Bidirectional dysregulation of synaptic glutamate signaling after transient metabolic failure
- Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, Germany
- Department of Physics, University of South Florida, United States
- German Center for Neurodegenerative Diseases (DZNE), Germany
Figures
Figure 1
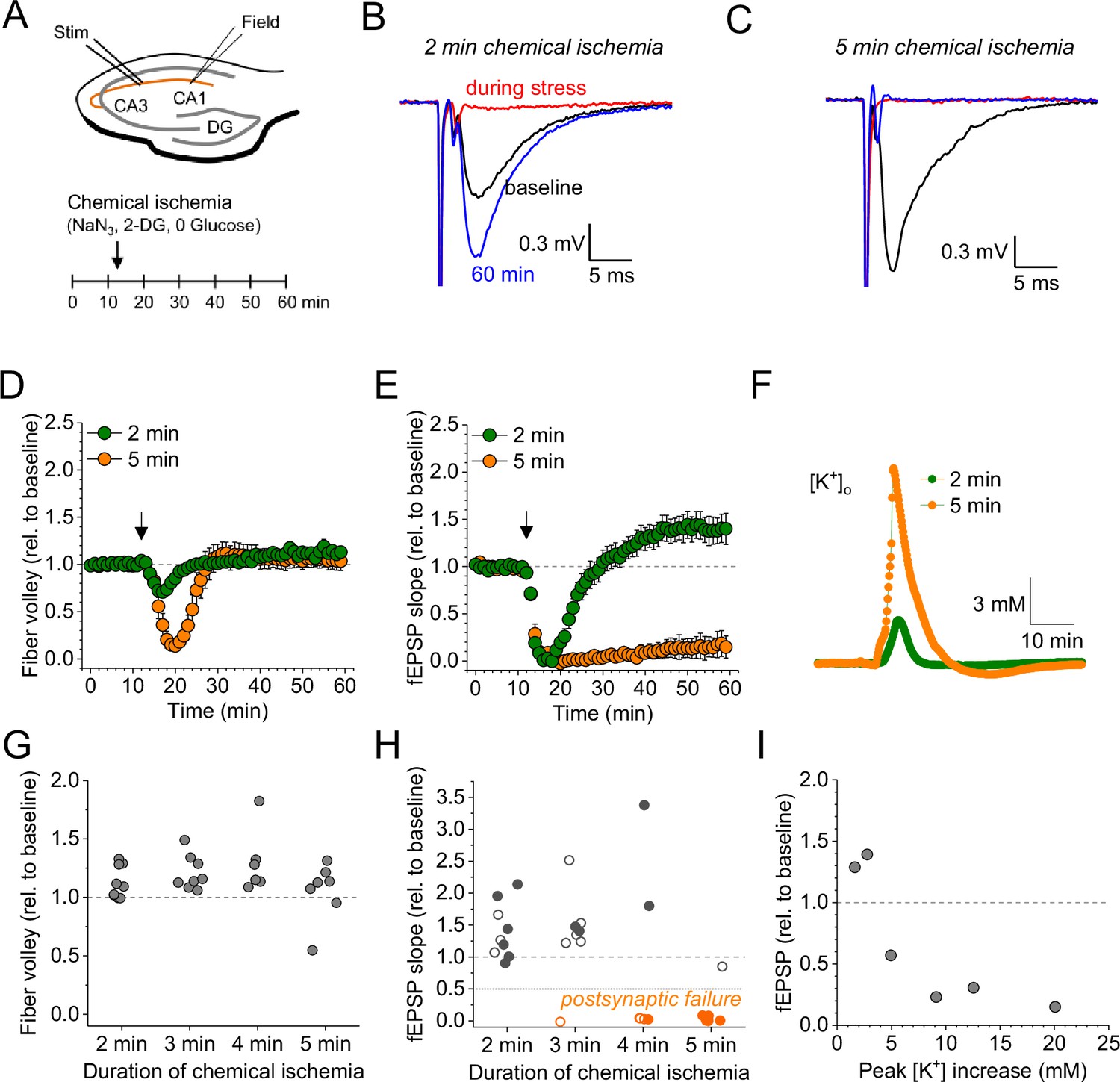
Duration-dependent and bidirectional effect of transient chemical ischemia on synaptic transmission.
