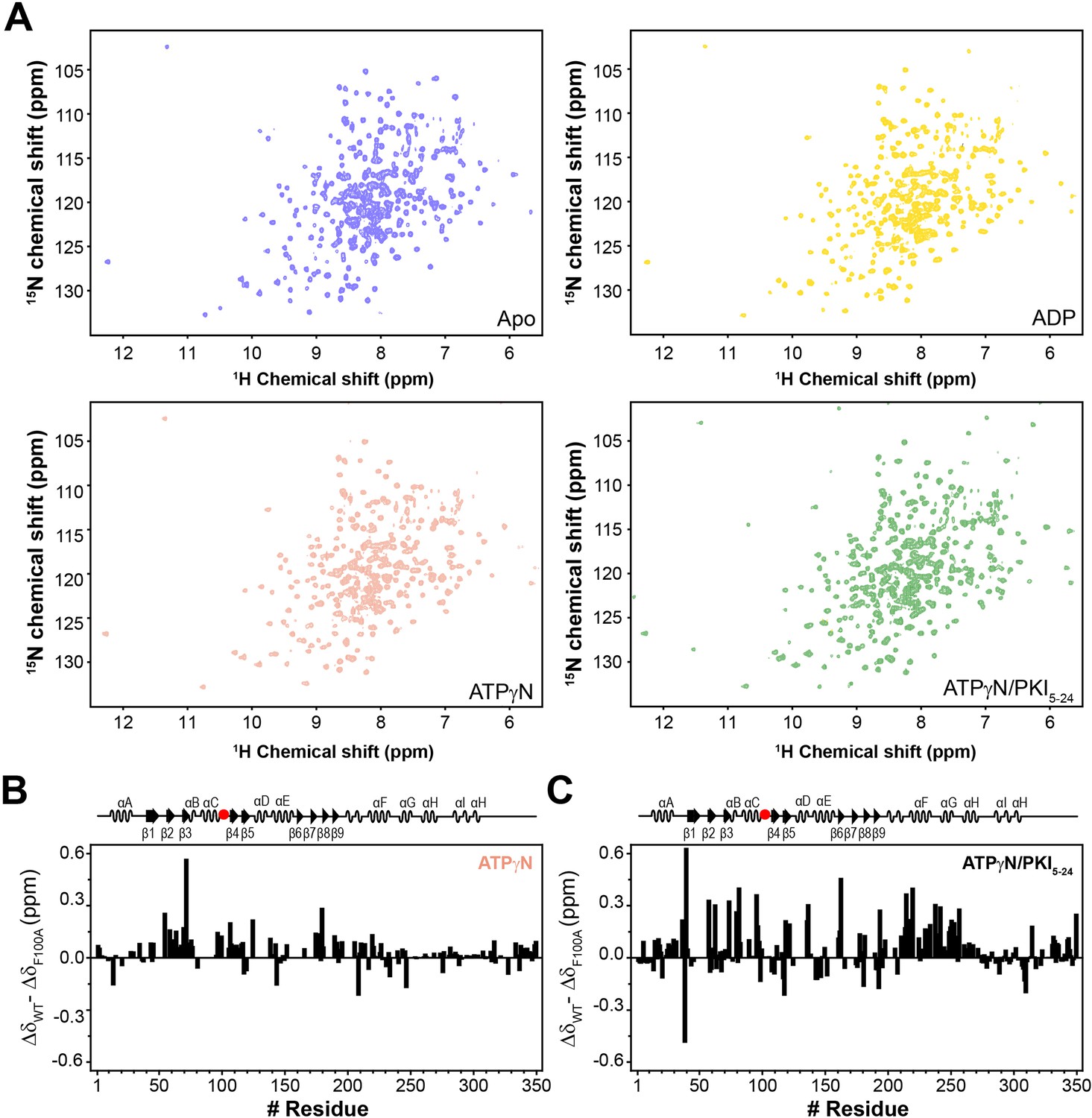
NMR fingerprints of PKA-CF100A.
(A) [1H,15N]-WADE-TROSY spectra of apo, ADP-, ATPγN-, and ATPγN/PKI5-24-bound PKA-CF100A. (B) Changes in the chemical shift perturbation (CSP) between PKA-CWT and PKA-CF100A bound to ATPγN. (C) Changes in CSP (ΔδWT − ΔδF100A) upon binding ATPγN and PKI5-24.
