Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo
- Division of Molecular and Cellular Biology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, United States
Figures
Figure 1 with 1 supplement

Elimination of eIF2A has no effect on bulk protein synthesis in the presence or absence of amino acid starvation.
(A) Polysome profiles of wild-type (WT) strain (BY4741) and eIF2AΔ mutant (F2247) untreated (i–ii) or treated with sulfometuron methyl (SM) (iii–iv). For (i–ii), cells were cultured in SC medium at 30°C to log-phase and treated with 50 μg/mL of cycloheximide 5 min prior to harvesting. For (iii–iv), cells were cultured in SC medium lacking Ile/Val and treated with 1 µg/mL of SM for 20 min before addition of cycloheximide. Cell extracts were resolved by sedimentation through sucrose density gradients and scanned continuously at 260 nm during fractionation. The plots show the A260 measured across the gradient with the top of the gradient on the left. (B) Schema of translational control of GCN4 mRNA, wherein translation of the main coding sequences (CDS) is induced by phosphorylation of eIF2α through a specialized ‘delayed reinitiation’ process mediated by four short upstream open-reading frames (uORFs). (See text for details.) (C) Genome browser view of ribosome profiling data for GCN4 mRNA. Tracks display RPF or mRNA reads mapped across the transcription unit, with the scales given in rpkm (reads per kilobase of transcript per million mapped reads). Data are presented for WT (blue) and eIF2∆ cells (purple) with or without SM treatment, as indicated. Each genotype/treatment includes two biological replicates, designated _a and _b. The main CDS is shown schematically in orange below the tracks and the four uORFs are in gray. The calculated values for log2∆TEWT+SM/WT and log2∆TEeIF2A∆+SM/eIF2A∆+SM and the respective false discovery rates (FDRs) are shown on the right.
Figure 1—figure supplement 1

High reproducibility between biological replicates of ribosome footprint profiling and RNA-seq analyses.
(A–H) Scatterplots depict the RPF (A, C, E, G) or mRNA (B, D, F, H) read densities for all expressed mRNAs across biological replicates of the wild-type (WT) (A, B), the eIF2AΔ mutant (C, D), sulfometuron methyl (SM)-treated WT (E, F), and SM-treated eIF2AΔ mutant (G, H). The read densities were calculated by mapping the reads to the coding sequences (CDS) of each gene and expressed as reads per million mapped reads (RPM) in individual libraries of biological replicates. The Pearson’s coefficient (r) is indicated in each plot, quantifying the degree of correlation between the replicate datasets.
Figure 2 with 1 supplement
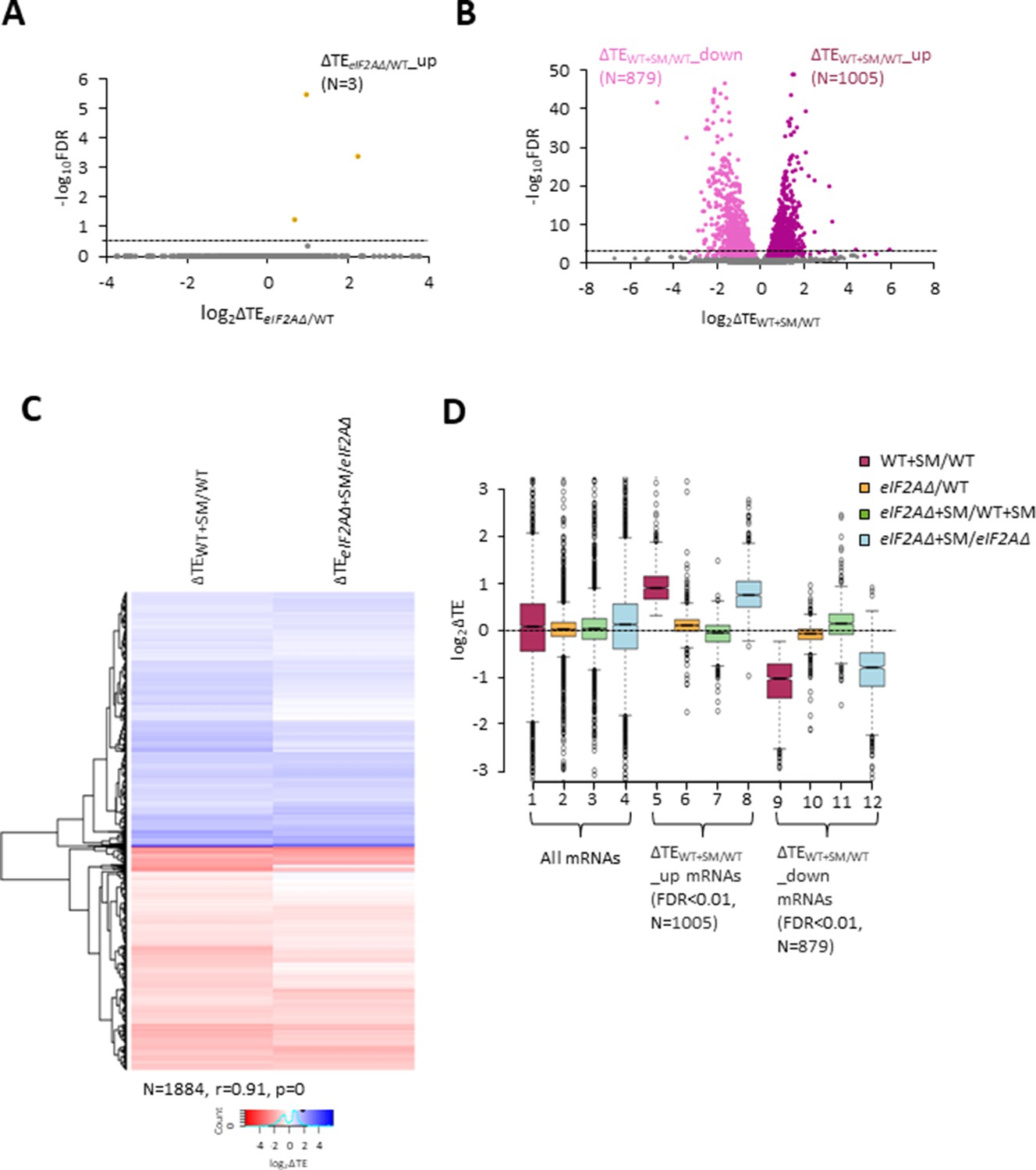
eIF2A is not critical for translation of any individual mRNAs in non-starved cells and has little impact on the reprogramming of translational efficiencies (TEs) conferred by amino acid starvation.