(A) Schematic of experimental design. Extracellular field potentials (Field) were recorded in the CA1 region in response to Schaffer collateral stimulation (Stim; paired pulses, 50 ms interstimulus interval, every 20 s). Arrow indicates the time point of application of a modified artificial cerebral spinal fluid (ACSF), inducing acute chemical ischemia, with 0 mM glucose, 2 mM deoxyglucose (2-DG), 5 mM sodium azide (NaN3) for 2–5 min. (B–C) Example traces for 2 min chemical ischemia (B) and 5 min chemical ischemia (C) (black, baseline; red, during chemical ischemia; blue, end of the experiment at 60 min). (D) Relative change of the axonal fiber volley amplitude compared to baseline (0–10 min) for 2 min of chemical ischemia (green) and 5 min of chemical ischemia (orange). Arrow indicates time point of application of the modified ACSF for chemical ischemia induction. (E) Same as in (D) but for the field excitatory postsynaptic potential (fEPSP) slope. (F) Example traces of extracellular [K+] recordings during 2 and 5 min of chemical ischemia. (G) Quantification of the relative change of the axonal fiber volley amplitude in the last 10 min of recordings (50–60 min) relative to baseline for 2, 3, 4, and 5 min (n=9, 8, 6, and 7, respectively) of chemical ischemia. (H) Same experiments as in (G) but analysis of the fEPSP slope. Graphs in H and G include results from recording shown in B–E. Persistent failure of synaptic transmission highlighted in orange. Filled circles correspond to recordings, in which astrocytes expressed iGluSnFR (see Figure 2 and text). (I) Change of fEPSP in the last 10 min of recordings (50–60 min) relative to the baseline plotted against maximum [K+] increase during chemical ischemia. Data are expressed as mean ± s.e.m.
Figure 2 with 4 supplements
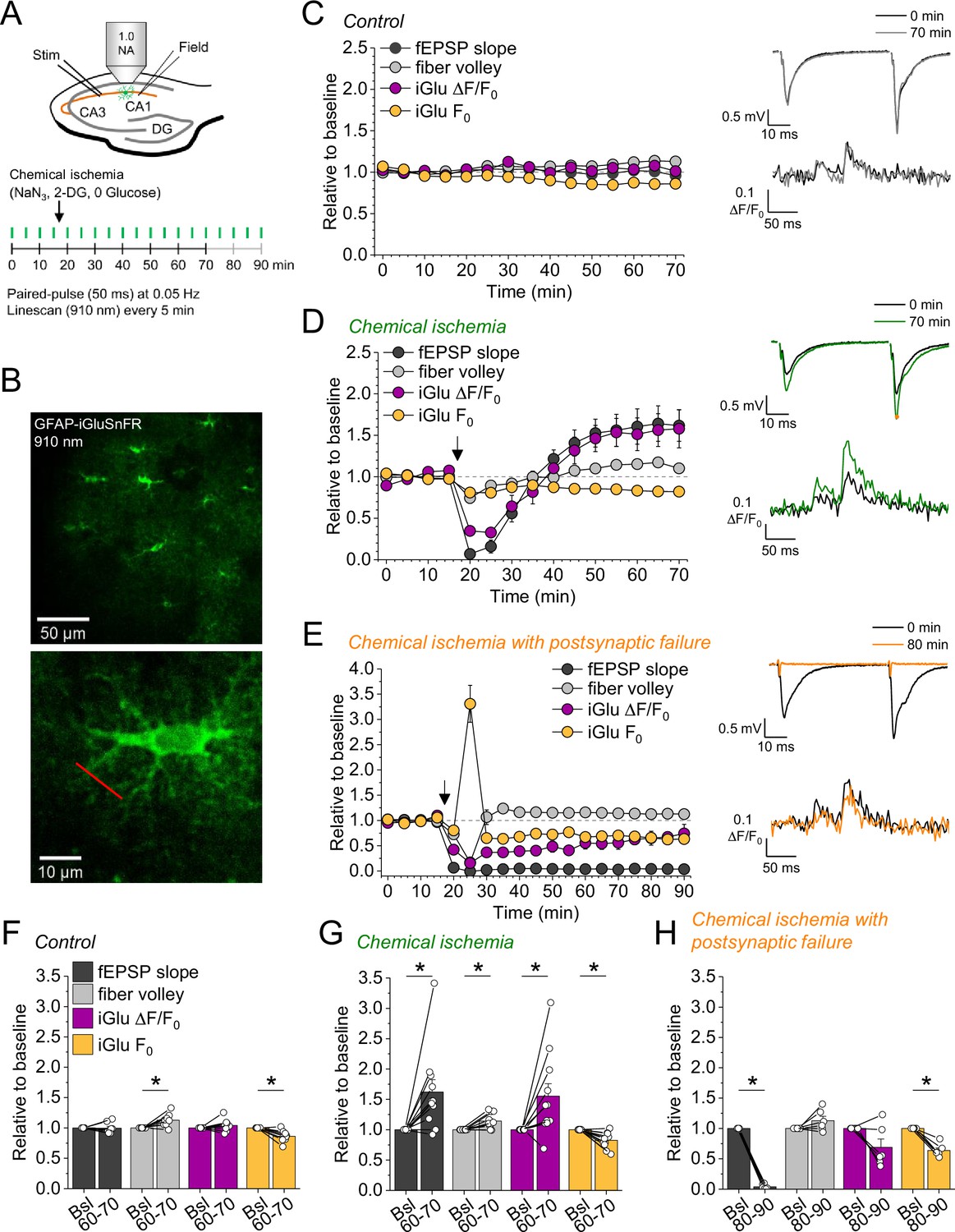