Figure 9—figure supplement 2
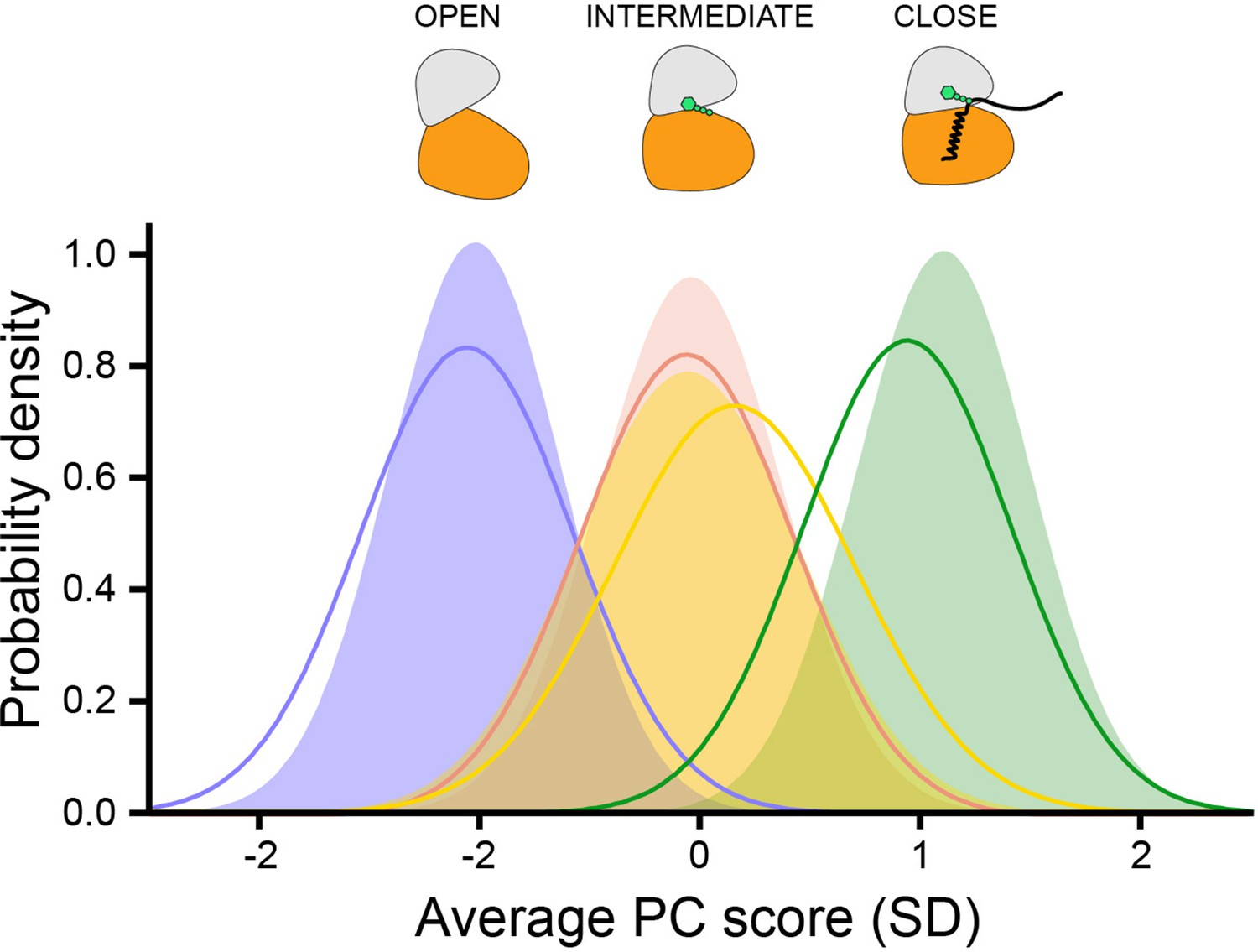
COordiNated ChemIcal Shifts bEhavior (CONCISE) plot showing the shifts of the probability distribution of the amide resonances as a function of nucleotides and substrate binding.
The per-residue chemical shift information is averaged into the average principal component (PC) score indicative of the position of each conformational state of the kinase along the open-to-closed equilibrium.
