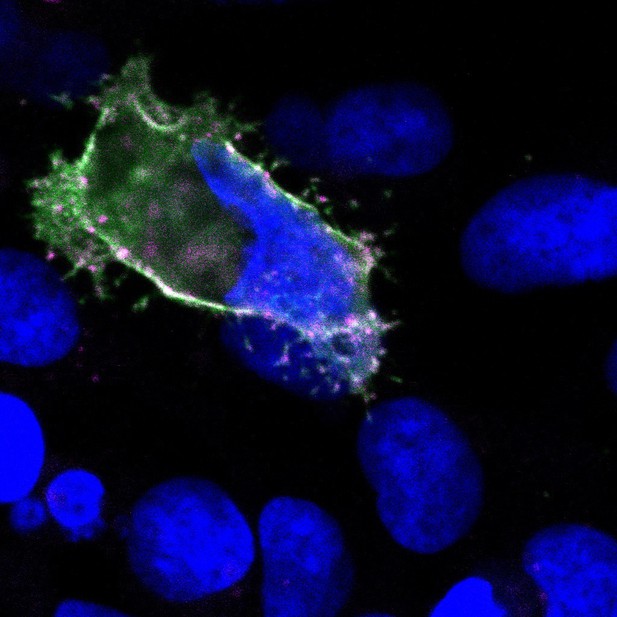
Image of a human kidney cell which has PC-1 (purple) and PC-2 (green) proteins inserted into its plasma membrane. Image credit: Markus Delling (CC BY 4.0)
On the surface of most animal and other eukaryotic cells are small rod-like protrusions known as primary cilia. Each cilium is encased by a specialized membrane which is enriched in protein complexes that help the cell sense its local environment. Some of these complexes help transport ions in out of the cell, while others act as receptors that receive chemical signals called ligands. A unique ion channel known as the polycystin complex is able to perform both of these roles as it contains a receptor called PC-1 in addition to an ion channel called PC-2.
Various mutations in the genes that code for PC-1 and PC-2 can result in autosomal dominant polycystic kidney disease (ADPKD), which is the most common monogenetic disease in humans. However, due to the small size of primary cilia – which are less than a thousandth of a millimeter thick – little is known about how polycystin complexes are regulated and how mutations lead to ADPKD. To overcome this barrier, Ha et al. modified kidney cells grown in the lab so that PC-1 and PC-2 form a working channel in the plasma membrane which surrounds the entire cell. As the body of a cell is around 10,000 times bigger than the cilium, this allowed the movement of ions across the polycystin complex to be studied using conventional techniques.
Experiments using this newly developed assay revealed that a region at one of the ends of the PC-1 protein, named the C-type lectin domain, is essential for stimulating polycystin complexes. Ha et al. found that this domain of PC-1 is able to cut itself from the protein complex. Further experiments showed that when fragments of PC-1, which contain the C-type lectin domain, are no longer bound to the membrane, they can activate the polycystin channels in cilia as well as the plasma membrane. This suggests that this region of PC-1 may also act as a secreted ligand that can activate other polycystin channels.
Some of the genetic mutations that cause ADPKD likely disrupt the activity of the polycystin complex and reduce its ability to transport ions across the cilia membrane. Therefore, the cell assay created in this study could be used to screen for small molecules that can restore the activity of these ion channels in patients with ADPKD.