Bidirectional dysregulation of glutamate release by transient chemical ischemia.
(A) Schematic of experimental design. Combined recording of extracellular field potentials (Field) in the CA1 region in response to Schaffer collateral stimulation (Stim; paired pulses, interstimulus interval of 50 ms, every 20 s) and two-photon excitation fluorescence line scan imaging (910 nm, 6× every 5 min) of the glutamate indicator iGluSnFR. (B) Top: example of virally induced iGluSnFR expression by astrocytes in the stratum radiatum of the CA1 region. Bottom: single iGluSnFR-expressing astrocyte and representative location of line scan (red line). (C–E) Left, field excitatory postsynaptic potential (fEPSP) slope, axonal fiber volley, iGluSnFR (iGlu) ΔF/F0 and iGlu resting fluorescence (F0) relative to baseline (0–10 min) for control (n=8) (C), chemical ischemia (n=11) (D), and chemical ischemia followed by postsynaptic failure of synaptic transmission (n=6) (E). Arrow indicates start of chemical ischemia. Right, example traces of fEPSP (top) and iGluSnFR ΔF/F0 (bottom, average of 6 scans) at the beginning and the end of the recording. The electrophysiological results in (D and E) are a subset from Figure 1. (F–H) Summary of parameters analyzed in (C–E) in the last 10 min of recording (60–70/80–90 min) compared to baseline (Bsl). (F) Control: fEPSP slope, p=0.894; fiber volley, p=0.012; iGlu ΔF/F0, p=0.310; iGlu F0, p=0.006; n=8, paired Student’s t-test. (G) Chemical ischemia: fEPSP slope, p=0.013; fiber volley, p=0.003; iGlu ΔF/F0, p=0.024; iGlu F0, p=0.001; n=11, paired Student’s t-test. (H) Chemical ischemia with postsynaptic failure: fEPSP slope, p<0.0001; fiber volley, p=0.134; iGlu ΔF/F0, p=0.073; iGlu F0, p=0.0004; n=6, paired Student’s t-test. Data are expressed as mean ± s.e.m.