(A) Volcano plot depicting the log2 ratios of TEs in eIF2AΔ versus wild-type (WT) cells (log2∆TEeIF2A∆/WT values) for each mRNA (x-axis) versus negative log10 of the false discovery rate (FDR) (y-axis) determined by DESeq2 analysis of ribosome profiling data for the 5340 mRNAs with evidence of translation. Genes showing a significant increase in TE in eIF2AΔ versus WT cells at FDR < 0.25 (∆TEeIF2A∆+SM/WT_up) are plotted in orange circles. The dotted line marks the 25% FDR threshold, below which all other 5337 mRNAs are plotted in gray. (B) Volcano plot as in (A) showing the log2 ratios of TEs in WT+ sulfometuron methyl (SM) cells versus WT cells (log2∆TEWT+SM/WT values) for the 5441 mRNAs with evidence of translation. The dotted line marks the 1% FDR threshold. Genes showing a significant increase (∆TEeIF2A∆+SM/WT_up) or decrease (∆TEWT+SM/WT_down) in TE in WT+SM versus WT cells at FDR < 0.01 are plotted in magenta and pink circles, respectively. (C) Hierarchical clustering analysis of log2∆TE values for the 1884 mRNAs (arrayed from top to bottom) that exhibit significant TE decreases or increases in SM-treated versus untreated WT cells at FDR < 0.01 (defined in (B)) conferred by SM treatment of WT cells (column 1) or SM treatment of eIF2AΔ cells (column 2), with the log2ΔTE values represented on a color scale ranging from 4 (dark blue) to –4 (dark red). The Pearson coefficient (r) and corresponding p-value for the correlation between log2∆TE values in the two columns are indicated below. (D) Notched box plots of log2ΔTE values for the indicated mutant/condition for all mRNAs (columns 1–4) or for the indicated mRNA groups identified in (B). The y-axis scale was expanded by excluding a few outliers from the plots.
-
Figure 2—source data 1
Spreadsheet tabulates the log2 ratios of translational efficiencies (TEs) in eIF2AΔ versus wild-type (WT) cells (log2∆TEeIF2A∆/WT values) for each mRNA and the corresponding false discovery rate (FDR) determined by DESeq2 analysis of ribosome profiling and parallel RNA-Seq data for the 5340 mRNAs with evidence of translation (Figure 2A).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig2-data1-v1.xlsx
-
Figure 2—source data 2
Spreadsheet tabulates the log2 ratios of translational efficiencies (TEs) in SM treated versus untreated WT cells (log2∆TEWT+SM/WT values) and the corresponding false discovery rate (FDR) determined by DESeq2 analysis of ribosome profiling and parallel RNA-Seq data for the 5441 mRNAs with evidence of translation (Figure 2, Figure 2—figure supplement 1).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig2-data2-v1.xlsx
-
Figure 2—source data 3
Spreadsheet tabulates the log2 ratios log2ΔTE values for the indicated mutant/condition for the indicated mRNA groups identified in Figure 2B and D.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig2-data3-v1.xlsx
Figure 2—figure supplement 1

Relative translational efficiency (TE) changes evoked by increased eIF2α phosphorylation in cells lacking eIF2A are broadly similar to relative TE changes conferred by increased eIF2α phosphorylation in wild-type (WT) cells.