Figure 10 with 1 supplement
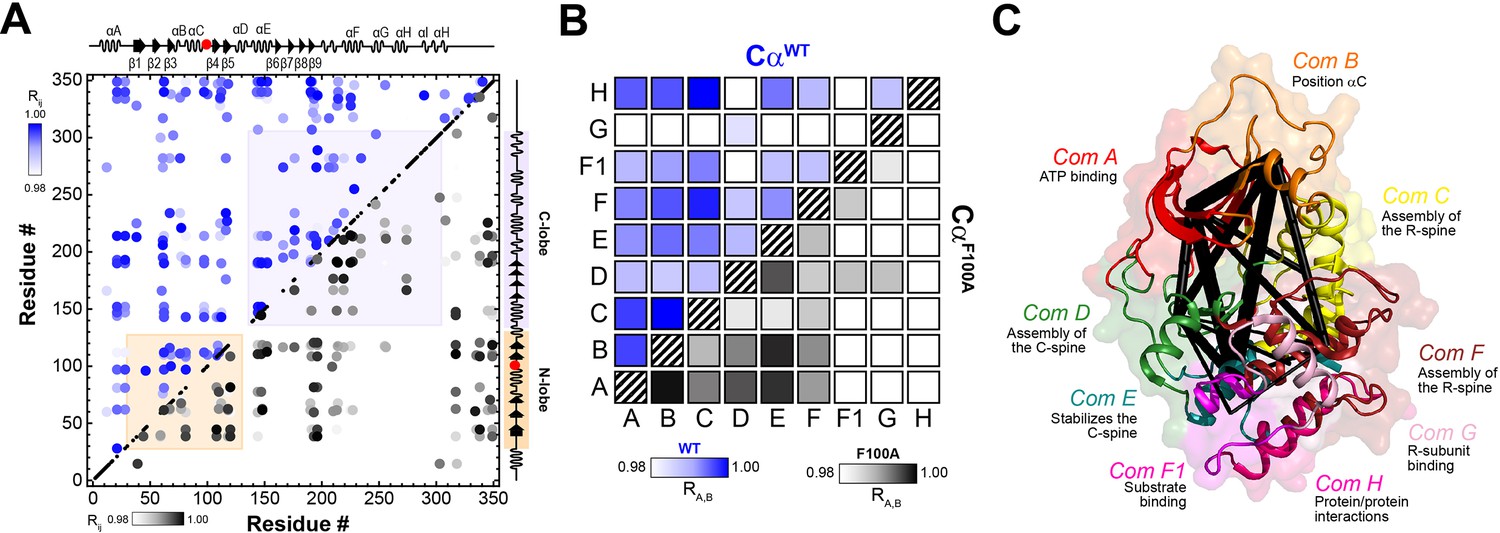
Correlated chemical shift changes reveal the uncoupling of the intramolecular allosteric network in PKA-CF100A.
(A) Comparison of the CHEmical Shift Covariance Analysis (CHESCA) matrices obtained from the analysis of the amide CS of PKA-CWT (blue correlations) and PKA-CF100A (black correlations). The correlations coefficients (Rij) were calculated using the apo, ADP-, ATPγN-, and ATPγN/PKI5-24-bound states. For clarity, only correlation with Rij > 0.98 are displayed. For the enlarged CHESCA map of F100A see Figure 10—figure supplement 1. The data for the PKA-CWT matrix were taken from Walker et al., 2019. (B) Community CHESCA analysis of PKA-CWT (blue correlations) and PKA-CF100A (black correlations). Only correlations with RA,B > 0.98 are shown. (C) Spider plot showing the extent of intramolecular correlations identified by the community CHESCA analysis for PKA-CF100A mapped onto the crystal structure (PDB: 4WB5). The thickness of each line in the spider plot indicates the extent of coupling between the communities.