Figure 2—figure supplement 1

Stimulus-response relationship of field excitatory postsynaptic potential (fEPSP) slope and iGluSnFR ΔF/F0 before and after chemical ischemia.
Further analyses of recordings presented in Figure 2. (A) fEPSP slope in response to increasing stimulation intensities before and after control recordings (left, n=8), recordings with chemical ischemia (middle, n=9), and chemical ischemia with postsynaptic failure (right, n=5). For each stimulation intensity two sweeps of a paired-pulse (50 ms, first and second pulse) protocol were recorded. (B) Same as in (A) but for iGluSnFR ΔF/F0 amplitude in response to first and second pulse. (C) Example traces of iGluSnFR ΔF/F0 recordings before (top row) and after (bottom row) control (left), chemical ischemia (middle), and chemical ischemia with postsynaptic failure (right) for 50, 100, 200, and 400 µA stimulation intensity (average of 2 scans/stimulation intensity). Data are expressed as mean ± s.e.m.
Figure 2—figure supplement 2

Repeated chemical ischemia.
In a single experiment we have been able to induce acute chemical ischemia twice with stable electrophysiological and imaging for more than 3 hr. Note the persistent potentiation of the field excitatory postsynaptic potential (fEPSP) slope and glutamate transients. (A) Time course of the indicated parameters (also see Figure 2) including the resting iGluSnFR fluorescence before the stimulus (F0) and its absolute change after the stimulus (ΔF). The two dips at 20 and 100 min indicate the two time points of chemical ischemia. (B) Example electrophysiological traces at the color-coded time points. (C) Example of iGluSnFR line scans at the color-coded time points.
Figure 2—figure supplement 3

Dynamic range of iGluSnFR is not affected by chemical ischemia without postsynaptic failure.
(A) Left, example of an iGluSnFR-expressing astrocyte in the stratum radiatum of the CA1 region and iontophoresis micropipette (red, dotted line) filled with glutamate (150 mM) and Alexa Fluor 633 (40 µM). Middle, example of two-photon excitation line scan imaging (910 nm) of iGluSnFR fluorescence right in front of the iontophoresis micropipette tip in the periphery of an astrocyte (left, white line) during glutamate ejection by a 100 nA current (250 ms) pulse to saturate the iGluSnFR sensor. Right, change in iGluSnFR fluorescence (ΔF/F0) of line scan (middle, white dotted box). (B) Quantification of maximum ΔF/F0 amplitude during a saturating iontophoretic glutamate application pulse for control, chemical ischemia, and chemical ischemia with postsynaptic failure (+post. failure). p=0.031, one-way ANOVA; control vs. chemical ischemia, p=0.641; control vs. chemical ischemia with postsynaptic failure. p=0.024; chemical ischemia vs. chemical ischemia with postsynaptic failure, p=0.119; n=18, 19, and 9 from left to right; post hoc Tukey test. Data are expressed as mean ± s.e.m.
Figure 2—figure supplement 4

The configuration of the extracellular space (ECS) is only affected after severe chemical ischemia.