(A) Volcano plot as in Figure 2A showing the log2 ratios of TEs in sulfometuron methyl (SM)-treated eIF2A∆ versus untreated eIF2AΔ cells (∆TEeIF2A∆+SM/eIF2A∆ values) for the 5426 mRNAs with evidence of translation. The dotted line marks the 1% false discovery rate (FDR) threshold. Genes showing a significant increase (∆TEeIF2A∆+SM/eIF2A∆_up) or decrease (∆TEeIF2A∆+SM/eIF2A∆_down) in TE in SM-treated eIF2AΔ versus eIF2AΔ mutant cells at FDR < 0.05 are plotted in dark and light blue circles, respectively. (B) Proportional Venn diagram showing overlap between the 1884 mRNAs identified in Figure 2B and 786 mRNAs identified in (A).
-
Figure 2—figure supplement 1—source data 1
Spreadsheet tabulates the log2 ratios of translational efficiencies (TEs) in sulfometuron methyl (SM)-treated eIF2AΔ versus untreated eIF2AΔ cells (∆TEeIF2A∆+SM/eIF2A∆ values) for each mRNA and the corresponding false discovery rate (FDR) determined by DESeq2 analysis of ribosome profiling and parallel RNA-Seq data for the 5426 mRNAs with evidence of translation (Figure 2—figure supplement 1).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig2-figsupp1-data1-v1.xlsx
Figure 3 with 1 supplement

Examination of a small group of mRNAs showing evidence of a conditional requirement for eIF2A when eIF2 is impaired.
(A) Volcano plot as in Figure 2A showing the log2 ratios of translational efficiencies (TEs) in eIF2AΔ cells treated with sulfometuron methyl (SM) versus wild-type (WT) cells treated with SM (log2∆TEeIF2A∆+SM/WT+SM values) for the 5482 mRNAs with evidence of translation. The dotted line marks the 25% false discovery rate (FDR) threshold. Genes exhibiting a significant increase (∆TEeIF2A∆+SM/WT+SM_up) or decrease (∆TEeIF2A∆+SM/WT+SM_down) at FDR < 0.25 are plotted in dark or light green circles, respectively. (B) Notched box plots of log2ΔTE values for the indicated mutant/condition for the 32 mRNAs in the group ∆TEeIF2A∆+SM/WT+SM_down defined in (A). The y-axis scale was expanded by excluding a few outliers from the plots. Statistical significance determined using the Mann–Whitney U test is indicated for the changes in column 4 compared to the changes observed for all mRNAs. (C) Hierarchical clustering analysis of log2∆TE values for the 32 mRNAs (arrayed from top to bottom) in the group defined in (A) for the four comparisons listed across the top, with log2ΔTE values represented on a color scale ranging from 4 (dark blue) to –4 (dark red). The systematic gene names are listed for all 32 mRNAs, and the common name is indicated for those genes subjected to LUC reporter analysis below. Genes marked with ‘#’s display the pattern of TE changes consistent with conditional stimulation by eIF2A when eIF2 function is reduced by phosphorylation. Only 17 of the 32 transcripts (marked with ‘#’) displayed the diagnostic pattern of an appreciable reduction in TE both on elimination of eIF2A in SM-treated cells and on SM treatment of cells lacking eIF2A (red or pink hues in columns 1–2) but either a lesser reduction, no change, or increase in TE on SM treatment of WT cells and on elimination of eIF2A from untreated cells (light pink, white or blue hues in columns 3–4). (D) Expression of LUC reporters in different strains/conditions constructed for selected candidate genes analyzed in (C). The schematic depicts reporter construct design wherein the native gene promoter, 5' UTR, and first 20 codons of the coding sequences (CDS) are fused to firefly luciferase coding sequences (F.LUC), followed by a modified RPL41A 3' UTR. Plasmid-borne reporter constructs were introduced into the WT and eIF2AΔ strains and three independent transformants were cultured in SC-Ura medium at 30°C to log phase (-SM) or treated with SM at 1 μg/mL after log-phase growth in SC-Ura/Ile/Val and cultured for an additional 6 hr before harvesting. Luciferase activities were quantified in whole-cell extracts (WCEs), normalized to total protein, and reported as fold change in relative light units (RLUs) per mg of protein, as means (± SEM) determined from the replicate transformants. The changes in luciferase activity plotted for each of the two comparisons depicted in the histogram were calculated as ratios of the appropriate mean activities. Results of Student’s t-tests of the differences in fold changes between the indicated mutations/conditions are indicated. (E) Determination of relative TEs for the native mRNAs of selected candidate genes analyzed in (C, D). Cells were cultured in the four conditions described in (D), WCEs were resolved by sedimentation through 10–50% sucrose gradients, and fractions were collected while scanning at 260 nm. Total RNA was extracted from 80S and polysome fractions, and the abundance of each target mRNA was quantified in each fraction by qRT-PCR, and normalized for (i) the amounts of 18S rRNA quantified for the same fractions and (ii) for the total amounts of monosomes/polysomes recovered in the gradient. The resulting normalized amounts of mRNA in each fraction were multiplied by the number of ribosomes per mRNA in that fraction, summed across all fractions, and divided by the input amount of mRNA in the WCEs, normalized to ACT1 mRNA, to yield the TE for that mRNA in each condition. (See ‘Materials and methods’ for further details.) The changes in TE conferred by SM treatment of WT or eIF2AΔ cells were calculated for each replicate culture, untreated or SM-treated, and the mean TE changes with standard error of the means (SEMs) were plotted for the indicated comparisons. The results of Student’s t-tests of the differences in mean TE changes are indicated.
