Genetics: MicroRNAs get to the heart of development
- Baylor College of Medicine, United States
- Baylor College of Medicine and the Texas Heart Institute, United States
The heart is the first organ to form in mammalian embryos. In mice, the heart starts to pump blood shortly after embryonic day 8 (reviewed in Chen and Wang, 2012), and this primitive circulatory system is vital for ensuring the proper development of the embryo. Many different cell types coexist within the heart, such as muscle cells, vascular cells and pacemaker cells (reviewed in Chien et al., 2008). These different types of cell arise from a common pool of progenitor cells, but the details of this process and how these cells are maintained within the mature heart have puzzled cardiovascular scientists. Now, in eLife, Kathryn Ivey, Deepak Srivastava and co-workers at the Gladstone Institute of Cardiovascular Disease—including Amy Heidersbach as first author—have offered some important clues about the underlying mechanisms (Heidersbach et al., 2013).
The development of the heart is regulated at both the transcriptional and post-transcriptional level; regulation at the post-transcriptional level involves small non-coding RNA molecules called microRNAs (miRs). These molecules, which are evolutionarily conserved, regulate gene expression by binding to specific sequences within the messenger RNAs and reducing their stability or preventing them from being translated into proteins. Precise miR activity is required for the heart to develop normally, and for it to be able to respond to challenges such as insufficient blood supply, and pressure overload (the increased stress that develops in the left ventricular wall when the heart pumps blood). Currently known cardiac enriched miRs include miR-1, 133, 206, 208 and 499 (reviewed in Chen and Wang, 2012), with miR-1 being the most abundant in the adult mouse heart.
MiR-1 is co-transcribed with miR-133a, and has two copies in the mouse genome, miR-1-1 on chromosome 2 and miR-1-2 on chromosome 18. Srivastava and co-workers have previously investigated the role of miR-1-2 during the development and maintenance of the heart (Zhao et al., 2007). In the current study, they have also involved its close relative, miR-1-1, in order to investigate the consequences of complete loss of miR-1 in the mammalian heart, and to identify any functions of miR-1 that are dependent on the amount of miR present.
Both miR-1-1 and miR-1-2 give rise to identical mature miR-1 species. Accordingly, targeted deletion of miR-1-1 results in a phenotype similar to that described for miR-1-2-null mice. However, the genetic background of the knock-out mice seems to affect the phenotype, as indicated by results from Heidersbach et al. and also those of another group who observed no phenotypic changes in miR-1-133 knock-out mice (Wystub et al., 2013). Nevertheless, both groups show that mice lacking all miR-1 copies (miR-1 null) die before weaning due to developmental defects. Heidersbach et al. observed abnormalities in mitochondria (the cell’s energy-producing organelles) as well as dysfunctional sarcomeres—the basic functional units of muscle fibres, which consist of thick filaments formed from the protein myosin and thin filaments made up of the protein actin.
Heidersbach et al. identified a gene called myosin light chain kinase (mlck) as being a direct target of miR-1. The mlck gene encodes an enzyme that adds phosphate groups to myosin molecules, and levels of this enzyme were increased in miR-1 null mice. However, this increased level of mlck was accompanied by a reduction, rather than an increase, in the phosphorylation of its substrate, the myosin light chain. This apparent contradiction was resolved by the discovery that miR-1 deletion leads to increased expression of an alternative form of MLCK, known as telokin, which lacks catalytic activity. Telokin is normally found only in smooth muscle—the type of weakly contractile muscle that lines the gut and the blood vessels—and its increased expression prevented MLCK kinase from phosphorylating myosin.
However, the fact that telokin alone was primarily induced—and not other forms of mlck, which all have regulatory sequences that are recognized by miR-1—implies that this might not be the whole story. Likewise, the dramatic increase in mlck expression (roughly fourfold) also suggests involvement of a more complex regulatory mechanism, since miRs generally change the expression level of their target messenger RNA by only 20–50% (reviewed in van Rooij and Olson, 2012).
When Heidersbach et al. analyzed the genes that were up-regulated in the hearts of miR-1 null mice, they found that levels of a protein called smMYOCD were dramatically increased. This protein, known as ‘smooth muscle version of Myocardin’, interacts with a transcription factor called SRF, which regulates the expression of genes that determine the characteristics of cardiac and smooth muscle. It turns out that smMYOCD is a direct target of miR-1. Moreover, the smMYOCD-SRF complex is more effective at promoting transcription of telokin than the cardiac version of Myocardin is. This completes a negative feedback loop that ensures that heart cells have a cardiac rather than smooth muscle phenotype: miR-1 suppresses smMYOCD, promoting the expression of the cardiac enzyme mlck, rather than smooth muscle genes such as telokin, resulting in phosphorylation of myosin and the formation of sarcomeres (Figure 1).