(A) Schematic of experimental design. Iontophoretically induced TMA+ transients (Iontophoresis, 30 s every 2 min) were detected with a TMA-sensitive microelectrode (TSM) in the CA1 region during control recordings, and recordings with chemical ischemia without and with postsynaptic failure. Schaffer collaterals were stimulated as before (Stim; paired pulse [50 ms], 0.05 Hz). (B–D) Left, relative change of ECS fraction and diffusivity (D*) compared to baseline (0–10 min) for control (B), chemical ischemia (C), and chemical ischemia with postsynaptic failure (D). Arrow indicates nominal start of chemical ischemia. Right, example traces of TMA+ transients during baseline and last 5 min of recording (65–70 min) with corresponding fits (red). (E) Quantification of the ECS fraction in the last 10 min of recording (60–70 min) compared to baseline (Bsl). Control: p=0.210, n=6; chemical ischemia: p=0.297, n=6; chemical ischemia with postsynaptic failure: p=0.0008, n=6; paired Student’s t-test. (F) As in (E) but for diffusivity (D*). Control: p=0.0063; chemical ischemia: p=0.0105; chemical ischemia with postsynaptic failure: p=0.0002; paired Student’s t-test. One-way ANOVA of effects after 60–70 min: p<0.0001. Post hoc Tukey tests: control 60–70 min vs. chemical ischemia 60–70 min, p=0.990; control 60–70 min vs. chemical ischemia with postsynaptic failure 60–70 min, p<0.0001; chemical ischemia 60–70 min vs. chemical ischemia with postsynaptic failure 60–70 min, p=0.0001. Data are expressed as mean ± s.e.m. TMA, tetramethylammonium.
Figure 3
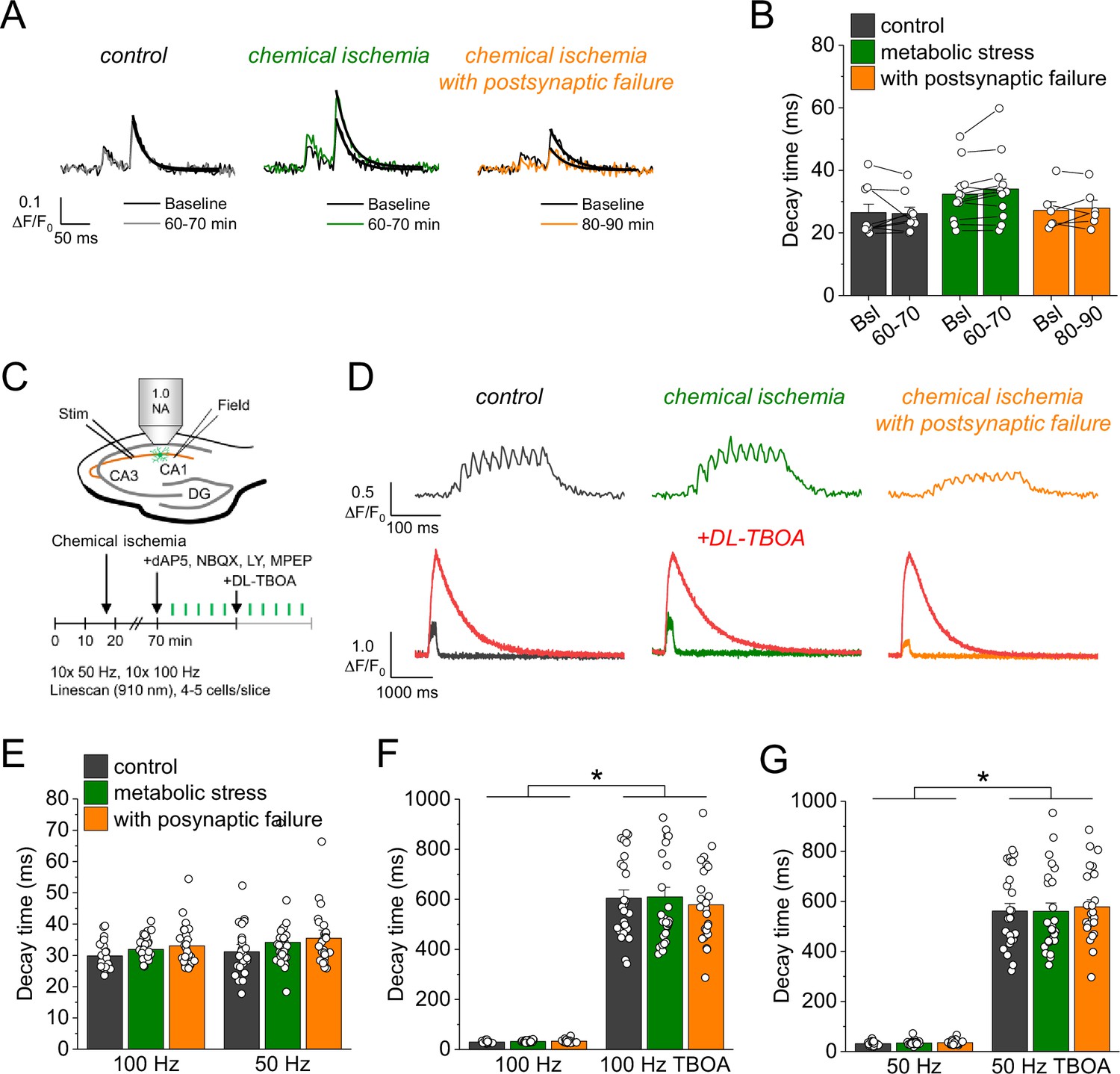
Glutamate clearance is not affected by transient chemical ischemia.