-
Figure 3—source data 1
Spreadsheet tabulates the log2 ratios of translational efficiencies (TEs) in eIF2AΔ cells treated with sulfometuron methyl (SM) versus wild-type (WT) cells treated with SM (log2∆TEeIF2A∆+SM/WT+SM values) for each mRNA and the corresponding false discovery rate (FDR) determined by DESeq2 analysis of ribosome profiling and parallel RNA-Seq data for the 5482 mRNAs with evidence of translation (Figure 3A).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig3-data1-v1.xlsx
-
Figure 3—source data 2
Spreadsheet tabulates the log2ΔTE values for the indicated mutant/condition for the 32 mRNAs in the group ∆TEeIF2A∆+SM/WT+SM_down defined in Figure 3A and B.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig3-data2-v1.xlsx
-
Figure 3—source data 3
Spreadsheet tabulates the log2ΔTE values for the indicated mutant/condition for the 32 mRNAs in the group ∆TEeIF2A∆+SM/WT+SM_down defined in Figure 3A and C.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig3-data3-v1.xlsx
-
Figure 3—source data 4
Spreadsheet tabulates the changes in luciferase activity expressed from F.LUC reporters calculated for sulfometuron methyl (SM)-treated versus untreated wild-type (WT) and SM-treated versus untreated eIF2AΔ for three biological replicates (Figure 3D).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig3-data4-v1.xlsx
-
Figure 3—source data 5
Spreadsheet 1, ‘raw CT values for mRNA from qRT’, tabulates the raw CT values for mRNA from qRT reactions for three biological replicates: a, b, and c. Spreadsheet 2, ‘Dilution factor exemplar WT_a’, provides an example file for one of the biological replicate WT (wild type) samples, illustrating how the dilution factor was calculated. Spreadsheet 3, ‘% 18S rRNA’, provides an example file illustrating how the the dilution factor was calculated. Spreadsheet 4, ‘Area % & Polysome norm factor’, tabulates following the parameters for all the biological replicates: area under peaks from UV trace; SUM 80S+polysomes; average SUM 80S+polysomes; polysome recovery normalisation factor; and % total area under 80S+polysomes from UV trace in each peak. Spreadsheet 5,’ ‘rRNA normalisation factor’, illustrates the calculations required to determine the rRNA normalization factor. Spreadsheet 6, ‘SAG1’, illustrates the calculations required to determine ∆Monosome-Polysome/total RNA (see ‘Materials and methods’ for details) (Figure 3E).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig3-data5-v1.xlsx
Figure 3—figure supplement 1

Representative separation of polysomes by sedimentation through a sucrose density gradient in the experiment depicted in Figure 3E.
The A260 values were determined continuously during fractionation of the gradient. Fractions pooled for isolation of RNA from 80S monosome or the various polysomal species are indicated by boxes.
Figure 4

eIF2A has little or no effect on the translation of three mRNAs reported to contain internal ribosome entry sites (IRESs).
Genome browser views of RPF and RNA reads from ribosome profiling data for (A) URE2 mRNA, (B) GIC1 mRNA, and (C) PAB1 mRNA presented as in Figure 1C. The calculated values for log2∆TEeIF2A∆/WT and log2∆TEeIF2A∆+SM/WT+SM with the respective false discovery rates (FDRs) are shown on the right. The scale is shown on the left. The region containing the URE2 IRES is enclosed in a dotted box, with the AUG start codon highlighted in red. Locations of the GIC1 and PAB1 IRESs have not been defined.
Figure 5

eIF2A plays little or no role in upstream open-reading frame (uORF)-mediated translational control of CPA1 or YAP2/CAD1 mRNA.
Figure 6 with 2 supplements

Minimal effects of eliminating eIF2A on translation of mRNAs harboring translated upstream open-reading frames (uORFs).