Figure 10—figure supplement 1
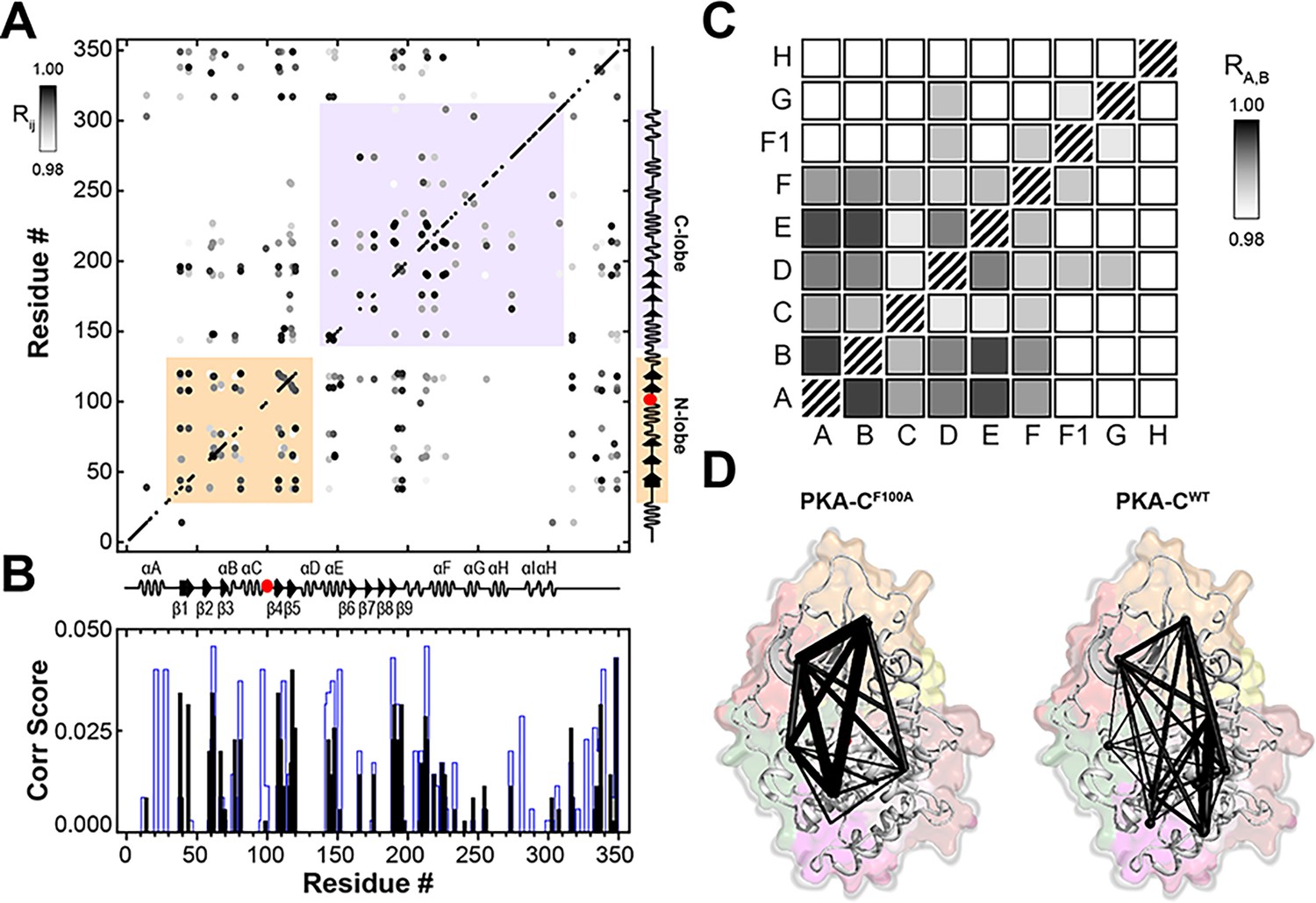
Intermolecular allosteric network of F100A mapped using CHEmical Shift Covariance Analysis (CHESCA) and community CHESCA.
(A) CHESCA matrix obtained from the amide chemical shift trajectories of PKA-CF100A in the apo, ADP-, ATPγN-, and ATPγN/PKI5-24-bound states. Only correlations with Rij > 0.98 are displayed. (B) Plot of the correlation scores versus residue calculated for PKA-CWT (blue) and PKA-CF100A (black). (C) Community CHESCA analysis of PKA-CF100A. Only correlations with RA,B > 0.98 are shown. (D) Spider plots indicating the correlated structural communities of PKA-CF100A and PKA-CWT plotted on their corresponding structures. The size of each node is independent of the number of residues it encompasses, and the weight of each line indicates the strength of coupling between the individual communities.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Homo sapiens) | PKA-Cα | PKA-C | Uniprot ID: P17612 | |
| Strain, strain background (Escherichia coli) | BL21(DE3) pLyss | Agilent | Cat. #200132 | Chemically competent cells |
| Sequence-based reagent | PKA-CF100A | This study | PCR primer (Forward) | tattctgcaagcggtgaacgccccgtttctggttaagctg |
| Sequence-based reagent | PKΙα 5-24 | Synthetic peptide | PKI5-24 | Chemically synthesized |
| Sequence-based reagent | Kemptide | Synthetic peptide | Kemptide | LRRASLG |
| Commercial assay or kit | QuikChange Lightning Multi Mutagenesis Kit | Agilent genomics | Cat #210519 | Commercial mutagenesis kit |
| Recombinant DNA reagent | PKA-CF100A | This study | PKA-CF100A | Single Ala mutant of PKA-C |
| Chemical compound, drug | AMP-PNP or ATPγN | Roche Applied Science | CAS 25612-73-1 | ATP analogous |
| Chemical compound, drug | ADP | Sigma-Aldrich | CAS 20398-34-9 | Nucleotide |
| Software, algorithm | TopSpin 4.1 | Bruker Inc | https://www.bruker.com/ | |
| Software, algorithm | NMRFAM-Sparky | NMRFAM | https://nmrfam.wisc.edu/nmrfam-sparky-distribution/ | |
| Software, algorithm | NMRPipe | Delaglio F., NIH | https://www.ibbr.umd.edu/nmrpipe/install.html | |
| Software, algorithm | POKY | Lee W. | https://sites.google.com/view/pokynmr | |
| Software, algorithm | COordiNated ChemIcal Shift bEhavior (CONCISE) | Veglia G. | https://conservancy.umn.edu/handle/11299/217206 https://conservancy.umn.edu/handle/11299/227294 | Matlab script |
| Software, algorithm | CHEmical Shift Covariance Analysis (CHESCA) | Melacini G. | https://academic.oup.com/bioinformatics/article/37/8/1176/5905475?login=true | NMRFAM-Sparky &POKY tools |
| Software, algorithm | PyMol | Schrödinger, LLC | https://pymol.org | |
| Software, algorithm | MatLab2022b | MathWorks | https://www.mathworks.com/products/matlab.html | |