(A) Example traces of iGluSnFR ΔF/F0 in response to paired-pulse stimulation (interstimulus interval 50 ms) during baseline (black) and last 10 min (60–70/80–90 min) of control (left, gray), chemical ischemia (middle, green), and chemical ischemia with postsynaptic failure (right, orange) recordings. Black line indicates exponential fit for the analysis of iGluSnFR decay time. (B) Quantification of iGluSnFR fluorescence decay time of the second pulse of the paired-pulse stimulation during the last 10 min of recording (60–70/80–90 min) compared to baseline (Bsl) for control, chemical ischemia, and chemical ischemia with postsynaptic failure. Control: p=0.860, n=9; chemical ischemia, p=0.062, n=12; chemical ischemia with postsynaptic failure, p=0.648, n=6; paired Student’s t-tests. (C) Schematic of experimental design. Combined recording of extracellular field potentials (Field) in the CA1 region in response to Schaffer collateral stimulation (Stim; paired pulse [50 ms], 0.05 Hz) and two-photon excitation fluorescence line scan imaging (910 nm) of iGluSnFR. After 70 min, D-AP5 (50 µM), NBQX (20 µM), LY341495 (50 µM), and MPEP (10 µM) were added to the recording solution and iGluSnFR fluorescence changes (ΔF/F0) were recorded in 4–5 cells in response to 10× 50 Hz and 10× 100 Hz stimulation before and after application of DL-TBOA (100 µM). (D) Example traces of iGluSnFR ΔF/F0 line scan recordings (average of 6 scans) in response to 10× 50 Hz stimulation (top row) after control (left), chemical ischemia (middle), and chemical ischemia with postsynaptic failure recordings (right). The same cells were tested again after block of glutamate transporters by DL-TBOA (bottom row, red traces together with ‘before’ traces from upper row on different timescale for comparison). (E) Quantification of iGluSnFR fluorescence decay time in response to 10× 100 Hz and 10× 50 Hz stimulation after control, chemical ischemia recording without and with postsynaptic failure. 100 Hz: p=0.060, 50 Hz: p=0.149; n=25, 29, and 25 cells for control/chemical ischemia/chemical ischemia with postsynaptic failure from 5, 6, and 5 independent experiments, respectively; Kruskal-Wallis ANOVA. (F) Quantification of iGluSnFR fluorescence decay time in response to 10× 100 Hz stimulation in the presence and absence of DL-TBOA (same cells as in E). p<0.0001 for control, chemical ischemia, and chemical ischemia with postsynaptic failure, paired sample Wilcoxon signed-rank tests. (G) As in F but for 10× 50 Hz stimulation. p<0.0001 for control, chemical ischemia, and chemical ischemia with postsynaptic failure, paired sample Wilcoxon signed-rank tests. Data are expressed as mean ± s.e.m.