(A) Notched box plots of log2ΔTE values for all mRNAs (for which translational efficiencies (TEs) could be determined from our ribosome profiling data) containing annotated AUG- or NCC-uORFs (i), conserved AUG- or NCC-uORFs (ii), or single functional inhibitory AUG-uORFs (iii), conferred by sulfometuron methyl (SM) treatment of wild-type (WT) cells (maroon), by the eIF2AΔ mutation in untreated cells (orange), or by the eIF2AΔ mutation in SM-treated cells (green). Statistical significance determined using the Mann–Whitney U test is indicated for selective comparisons of changes observed for the indicated groups in comparison to the changes for all mRNAs. A few outliers were omitted from the plots to expand the y-axis scale. (B) Notched box plots as in (A) for the subsets of the same mRNA groups analyzed there exhibiting >1.41-fold increases in TE in SM-treated versus untreated WT cells. A few outliers were omitted from the plots to expand the y-axis scale. Statistical significance determined as in (A). (C) Notched box plots as in (A, B) for the subsets of the mRNA groups analyzed there exhibiting >1.41-fold decreases in TE in SM-treated eIF2AΔ versus SM-treated WT cells. A few outliers were omitted from the plots to expand the y-axis scale. Statistical significance determined as in (A). (D) Proportional Venn diagram showing overlap between the 17 mRNAs identified in Figure 3A showing evidence for a conditional requirement for eIF2A when eIF2 function is reduced by SM (marked in Figure 3A with ‘#’s) and the 514 mRNAs bearing functional AUG or NCC-uORFs.
-
Figure 6—source data 1
Spreadsheet 1–3, ‘Figure 6A(i)–(iii)’, tabulates the lists and log2ΔTE values for the indicated mutant/condition for the annotated AUG- or NCC-uORFs (Figure 6A(i)), conserved AUG- or NCC-uORFs (Figure 6A(ii)), or single functional inhibitory AUG-uORFs (Figure 6A(iii)) (Figure 6A).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data1-v1.xlsx
-
Figure 6—source data 2
Spreadsheet 1–3, Figure 6B(i)–(ii), tabulates the lists and log2ΔTE values as in Figure 6—source data 1 for the subsets of the same mRNA groups analyzed there exhibiting >1.41-fold increases in translational efficiency (TE) in sulfometuron methyl (SM)-treated versus untreated wild-type (WT) cells (Figure 6B).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data2-v1.xlsx
-
Figure 6—source data 3
Spreadsheet 1–3, ‘Figure 6C(i)–(iii)’, tabulates the lists and log2ΔTE values as in Figure 6—source data 1 for the subsets of the same mRNA groups analyzed there exhibiting >1.41-fold decreases in translational efficiency (TE) in sulfometuron methyl (SM)-treated eIF2AΔ versus SM-treated wild-type (WT) cells (Figure 6C).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data3-v1.xlsx
-
Figure 6—source data 4
Spreadsheet tabulates lists of the 514 mRNAs bearing functional AUG or NCC-uORFs, 17 mRNAs identified in Figure 3A showing evidence for a conditional requirement for eIF2A when eIF2 function is reduced by sulfometuron methyl (SM), that is, ∆TEeIF2A∆+SM/WT+SM_down* (N = 17) group, and overlap of mRNAs with functional AUG- or NCC-uORFs (N = 514) and ∆TEeIF2A∆+SM/WT+SM_down (N = 17) (Figure 6D).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data4-v1.xlsx
-
Figure 6—source data 5
Spreadsheet tabulates the list of annotated AUG- or NCC-uORFs, chromosome coordinates, start codon of uORF, distances of the uORF AUG from the 5′ end of the mRNA and the main coding sequences (CDS) start codon, and the gene name.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data5-v1.xlsx
-
Figure 6—source data 6
Spreadsheet tabulates the list of conserved AUG- or NCC-uORFs, chromosome coordinates, start codon of uORF, distances of the uORF AUG from the 5′ end of the mRNA and the main coding sequences (CDS) start codon, and the gene name.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data6-v1.xlsx
-
Figure 6—source data 7
Spreadsheet tabulates the list of the functional uORFs, chromosome coordinates, start codon of uORF, distances of the uORF AUG from the 5′ end of the mRNA and the main coding sequences (CDS) start codon, and the gene name (May et al., 2023).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-data7-v1.xlsx
Figure 6—figure supplement 1

eIF2A plays a minimal in regulating upstream open-reading frame (uORF)-mediated translation.
(A, B) Smoothed scatterplots displaying the relationship between log2RROWT (x-axis) and log2RROeIF2A∆ (y-axis) for all mRNAs containing annotated AUG- or NCC-uORFs (A) or evolutionarily conserved AUG- or NCC-uORFs (B) in wild-type (WT) versus eIF2AΔ cells without sulfometuron methyl (SM) treatment. No mRNAs showed ≥2-fold changes in RRO in the eIF2AΔ mutant versus WT cells at false discovery rate (FDR) < 0.5. (C, D) Smoothed scatterplots displaying the relationship between log2RROWT+SM (x-axis) versus log2RROeIF2A∆+SM (y-axis) for the same mRNAs analyzed in (A, B) but in the presence of SM. Again, no mRNAs showed ≥2-fold changes in RRO in the eIF2AΔ mutant versus WT cells at FDR < 0.5. (E–H) Notched box plot displaying log2RRO values for all mRNAs containing annotated AUG- or NCC-uORFs (E, G) or evolutionarily conserved AUG- or NCC-uORFs (F, H) in untreated WT and eIF2AΔ mutant (E, F) or SM-treated WT and eIF2AΔ mutant (G, H). The y-axis scale was expanded by omitting a few outliers. Statistical significance determined using the Mann–Whitney U test is shown for the bracketed comparisons in panels (E, G).