| Software, algorithm | GraphPad Prism 9 | GraphPad Software Inc | https://www.graphpad.com/ | |
| Software, algorithm | GROMACS 4.6 | Hess B et al. | http://www.gromacs.org/ | |
| Software, algorithm | CHARMM36a1 | Best et al., 2012 | https://doi.org/10.1021/ct300400x | |
| Software, algorithm | PLUMED 2.1.1 | Bonomi et al., 2009 | https://www.plumed.org/doc-v2.5/user-doc/html/_c_h_a_n_g_e_s-2-1.html | |
| Software, algorithm | ALMOST 2.1 | Kohlhoff et al., 2009. | https://svn://svn.code.sf.net/p/almost/code/almost-code | |
| Software, algorithm | METAGUI | Biarnés et al., 2012 | https://www.sciencedirect.com/science/article/pii/S0010465511003079 | |
| Software, algorithm | MDTraj | McGibbon et al., 2015 | https://www.sciencedirect.com/science/article/pii/S0006349515008267 | |
| Software, algorithm | SPARTA+ | Shen and Bax, 2010 | https://spin.niddk.bov/bax/software/SPARTA+/ | |
| Software, algorithm | MSMbuilder | Harrigan et al., 2017; | https://msmbuilder.org/ | |
| Software, algorithm | Mutinf | McClendon et al., 2012 | https://simtk.org/projects/mutinf/ |
Additional files
-
Supplementary file 1
Kinetic parameters of Kemptide phosphorylation by PKA-CWT and PKA-CF100A obtained from coupled assays.
The KM and Vmax values were obtained from a nonlinear least squares analysis of the concentration-dependent initial phosphorylation rates. Errors in the kcat/KM ratios were propagated from the individual errors in KM and kcat.
- https://cdn.elifesciences.org/articles/91506/elife-91506-supp1-v1.docx
-
Supplementary file 2
Changes in enthalpy, entropy, free energy, and dissociation constants for nucleotide binding to PKA-CWT and PKA-CF100A.
All errors were calculated from triplicate measurements. Values for PKA-CWT are taken from Walker et al., 2019
- https://cdn.elifesciences.org/articles/91506/elife-91506-supp2-v1.docx
-
Supplementary file 3
Changes in enthalpy, entropy, free energy, and dissociation constants for PKI5-24 binding to apo and ATPγN - saturated PKA-CWT and PKA-CF100A.
All errors were derived from triplicate measurements. The error for the cooperativity coefficient (σ) was propagated from the errors in Kd. Values for PKA-CWT were originally published in Walker et al., 2019.
- https://cdn.elifesciences.org/articles/91506/elife-91506-supp3-v1.docx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/91506/elife-91506-mdarchecklist1-v1.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The αC-β4 loop controls the allosteric cooperativity between nucleotide and substrate in the catalytic subunit of protein kinase A
eLife 12:RP91506.
https://doi.org/10.7554/eLife.91506.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}