Figure 4 with 2 supplements
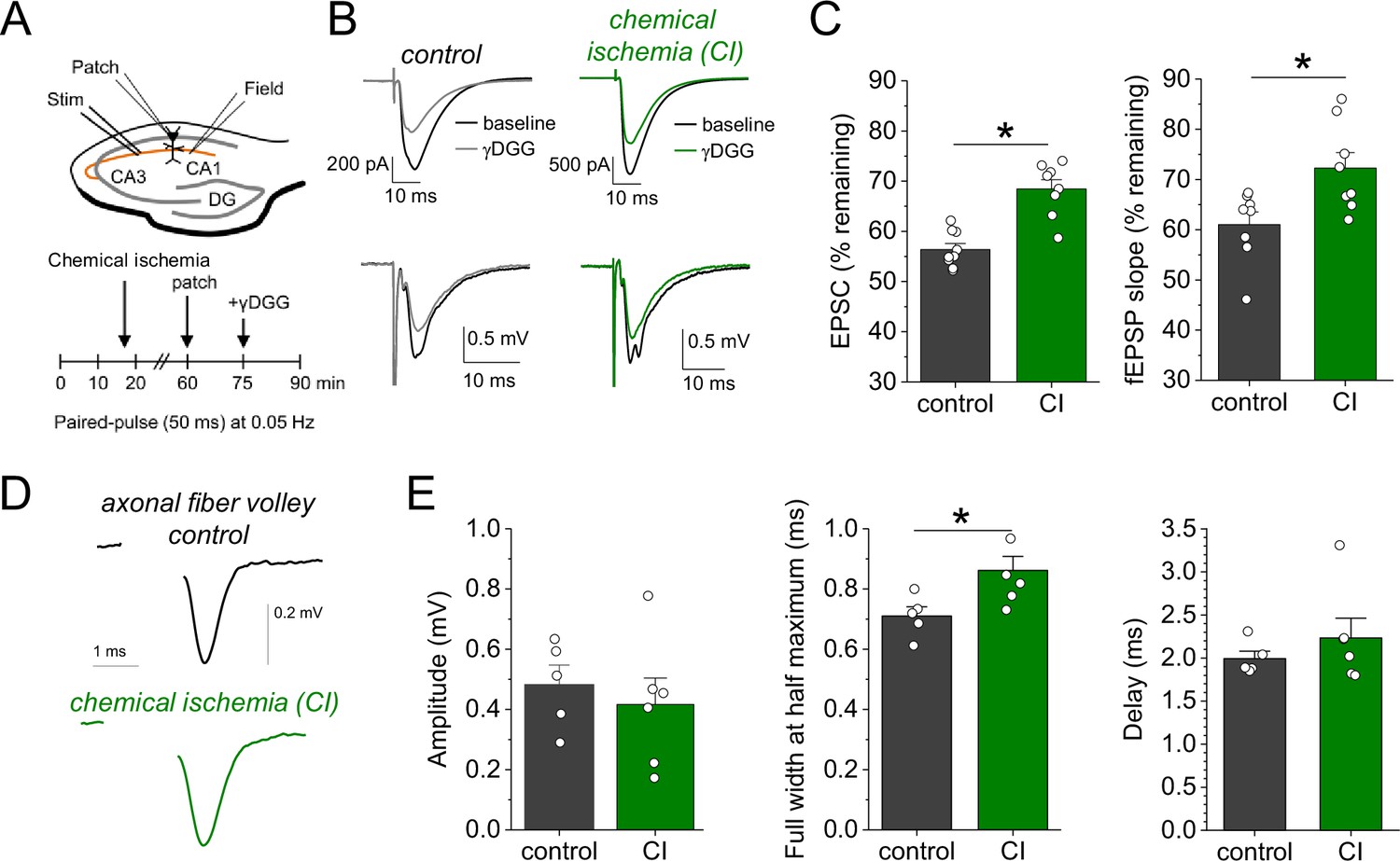
Chemical ischemia increases synaptic glutamate release.