-
Figure 6—figure supplement 1—source data 1
Spreadsheet 1 tabulates the log2 ratios of the following parameters for all the expressed annotated uAUG or NCC-uORFs listed in column A in eIF2AΔ versus wild-type (WT) cells: relative ribosome occupancy (RRO) in eIF2AΔ versus WT cells (RRO Change), RRO for WT, and RRO of eIF2AΔ (Figure 6—figure supplement 1A and E).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-figsupp1-data1-v1.xlsx
-
Figure 6—figure supplement 1—source data 2
Spreadsheet 1 tabulates the log2 ratios of the following parameters for all the evolutionarily conserved expressed uAUG or NCC-uORFs listed in column A in eIF2AΔ versus wild-type (WT) cells: relative ribosome occupancy (RRO) in eIF2AΔ versus WT cells (RRO Change), RRO for WT, and RRO of eIF2AΔ (Figure 6—figure supplement 1B and F).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-figsupp1-data2-v1.xlsx
-
Figure 6—figure supplement 1—source data 3
Spreadsheet 1 tabulates the log2 ratios of the following parameters for all the expressed annotated uAUG or NCC-uORFs listed in column A in sulfometuron methyl (SM)-treated wild-type (WT) and SM-treated eIF2AΔ mutant: relative ribosome occupancy (RRO) in eIF2AΔ treated with SM versus WT cells treated with SM (RRO Change), RRO for WT treated with SM, and RRO of eIF2AΔ treated with SM (Figure 6—figure supplement 1C and G).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-figsupp1-data3-v1.xlsx
-
Figure 6—figure supplement 1—source data 4
Spreadsheet 1 tabulates the log2 ratios of the following parameters for all the evolutionarily conserved expressed uAUG or NCC-uORFs listed in column A in eIF2AΔ treated with sulfometuron methyl (SM) versus wild-type (WT) cells treated with SM: relative ribosome occupancy (RRO) in eIF2AΔ cells treated with SM versus WT cells treated with SM (RRO Change), RRO for WT treated with SM, and RRO of eIF2AΔ treated with SM (Figure 6—figure supplement 1D and H).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-figsupp1-data4-v1.xlsx
-
Figure 6—figure supplement 1—source data 5
Spreadsheet 1 tabulates the β-galactosidase activities in units of nmol of ONPG cleaved per mg of protein per min in wild-type (WT) and eIF2AΔ strains.
Additionally, the spreadsheet provides the fold change of means and the associated ± SEMs (standard error of the means) of these activities. These values have been calculated based on data obtained from three independent transformants for each condition (Figure 6—figure supplement 2B).