Figure 1
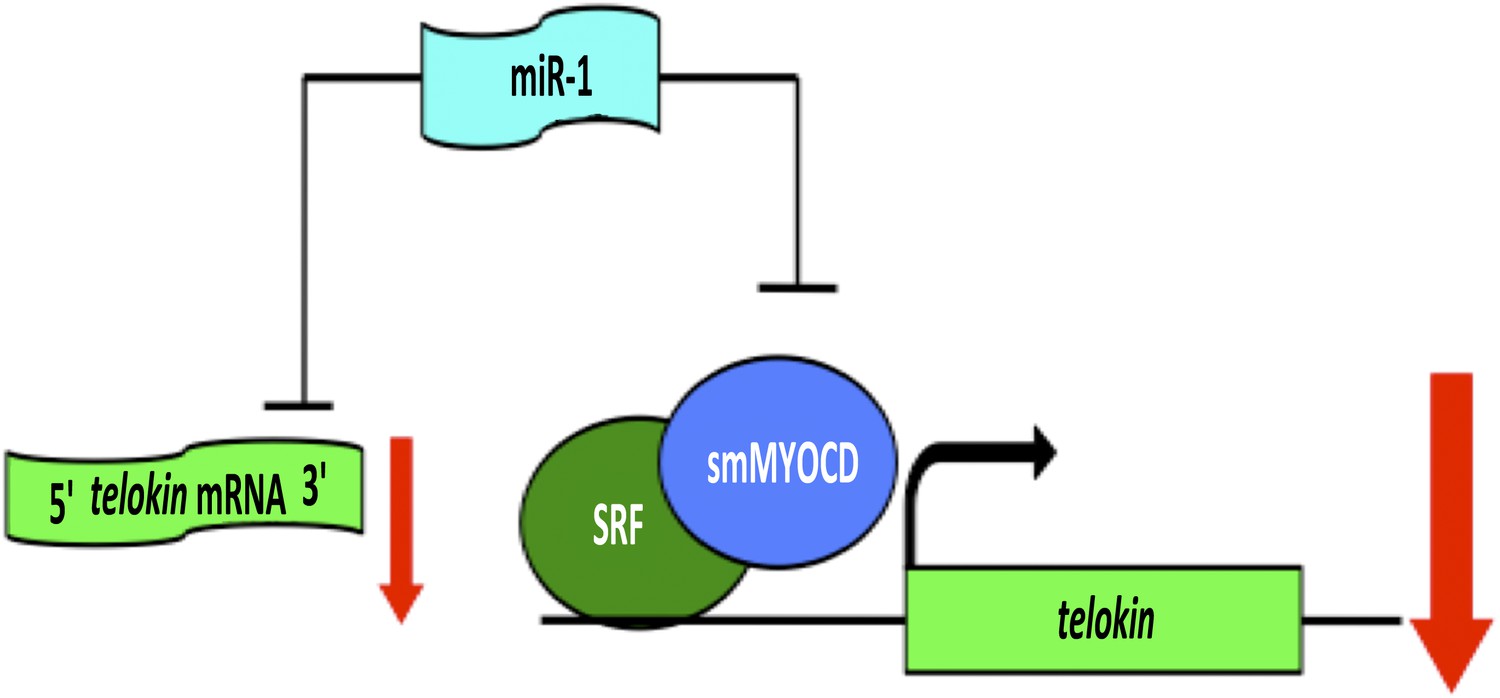
A network of genes regulated by a microRNA controls muscle development within the heart.
The formation of highly contractile cardiac muscle, as opposed to weakly contractile smooth muscle, depends on the inhibition of a protein called telokin by a microRNA called miR-1. In addition to directly inhibiting the expression of telokin (left), miR-1 also inhibits the expression of telokin indirectly by reducing the expression of a protein called smMYOCD that, working with a transcription factor called SRF, activates the transcription of telokin (right). This combination of two different regulatory mechanisms guarantees that telokin will be inhibited in heart cells, ensuring the formation of highly contractile cardiac muscle.
This impressive feat of molecular sleuthing by Heidersbach et al. raises a number of important points that go beyond this specific story. Theoretically, miRs can target hundreds of genes across the genome due to the short target sequences (6–8 nucleotides) they recognize. The working model built by Heidersbach et al. illustrates how networks of genes regulated by miRs can indirectly control the expression of a critical gene in any tissue (Figure 1). Direct miR-mediated repression of a transcriptional activator for a target gene amplifies the influence of that miR on its target. In the example described by Heidersbach et al., the feedback loop comprised of miR-1, SRF and smMYOCD guarantees the sensitive and powerful regulation of telokin in response to environmental and pathological stimuli, and probably makes the heart more robust.