(A) Schematic of experimental design. Extracellular field potentials (Field) were recorded in the CA1 region in response to Schaffer collateral stimulation (Stim; paired pulse [50 ms], 0.05 Hz). After 60 min, a whole-cell patch clamp recording from a CA1 pyramidal neuron was started. After recording baseline excitatory postsynaptic current (EPSC) responses for 15 min, γ-D-glutamylglycine (γ-DGG) (1 mM) was added to the extracellular solution. (B) Example traces of EPSCs (top) and field excitatory postsynaptic potentials (fEPSPs) (bottom) before (black) and after γ-DGG application for control (left, gray) and chemical ischemia (right, green) recordings. (C) Quantification of the remaining EPSC amplitude (left) and fEPSP slope (right) (in % of the baseline value) after γ-DGG application for control (n=9) and chemical ischemia (n=8) recordings. EPSC, p<0.0001; fEPSP, p=0.014; paired two-sample Student’s t-test. Data are expressed as mean ± s.e.m. (D) Example of axonal fiber volleys from experiments shown in Figure 3C–E. Stimulus artifact removed for clarity. (E) Comparison of the fiber volley amplitude (left, p=0.57), fiber volley half width at half maximum (middle, p=0.026), and fiber volley delay (time between onset of the stimulus and fiber volley peak, right, p=0.39). n=5 (control) and 6 (chemical ischemia), unpaired Student’s t-tests.
Figure 4—figure supplement 1

Effect of transient chemical ischemia on short-term synaptic plasticity.
(A) Example traces of field excitatory postsynaptic potentials (fEPSPs) (top) and iGluSnFR ΔF/F0 fluorescence (bottom, average of 6 scans) before (black) and after (green) chemical ischemia without postsynaptic failure peak-scaled to baseline trace (same examples as in Figure 2D but peak-scaled). (B) Quantification of paired-pulse ratio (PPR) of the fEPSP slope (second fEPSP slope/first fEPSP slope) during the last 10 min of the recording (60–70 min) compared to baseline (Bsl) for control and chemical ischemia. Loss of the fEPSP in postsynaptic failure prevents analysis in these recordings. Control: p=0.082, n=12; chemical ischemia, p=0.033, n=14; paired Student’s t-tests. (C) Same as in (B) but for PPR of iGluSnFR ΔF/F0 amplitudes. Control: p=0.256, n=9; chemical ischemia, p=0.065, n=12; chemical ischemia with postsynaptic failure, p=0.454, n=5; paired Student’s t-tests. (D) Relative change of PPR compared to baseline for fEPSP slopes and iGluSnFR ΔF/F0 amplitudes for control and chemical ischemia. fEPSP slope: p=0.127, n=12 and 14; iGlu, p=0.933, n=9 and 12; two-sample Student’s t-test (with Welch correction for unequal variances for the fEPSP slopes). Data are expressed as mean ± s.e.m. We conclude that there is no relevant change of short-term synaptic plasticity due to chemical ischemia because the reduction of the electrophysiological PPR compared to baseline is not statistically different from that observed in control recordings (B, D) and there is no statistically significant change of the PPR of iGluSnFR transients (C, D).
Figure 4—figure supplement 2

Reduced glutamate concentration in synaptic vesicles in conditions typical of chemical ischemia.
Evolution of the number of glutamate molecules in synaptic vesicles under normal condition (control, black), reduced cytosolic pH (pHC, red), higher Na+ and Cl- and lower K+ concentration in the cytoplasm (blue), lower extracellular pH (pHE, green), and reduced vesicular ATPase (VATPase) activity (pink). For illustration, the effect of a higher pHC is displayed (cyan), which is however not expected to occur in chemical ischemia.
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Bidirectional dysregulation of synaptic glutamate signaling after transient metabolic failure
eLife 13:RP98834.
https://doi.org/10.7554/eLife.98834.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}