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig6-figsupp1-data5-v1.xlsx
Figure 6—figure supplement 2
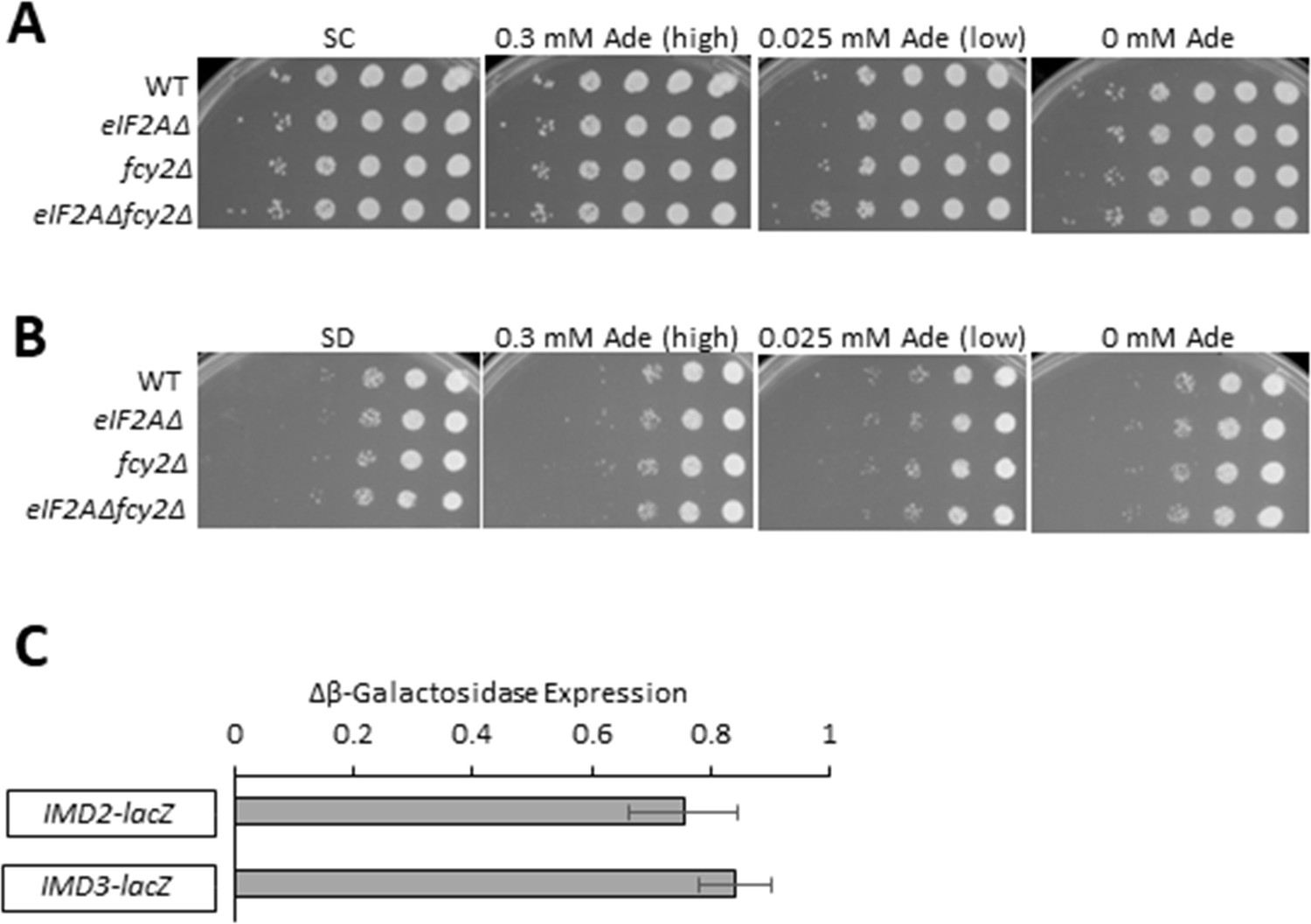
Lack of genetic interaction between eIF2A and the purine salvage pathway.
(A, B) Cell spotting assays were performed on synthetic complete (SC) plates (A) or synthetic minimal (SD) plates (B) to assess the growth of wild-type (WT), eIF2AΔ, fcy2Δ, and eIF2AΔ fcy2Δ strains. Ten-fold serial dilutions of saturated cultures were applied to SC or SD plates supplemented with the indicated concentrations of adenine and incubated at 30°C for 2 d. (C) WT and eIF2AΔ strains were transformed with the indicated lacZ reporter plasmids. Transformants were grown in SC-Ura (containing 0.015 mM adenine) to saturation and diluted into the same (fresh) medium and grown for 6 hr to A600 of ∼1.0. Whole-cell extracts (WCEs) were prepared and assayed for β-galactosidase activities, expressed in units of nmol of ONPG cleaved per mg of protein per min. The results represent the fold change of means (± SEMs) of activities (eIF2AΔ/WT) calculated from three independent transformants of each strain. The IMD2-lacZ reporter contains IMD2 coding sequences along with 1186 bp of 5’ non-coding sequences while the IMD3-lacZ reporter contains IMD3 coding sequences and 555 bp of 5’ non-coding DNA.
Figure 7

eIF2A plays no major role in stimulating translation elongation for particular tripeptide motifs.
(A) Scatterplot of average pause scores for 8006 tripeptide motifs, comparing the two biological replicates of ribosome profiling data for eIF2A∆ versus wild-type (WT) cells. Each dot on the plot represents a tripeptide motif. Pause scores were computed using a shift value of 18 nt from the 3′-end of the footprint, positioning the first codon of the tripeptide motif in the E site. (B) Scatterplot of average pause scores for 6267 tripeptide motifs, comparing the two biological replicates of sulfometuron methyl (SM)-treated versus untreated WT cells. All 351 detected motifs with valine codons in the A site are highlighted in red. Pause scores were computed as in (A).
-
Figure 7—source data 1
Spreadsheet 1 tabulates the pause scores for tripeptide motifs in eIF2AΔ cell, wild-type (WT) cells, and SM-treated WT cells.