Heidersbach et al. also reveal the importance of computational methods to unwind the complex web woven by miRs in organ development and homeostasis. Future progress in this area will require creative computational approaches to help discover new mechanisms and principles of miR-regulated gene expression.
References
-
microRNAs in cardiovascular developmentJ Mol Cell Cardiol 52:949–957.https://doi.org/10.1016/j.yjmcc.2012.01.012
-
MicroRNA therapeutics for cardiovascular disease: opportunities and obstaclesNat Rev Drug Discov 11:860–872.https://doi.org/10.1038/nrd3864
Article and author information
Author details
Publication history
Copyright
© 2013, Tao and Martin
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 445
- views
-
- 45
- downloads
-
- 12
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Genetics: MicroRNAs get to the heart of development
eLife 2:e01710.
https://doi.org/10.7554/eLife.01710
Further reading
-
- Chromosomes and Gene Expression
- Developmental Biology
Differentiation of female germline stem cells into a mature oocyte includes the expression of RNAs and proteins that drive early embryonic development in Drosophila. We have little insight into what activates the expression of these maternal factors. One candidate is the zinc-finger protein OVO. OVO is required for female germline viability and has been shown to positively regulate its own expression, as well as a downstream target, ovarian tumor, by binding to the transcriptional start site (TSS). To find additional OVO targets in the female germline and further elucidate OVO’s role in oocyte development, we performed ChIP-seq to determine genome-wide OVO occupancy, as well as RNA-seq comparing hypomorphic and wild type rescue ovo alleles. OVO preferentially binds in close proximity to target TSSs genome-wide, is associated with open chromatin, transcriptionally active histone marks, and OVO-dependent expression. Motif enrichment analysis on OVO ChIP peaks identified a 5’-TAACNGT-3’ OVO DNA binding motif spatially enriched near TSSs. However, the OVO DNA binding motif does not exhibit precise motif spacing relative to the TSS characteristic of RNA polymerase II complex binding core promoter elements. Integrated genomics analysis showed that 525 genes that are bound and increase in expression downstream of OVO are known to be essential maternally expressed genes. These include genes involved in anterior/posterior/germ plasm specification (bcd, exu, swa, osk, nos, aub, pgc, gcl), egg activation (png, plu, gnu, wisp, C(3)g, mtrm), translational regulation (cup, orb, bru1, me31B), and vitelline membrane formation (fs(1)N, fs(1)M3, clos). This suggests that OVO is a master transcriptional regulator of oocyte development and is responsible for the expression of structural components of the egg as well as maternally provided RNAs that are required for early embryonic development.
-
- Developmental Biology
Over the past several decades, a trend toward delayed childbirth has led to increases in parental age at the time of conception. Sperm epigenome undergoes age-dependent changes increasing risks of adverse conditions in offspring conceived by fathers of advanced age. The mechanism(s) linking paternal age with epigenetic changes in sperm remain unknown. The sperm epigenome is shaped in a compartment protected by the blood-testes barrier (BTB) known to deteriorate with age. Permeability of the BTB is regulated by the balance of two mTOR complexes in Sertoli cells where mTOR complex 1 (mTORC1) promotes the opening of the BTB and mTOR complex 2 (mTORC2) promotes its integrity. We hypothesized that this balance is also responsible for age-dependent changes in the sperm epigenome. To test this hypothesis, we analyzed reproductive outcomes, including sperm DNA methylation in transgenic mice with Sertoli cell-specific suppression of mTORC1 (Rptor KO) or mTORC2 (Rictor KO). mTORC2 suppression accelerated aging of the sperm DNA methylome and resulted in a reproductive phenotype concordant with older age, including decreased testes weight and sperm counts, and increased percent of morphologically abnormal spermatozoa and mitochondrial DNA copy number. Suppression of mTORC1 resulted in the shift of DNA methylome in sperm opposite to the shift associated with physiological aging – sperm DNA methylome rejuvenation and mild changes in sperm parameters. These results demonstrate for the first time that the balance of mTOR complexes in Sertoli cells regulates the rate of sperm epigenetic aging. Thus, mTOR pathway in Sertoli cells may be used as a novel target of therapeutic interventions to rejuvenate the sperm epigenome in advanced-age fathers.




{kind=link}