- https://cdn.elifesciences.org/articles/92916/elife-92916-fig7-data1-v1.xlsx
Tables
Table 1
Yeast strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | GE Healthcare |
| BY4742 | MATα his3Δ1 leu2Δ0 lysΔ2 ura3Δ0 | GE Healthcare |
| F2247/4684 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ygr054wΔ::kanMX4 | GE Healthcare |
| F2379/191 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 fcy2Δ::kanMX4 | GE Healthcare |
| YSG1 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ygr054wΔ::hphMX4 | This study |
| YSG3 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 fcy2Δ::kanMX4 ygr054wΔ::hphMX4 | This study |
Table 2
Plasmids used in this study.
| Name | Description | Source/reference | ||
|---|---|---|---|---|
| p6586 | IMD2-lacZ | Escobar-Henriques and Daignan-Fornier, 2001 | ||
| p6587 | IMD3-lacZ | Escobar-Henriques and Daignan-Fornier, 2001 | ||
| p4430 | hphMX4 | Goldstein and McCusker, 1999 | ||
| F.LUC reporter plasmids containing native promoter and 5' UTR (nucleotides (nt) indicated) and first 20 codons of the main CDSs of the indicated genes | ||||
| p6593 | 600 nt 5' ncDNA + first 20 codons of SVL3 in pRS416 | This study | ||
| p6594 | 600 nt 5' nc DNA + first 20 codons of NET1 in pRS416 | This study | ||
| p6595 | 1080 nt 5' ncDNA + first 20 codons of RAD9 in pRS416 | This study | ||
| p6596 | 1080 nt 5' ncDNA + first 20 codons of SAG1 in pRS416 | This study | ||
| p6597 | 300 nt 5' ncDNA + first 20 codons of PUS7 in pRS416 | This study | ||
| p6598 | 720 nt 5' ncDNA + first 20 codons of DBF4 in pRS416 | This study | ||
| p6599 | 420 nt 5' ncDNA + first 20 codons of PBP4 in pRS416 | This study | ||
| p6600 | 600 nt 5' ncDNA + first 20 codons of HKR1 in pRS416 | This study | ||
| p6601 | 840 nt 5' ncDNA + first 20 codons of MET4 in pRS416 | This study | ||
| p6602 | 360 nt 5' ncDNA + first 20 codons of CHS5 in pRS416 | This study | ||
| p6603 | 540 nt 5' ncDNA + first 20 codons of DNA2 in pRS416 | This study | ||
| p6604 | 420 nt 5' ncDNA + first 20 codons of NSI1 in pRS416 | This study | ||
Table 3
Primers used in this study.
| Primer name | Primer sequence 5′ – 3′ |
|---|---|
| Primers for qRT-PCR | |
| Firefly Fwd | GTGTTGGGCGCGTTATTTATC |
| Firefly Rev | TAGGCTGCGAAATGTTCATACT |
| CHS5 Fwd | CTGTGGAGGATGCCAATGAA |
| CHS5 Rev | AGAGGCAATGTCGGTAGTAAAC |
| DBF4 Fwd | GCAAGGCAAGAAACTGAAGAAG |
| DBF4 Rev | GATGTGCACCACTTGCTTTG |
| HRK1 Fwd | GCCATACAGCTCTGTCCATT |
| HRK1 Rev | AGAGGAAACGGATGCAGATAAG |
| NET1 Fwd | TGCTTCAGCTTCTTCCTCTTC |
| NET1 Rev | GGTTACGGGTGGTTTCCTATT |
| SAG1 Fwd | CCATCCAGTCCCTCATCTTATAC |
| SAG1 Rev | TGGCACAGAAGGCGTAAA |
| SVL3 Fwd | CCTCCTACAACCTCTGTTTCAG |
| SVL3 Rev | GGTACTAGGCGAAGCCATATTC |
| 18S rRNA Fwd | TCACCAGGTCCAGACACAATAAG |
| 18S rRNA Rev | TCTCGTTCGTTATCGCAATTAAGC |
| ACT1 Fwd | TGTGTAAAGCCGGTTTTGCC |
| ACT1 Rev | GATACCTCTCTTGGATTGAGCTTC |
| Primers used for deletion verification and construction of double deletion strains | |
| KanB | CTGCAGCGAGGAGCCGTAAT |
| KanC | TGATTTTGATGACGAGCGTAAT |
| Hyg Fwd | CGGATCCCCGGGTTAATTAA |
| HygC Rev | GAATTCGAGCTCGTTTAAAC |
| Hyg-B | TTTCGATCAGAAACTTCTCGACA |
| Hyg-C | TGCTCCGCATTGGTCTTGACC |
| eIF2A-A | TTCAGCTTCATAGCGATTTATTTTC |
| eIF2A-B | CATATGATTAATTGAGCCGGTTTAC |
| eIF2A-C | AGAGTACATAAGTCAACACCCAAGC |
| eIF2A-D | GACACTCCATATTCATTTATTGCCT |
| FCY2-A | TATCATTTCCGCTTATCTGACTTCT |
| FCY2-B | CTAACCTTAACACCAACTTCCTCAA |
| FCY2-C | TTGAGGAAGTTGGTGTTAAGGTTAG |
| FCY2-D | AATCAGCAGATTCCATCAAAAGTAG |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Yeast eIF2A has a minimal role in translation initiation and uORF-mediated translational control in vivo
eLife 12:RP92916.
https://doi.org/10.7554/eLife.92916.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}